Introduction
Hepatitis B virus (HBV) infection is a significant
health problem worldwide, with approximately 325 million people
living with chronic HBV infection (CHB) (1). Approximately 10% of people carrying
HBV die as a direct consequence of persistent viral infection
(2). The chronic stage of this
disease (i.e., CHB) can progress into liver cirrhosis (LC) or
hepatocellular carcinoma (HCC) under the influence of several
factors, including HBV DNA levels, genomic mutations, viral
genotype, or viral subtype (3–5).
Better understanding of these factors, especially the dynamic
variations in HBV, should allow researchers to develop an effective
strategy to reduce the burden of HBV infection.
HBV is a partially double-stranded DNA virus with
four main overlapping open reading frames (ORFs) that encode the
polymerase, surface, X, and pre-core/core proteins (6–9). The
high mutation rate of HBV is a consequence of the lack of
proofreading function of its polymerase gene (8). The estimated mutation rate in
hepadnaviral genomes is 2×104 base
substitutions/site/year (2,8). The
high mutation rate of this virus produces diverse viral
populations, commonly known as quasispecies (10,11).
Many studies have indicated that the emergence of mutation-derived
quasispecies is correlated with clinical outcomes and disease
progression (9,10,12).
Viral quasispecies usually differentiate into minor
(1–5% of the total population), intermediate (5–20% of the total
population), and major (>20% of the entire population)
populations (9). If variants in
the major population account for >80% of quasispecies, it is
likely that the modified mutants will ultimately replace the
wild-type strain in a process that may take 50 years. Some of the
variations are probably present at the time of HBV infection
(9,13,14).
Some variants identified in the major population are relatively
stable in the absence of the wild-type strain (13,14).
However, if modifications in 20–80% of the entire population, it is
likely that there is a mixture of wild-type and variant strains in
the viral population that may be selected as a consequence of
antiviral drugs and the host's immune responses (9,15).
It was reported that a high frequency of variants is probably
driven by interactions between the host's immune responses and the
virus itself during disease progression (9).
In Indonesia, variations in the HBV genome,
including changes in BCP (A1762T, G1764A, and T1753V), the pre-S
region, and the major hydrophilic region, have been correlated with
advanced liver disease (ALD) (3,10,16,17).
However, no studies have investigated whether variations in HBV
quasispecies are associated with the progression of ALD. In the
present study, we used high-throughput sequencing to detect
whole-genome quasispecies variations in Indonesian patients with
different types of liver disease. This study sought to determine
the nature of the quasispecies variations across the entire HBV
genome in vivo and whether they were related to the clinical
diagnoses.
Materials and methods
Subjects
Twelve hepatitis B surface antigen (HBsAg)-positive
patients were enrolled and their sera were collected at the General
Hospital of Surabaya and Hajj Hospital (Surabaya, Indonesia). Eight
of the patients had LC and/or HCC (ALD group) and four patients had
CHB (CHB group). Chronic hepatitis was defined as being positive
for HBsAg for >6 months with a raised or normal alanine
aminotransferase (ALT) level in patients who did not meet the
diagnostic criteria for LC or HCC. LC was diagnosed by either
histology (stage IV fibrosis) or clinical evidence of cirrhosis as
detected by liver biopsy, ultrasonography, computed tomography
(CT), or magnetic resonance imaging (MRI). HCC was diagnosed by at
least one of the following: positive liver biopsy, elevated
α-fetoprotein levels, and imaging findings (ultrasound, CT, or
MRI). All of the patients were treatment-naïve. None of the
patients were co-infected with human immunodeficiency virus,
hepatitis C virus, or hepatitis D virus.
Serologic testing
HBV DNA was detected with a TaqMan PCR assay
(COBAS® AmpliPrep/COBAS® TaqMan®
HBV tests; Roche Molecular Systems, Pleasanton, CA, USA) with a
lower limit of detection of 2.1 log copies/ml. The HBsAg titer was
quantified with a Lumipulse® HBsAg-HQ assay (Lumipulse,
Fujirebio, Tokyo, Japan). Hepatitis B e antigen (HBeAg) levels were
determined using a chemiluminescence immunoassay (Architect HBeAg;
Abbot Japan Co., Ltd., Tokyo, Japan). ALT and aspartate
aminotransferase (AST) levels were measured using standard
procedures.
DNA extraction and PCR
HBV DNA was extracted from 200 µl of serum using
QIAamp DNA Blood Mini kits (Qiagen, Tokyo, Japan). The whole genome
(four genes) was amplified using the three primer pairs: W1F
[GATTCCTGCTCAAGG AACC, nucleotides (nt) 529–547] and W1R (GCCTACAGC
CTCCTAGTAC, nt 1770–1788); W2F (ACTGGGAGGAGT TGGGGGAG, nt
1729–1748) and W2R (GCTGTAGCTCTT GTTCCCAAG, nt 2827–2847); and
HB10F (CGCAGAGAT CTCAATCTCGG, nt 2417–2437) and HB1R (GAAACATAG
AGGTGCCTTGAGCAG, nt 557–534). The PCR protocol comprised
pre-denaturation at 95°C for 5 min, followed by 30 cycles of
denaturation at 95°C for 1 min, annealing at 55°C for 1 min, and
extension at 72°C for 1.5 min, and finally post-extension at 72°C
for 5 min. The PCR products were analyzed on 2% agarose gel
electrophoresis and visualized with ethidium bromide under a UV
transilluminator. The target bands were purified with a QIAquick
Gel Extraction kit (Qiagen). The purified PCR products were
analyzed by direct sequencing and high-throughput sequencing to
identify viral genotypes and viral quasispecies, respectively.
Direct sequencing and HBV
genotyping
The PCR products were purified using ExoSAP-IT (USB
Corporation, Cleveland, OH, USA) and sequenced on an ABI Prism
3100-Avant genetic analyzer (Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) using the BigDye Terminator
version 3.1 cycle sequencing kit. The nt sequences obtained by
direct sequencing were aligned using ClustalW (http://www.genome.jp/tools-bin/clustalw)
and compared to reference sequences (genotypes A-G) retrieved from
the National Centre for Biotechnology Information (https://www.ncbi.nlm.nih.gov). A phylogenetic tree was
constructed using the minimum evolution and maximum likelihood
algorithm with MEGA 5.2 software (http://www.megasoftware.net) to determine HBV
genotypes. The viral subtypes were determined by S region analysis,
as previously described (18).
Next-generation sequencing
A next-generation sequencer (Genome Analyzer;
Illumina, Inc., San Diego, CA, USA) was used to detect variants in
the whole genomes of HBV quasispecies. The concentrations of the
purified PCR products were measured with a Qubit dsDNA HS Assay kit
(Invitrogen; Thermo Fisher Scientific, Inc.). Next, a PCR product
library encompassing the whole genome was prepared using the
Nextera XT DNA Sample Prep kit (Illumina, Inc.). The PCR products
were uniformly sheared into 500 bp fragments with the kits, and the
PCR product libraries were mixed with 1% 8 pM PhiX sequencing
control and run on a MiSeq sequencer (Illumina, Inc.) for
paired-end targeted sequencing. Finally, the fluorescent signals
were detected and analyzed using MiSeq Control Software (Illumina,
Inc.). The resulting images were used to produce sequence data in
the FASTQ format (11,15).
Sequence read mapping and data
analysis
Illumina, Inc. paired-end sequencing generates
overlapping read pairs (ORPs) from relatively short fragment
sequence libraries combined with relatively long reads. Quality
checks and data trimming were performed before assembling the
sequences using Genomics Workbench version 6.0.1 software (CLC bio,
Aarhus, Denmark). The sequencing results used in this study had
read quality scores of 30 (Q30), according to the manufacturer's
requirements. We used Q30 filtering and ORPs in order to eliminate
false-positive variants generated by PCR errors during the
sequencing process and to recover sequencing errors. The sequence
reads could therefore be used to detect viral variants occurring at
a low abundance with a high level of confidence. The aligned
sequence reads were mapped against the reference HBV genome of
genotype B3 and subtype adw (GeneBank accession no. AB713527). We
then determined the prevalence of each viral quasispecies within
the population. To achieve this, we used the setting ‘read
conflict’ in Genomics Workbench. Any conflicts between the sequence
reads were annotated on the consensus sequence after mapping was
complete (9,15).
Variants were defined as nt modifications that
resulted in an amino acid change (9,10,15,19).
The percentage variation (mutation frequency) was determined as the
proportion of nt changes in the total sequence reads that altered
amino acids. All nts with quasispecies of ≥1% of the entire viral
population were selected for analysis. A similar cut-off value was
used in previous studies (9,10,11,15).
Statistical analysis
All statistical analyses were performed with SPSS
software version 22 (IBM Corporation, Armonk, NY, USA). The
Kolmogorov-Smirnov and Shapiro-Wilk tests were used to assess the
normality of distribution of each variable. Differences between
groups were determined using the nonparametric Mann-Whitney U test
(for non-normally distributed variables) and Student's t-test (for
normally distributed variables). P<0.05 was considered to
indicate a statistically significant difference.
Results
Patient characteristics
The clinical characteristics of the patients with
CHB and ALD are shown in Table I.
There were no significant differences between the CHB and ALD
groups in terms of age (53.5 vs. 53.13 years, respectively;
P=0.922), AST levels (151.5 vs. 147.9 IU/l, respectively; P=0.836),
or ALT levels (86.75 vs. 95.57 IU/l, respectively; P=0.956).
However, the HBsAg titer was greater in the CHB group than in the
ALD group (461,300 vs. 1,491 IU/ml, respectively, P=0.156). There
were more males than females in both groups (Table I).
 | Table I.Clinical characteristics of
Indonesian patients according to their clinical diagnosis. |
Table I.
Clinical characteristics of
Indonesian patients according to their clinical diagnosis.
| Clin | Code | Sex/age
(years) | HBsAg (IU/ml) | ALT (IU/l) | AST (IU/l) |
Genotype/subtype | Mapping reads | Average
coverage | HBeAg (IU/ml) | Viral load
(logU/ml) |
|---|
| CHB | CHB1 | M/50 | 788 | 50 | 40 | B3/adw1 | 357894 | 80563 | 0.5 | 6.59 |
|
| CHB2 | M/56 | 39950.9 | 223 | 29 | B3/adw1 | 368495 | 11057 | 1370 | 8.88 |
|
| CHB3 | M/62 | 1801438 | 46 | 45 | B3/adw1 | 228351 | 81605 | 0.5 | 6.41 |
|
| CHB4 | F/46 | 3023.19 | 28 | 492 | B3/adw1 | 314734 | 9515 | 102 | 8.23 |
|
| Mean ± SD | 53.5±7 |
461,300±893,605 | 86.75±91.34 | 151.5±227.098 |
|
317,368.5±63,737 |
45,769.92±40,981.3 | 368.25±669.54 | 7.527±1.22 |
| ALD | ALD1 | F/52 | 3376 | 109 | 246 | B3/adw1 | 395270 | 12059 | 204 | 7.45 |
|
| ALD2 | M/57 | 208 | – | – | B3/adw1 | 242736 | 7155 | 0.5 | 4.61 |
|
| ALD3 | M/52 | 1088 | 149 | 203 | B3/adw1 | 314669 | 115479 | 0.5 | 6.32 |
|
| ALD4 | M/40 | 810 | 274 | 393 | B3/adw1 | 291658 | 94001 | 54 | 7.18 |
|
| ALD5 | M/35 | 722 | 42 | 32 | B3/adw1 | 342035 | 10401 | 120 | 7.18 |
|
| ALD6 | F/71 | 455 | 20 | 17 | B3/adw1 | 334186 | 79841 | 0.5 | 6.14 |
|
| ALD7 | M/71 | 1071 | 38 | 65 | B3/adw1 | 303581 | 100591 | 161 | 7.28 |
|
| ALD8 | M/47 | 4204 | 37 | 79 | B3/adw1 | 366147 | 11006 | 0.5 | 8.21 |
|
| Mean ± SD | 53.13±13.81 | 1,492±1465 | 83.63±91.18 | 147.9±386.485 |
|
323,785.25±46,962 |
53,921.85±47,807.49 | 67.625±83.04 | 6.796±1.09 |
Based on direct sequencing of the whole genome and
the S gene, all of the samples belonged to HBV genotype B3
(phylogenetic tree not shown) and subtype adw1, respectively.
High-throughput sequencing of HBV
variants
The mean coverage depth per nt after quality control
(ORP and Q30) was 45,769.92-fold in the CHB group and
53,921.85-fold in the ALD group. The number of mapping reads was
317,368.5±63,737 and 323,785.25±46,962 in the CHB and ALD groups,
respectively (Table I). Mapping
reads represent the nt annotation of the consensus sequence. The
rate of gene variation represents the probability of change per
position (9). The quasispecies
variants detected in ≥1% of the total viral population were
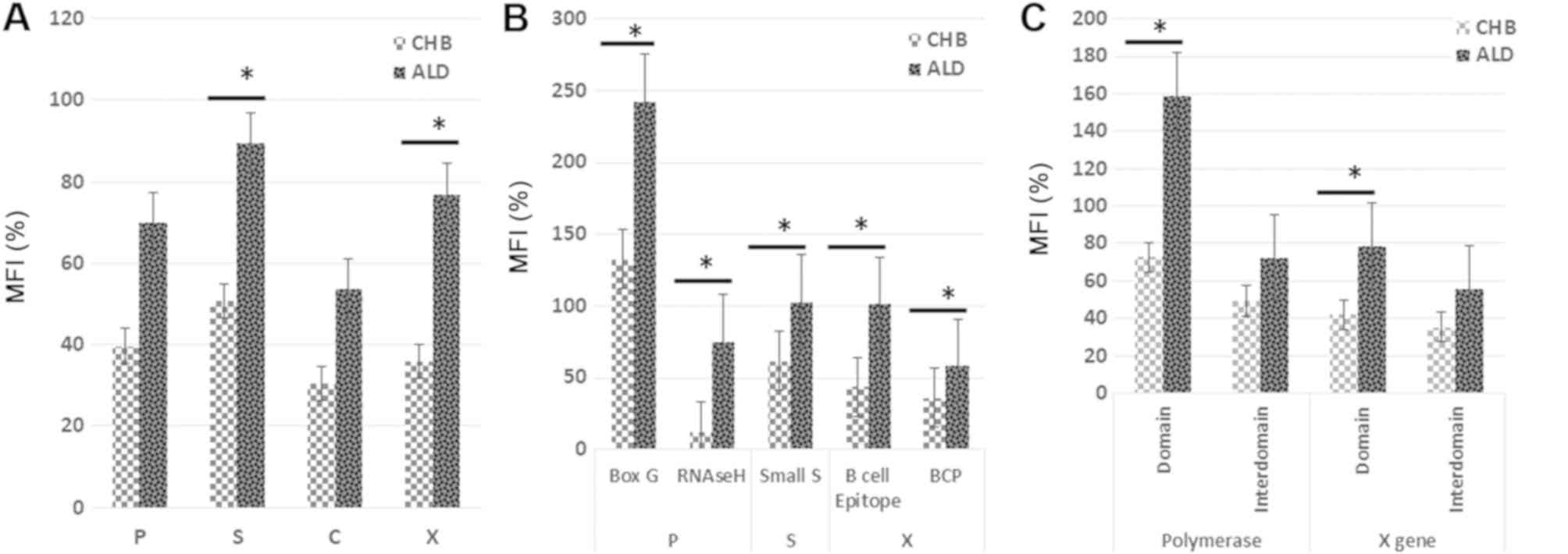
examined in four ORFs. The frequencies of modifications in the S
gene (P=0.047) and X gene (P=0.044) were significantly greater in
the ALD group than in the CHB group (Fig. 1A). The mutation rate in the S
region was 89.53 and 50.69% in the ALD and CHB groups,
respectively. The mutation rate in the X region was 76.95 and
35.88% in the ALD and CHB groups, respectively.
Variations in specific ORFs and
domains
Further analysis revealed that the substitution rate
in specific ORFs was significantly greater in the ALD group in RT
box G (P=0.048), RNAseH (P=0.038), small S region (P=0.031), BCP
(P=0.044) and B-cell epitope (P=0.042; Fig. 1B). Because the P-values indicate
the order of significance for each ORF relative to the progression
of severe disease, it seems that the small S region is the most
strongly affected, followed by RNAseH, B-cell epitope, BCP, and RT
box G. These specific ORFs might be strongly affected by the
duration of infection, from chronic infection to the development of
severe liver disease. Fig. 1C
shows the quasispecies variants in the domain, a specific ORF in
the same gene, and the inter-domain, the region between two domains
(ORFs). Box G and RNAseH were classified as the polymerase domain,
while B cell epitope and BCP were classified as X domain. The
region between RT box G and RNAseH and the region between B cell
epitope and BCP were classified as inter-domains. The mutation
frequency in the P domain (72.52 vs. 158.70%, P=0.037) and the X
domain (41.94 vs. 78.61%, P=0.015) were significantly greater in
the ALD group than in the CHB group. By contrast, the mutation
frequency in the inter-domain was not significantly different
between the CHB group and the ALD group in either the P gene (49.11
vs. 72.38%, P=0.109) or the X gene (35.33 vs. 55.83%, P=0.214).
Mutations associated with progression
to ALD
The quasispecies variants for each amino acid were
compared between the CHB and ALD groups to understand the potential
role of these variants on disease progression. The nt positions
showing significant differences in quasispecies are shown in
Table II. Because of the large
number of mutations detected by the high-throughput sequencing
method, we have only shown the mutations that were statistically
significant. Several variations are already known, including
rtV30F, rtD31V, pV697E, sK24Q, sE164A, sW196* I127N, K130M, and
V131I. The present study confirmed that during disease progression,
the proportion of quasispecies variants increased for most of the
mutations, except for W196* in the small S gene (Table II).
| Table II.Amino acid variants and viral
populations in the whole genome. |
Table II.
Amino acid variants and viral
populations in the whole genome.
|
| Quasispecies (mean
± SD) |
| P-value |
|
|
|
|---|
|
|
|
|
|
|
|
|
|---|
| Mutation | CHB | ALD | N | a | b | c | Position | Clinical
impact |
|---|
|
rtV27G/D/A | 12.893±7.882 | 33.115±6.246 | 0.225 | 0.001 |
| ↑ | RT box G | g(30) |
|
rtL29F/P/I/R | 20.191±7.168 | 27.165±3.107 | 0.008 |
| 0.048 | ↑ |
| – |
|
rtV30F/A/G | 8.911±3.232 | 23.045±5.079 | 0.932 | 0.000 |
| ↑ |
| h(11)(45) |
|
rtD31V/Y/G | 5.277±0.263 | 35.860±7.371 | 0.050 |
| 0.000 | ↑ |
| h(11)(45) |
|
rtK32T/Q/N | 2.583±0.416 | 4.601±1.767 | 0.073 | 0.015 |
| ↑ |
| – |
|
rtP34S/T | 3.545±0.293 | 9.064±1.651 | 0.120 | 0.000 |
| ↑ |
| – |
|
rtH35Y/Q/L/P | 5.333±2.028 | 22.320±9.064 | 0.181 | 0.001 |
| ↑ |
| g(30) |
|
rtN36T/K/H | 6.215±0.947 | 18.859±2.675 | 0.041 |
| 0.000 | ↑ |
| g(30) |
|
V697E/G/A | 1.170±0.122 | 2.852±1.407 | 0.041 |
| 0.032 | ↑ | RNAseH | i(46)(47) |
|
T702P/A/S | 1.499±0.380 | 3.888±1.847 | 0.043 |
| 0.013 | ↑ |
| – |
| I710L/M | 1.005±0.007 | 1.526±0.384 | 0.431 | 0.002 |
| ↑ |
| – |
|
E729K/A/D | 1.419±0.276 | 3.699±0.990 | 0.848 | 0.015 |
| ↑ |
| – |
| K743T/N | 1.730±1.181 | 9.928±9.252 | 0.347 | 0.029 |
| ↑ |
| – |
|
L21F/S/V/W | 6.062±3.041 | 17.180±2.449 | 0.741 | 0.000 |
| ↑ | small S | (11) |
|
T23I/P/K/S/R | 3.840±0.646 | 23.971±5.705 | 0.590 | 0.000 |
| ↑ |
| (11) |
|
K24Q/T/N | 1.112±0.323 | 1.563±0.323 | 0.499 | 0.038 |
| ↑ |
| (23)(6) |
|
I25T/N/S | 10.263±2.000 | 23.086±2.591 | 0.154 | 0.000 |
| ↑ |
| (11) |
| E164A | 1.174±0.113 | 1.496±0.236 | 0.920 | 0.050 |
| ↑ |
| j,k(17)(48)(49)(40) |
| W196* | 2.602±0.994 | 1.481±0.387 | 0.699 | 0.009 |
| ↓ |
| f,k(40)(39) |
| V224A | 1.375±0.345 | 3.612±4.568 | 0.001 |
| 0.032 | ↑ |
| – |
|
E24D/A/K | 1.369±0.308 | 2.262±1.274 | 0.501 | 0.021 |
| ↑ | B cell epitope | – |
| A40G | 1.179±0.115 | 10.865±12.658 | 0.501 | 0.029 |
| ↑ |
| – |
| I127N | 11.464±11.221 | 45.699±46.565 | 0.007 |
| 0.022 | ↑ | BCP | d,e(50)(51) |
| K130M | 2.103±1.315 | 81.133±34.246 | 0.000 |
| 0.014 | ↑ |
| d,e(50)(51) |
| V131I | 9.929±20.363 | 44.103±40.632 | 0.060 | 0.046 |
| ↑ |
| d,e(50)(51) |
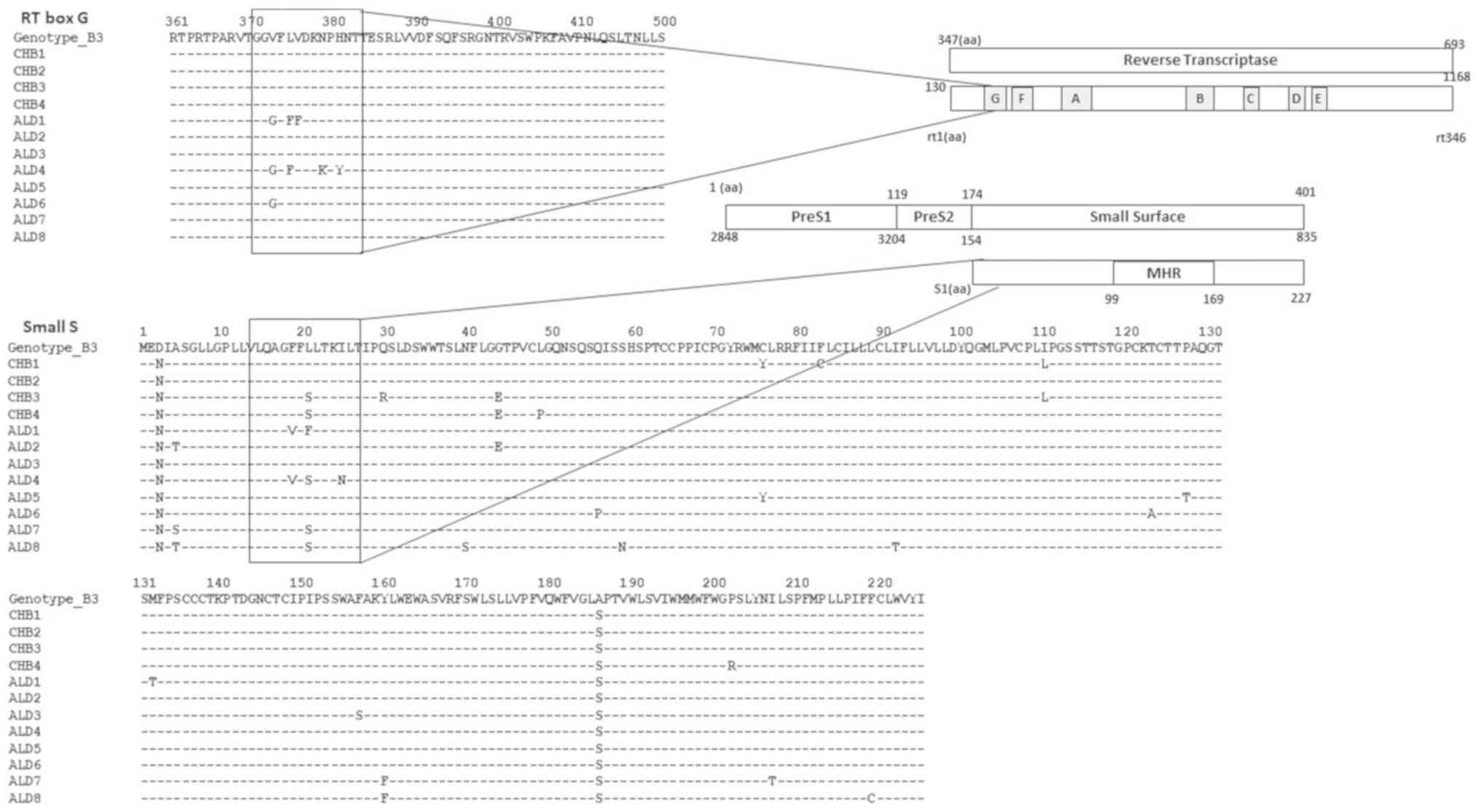
RT box G and small S gene
overlapping
This study found high rate of variants in the RT box
G with emergence of major mutations (Fig. 2), even though the P gene is the
most conserved gene in the HBV genome. The overlapping RT box G
influenced the variations and in the small S gene and introduced
major mutations in the small S gene. Major mutations with
quasispecies in >20% of the population can usually be confirmed
by direct sequencing, whereas mutations with quasispecies in
<20% are only detected by high-throughput sequencing.
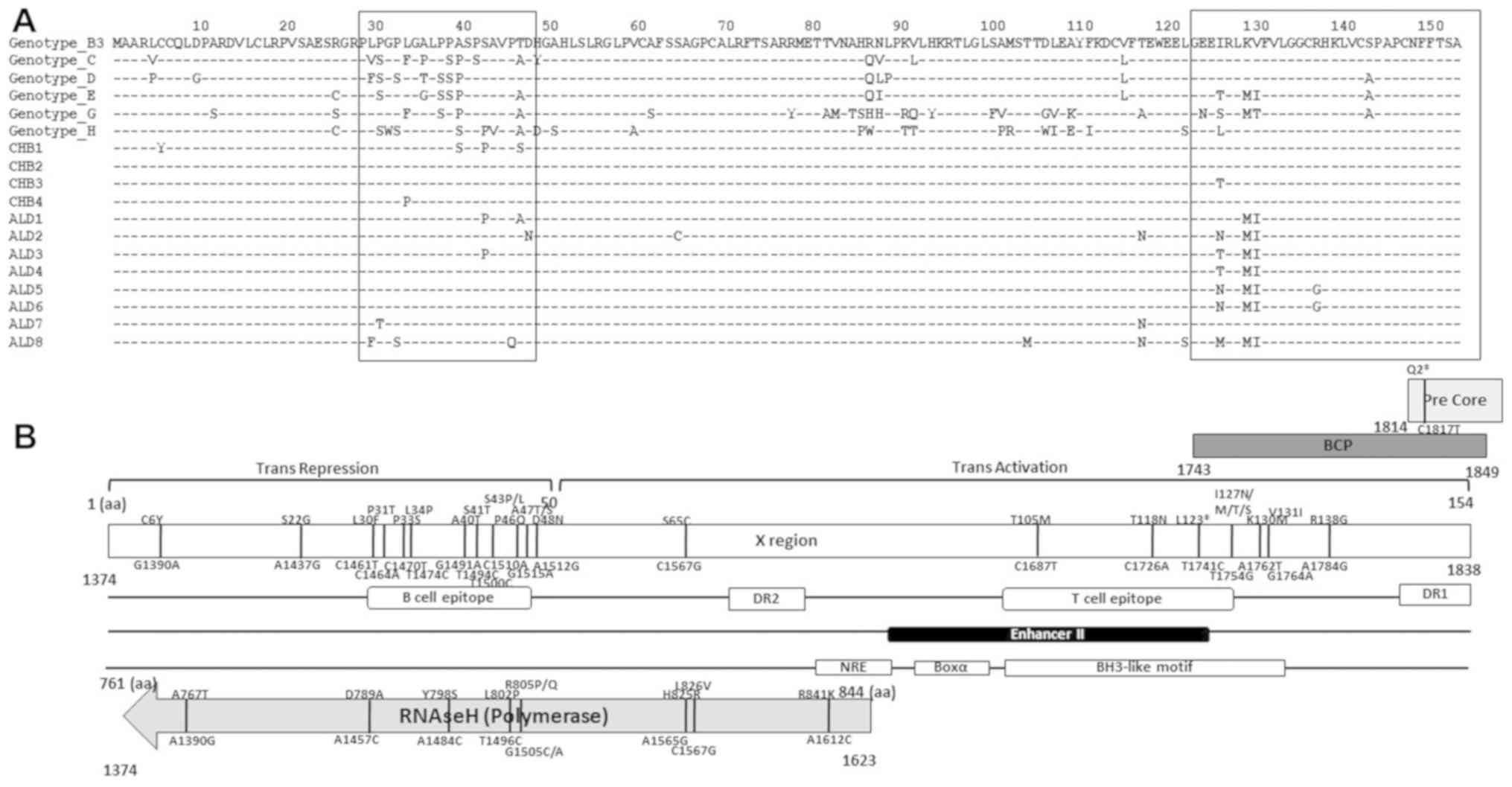
X gene alignment and mutation
mapping
Fig. 3A shows the
alignment of the HBV X protein (HBx). The quasispecies variants
accumulated in the B-cell epitope and BCP region, in particular.
Fig. 3B shows that the significant
mutations within the overlapping X gene and RNAseH, and mutations
in the pre-Core gene influenced other variations in each ORF
(20).
Discussion
This study demonstrated that all the HBV strains
isolated in Java, especially in Surabaya, belong to genotype B3.
These results are consistent with previous studies showing that HBV
genotype B3 and subtype adw are predominant in Indonesia and South
East Asia (3,15,21,22).
All of the HBV strains identified in this study belonged to subtype
adw1, which is the predominant HBV subtype in Sumatra and Java
(9,23). This subtype was strongly associated
with surface gene variations and immune escape mutants in patients
with HCC (3,9,23).
Surface gene variations could modify the antigenicity of HBsAg and
allow the virus to evade neutralizing anti-HBs, thus facilitating
progression to severe disease (4,9,13,24).
It was reported that the HBsAg titer decreases during disease
progression but it may also increase during the re-activation stage
of chronic infection (25).
However, the HBsAg titer was not significantly different between
the two groups in the present study (Table I). In this study, we ruled out age,
sex, and other serological markers (ALT, AST, and HBeAg) as
influencing factors because there were no significant differences
in these variables between the CHB and ALD groups. Because of the
comparable characteristics, we suggest that the progression towards
severe liver disease was influenced by the duration of infection
and quasispecies variation. The duration of infection indicates
whether exposure occurred as a consequence of vertical or
horizontal transmission (9).
In a population with high endemic HBV infection such
as Indonesia, it is essential to understand HBV quasispecies
variation in order to develop vaccines, immunodiagnostic reagents,
and effective therapeutic strategies (26). Recently, several research groups in
various countries have used high-throughput sequencing to analyze
HBV variations. It was reported that quasispecies variations in the
RT 1 motif region and the small S region were detected shortly
after treatment with nt analogs (11). Next-generation sequencing was also
compared with clone-based sequencing to analyze reverse
transcriptase quasispecies complexity in China (27,28).
The authors concluded that next-generation sequencing was
clinically valuable for predicting treatment efficacy (28). Deep sequencing was useful to detect
minor quasispecies variants in relation to emergence of drug
resistance in Italy (29). The
same method was also used to detect the accumulation of
quasispecies variations in the ‘a determinant’ region in patients
with vertically transmitted HBV infection in California, USA
(30). Owing to its ability to
identify quasispecies variants occurring at a low abundance, the
high-throughput screening method used here is more sensitive and
represents a more useful tool than conventional direct sequencing
(9,15,31).
By using high-throughput methods, it is possible to determine the
actual prevalence of viral quasispecies variants that might be
clinically relevant. Based on the present findings, high-throughput
sequencing showed strong sensitivity for quasispecies variants
occurring in <20% of the population. We observed a high
variation in naturally occurring quasispecies in this population of
Indonesian HBV patients, and most of these quasispecies increased
during disease progression.
The present study revealed that the quasispecies
variations mainly accumulated in the S and X regions of the HBV
genome in patients with severe liver diseases. Variations in the S
region can impair the secretion of HBsAg and virions. Furthermore,
the accumulation of HBsAg or virions in hepatocytes can induce
endoplasmic reticulum stress, exacerbate inflammation, and
eventually lead to more severe liver disease (9). By contrast, variations in the X
region are primarily associated with activation of signal
transduction. The X protein is a nuclear coactivator that activates
signal transduction by several pathways, including the s (NF-κB)
signaling pathway. NF-κB is necessary for cell growth and
viability. Recent studies showed the activation of NF-κB could
prevent apoptosis (32–34). It was suggested that the activation
of NF-κB by X protein might promote the survival of HBV-infected
hepatocytes and could contribute to hepatocarcinogenesis (7,32,33).
In this study, K130M and V131I were predominant in patients with
ALD. Wild-type HBx protein was reported to activate
hypoxia-inducible factor-1α (HIF-1α), which could contribute to HCC
progression (35,36). Thus, the variations K130M and V131I
in X protein might upregulate HIF-1α function.
In the present study, we observed a trend for
increased frequencies of rtV27G, rtL29F, rtV30F, rtD31V, rtK32T,
rtP34S, rtH35Y, and rtN36T during the period of infection.
Interestingly, these mutations accumulated in RT Box G, especially
the RT-1 motif, which is reportedly related to protein priming and
an epitope on the polymerase that is targeted by antibodies
(37). It was suggested that these
mutations might enhance viral replication activity and thus affect
the clinical outcome (37). The
frequencies of the variants pV697E, pT702P, p710L, pE729K, and
pK743T were also in the ALD group. These mutations were located in
the RNAseH domain and were predicted to influence pgRNA degradation
and enhance viral replication function. The frequencies of the
variations sL21S/F, sT23I, sK24Q, sI25T, sE164A, and sV224A in the
small S gene were also increased in the ALD group. These mutations
altered the structural integrity of S protein and reduced its
binding affinity to the anti-HBs antibody, to yield immune escape
mutants. By contrast, the frequency of the vaccine escape variant
sW196*, which is reportedly related to immune therapy failure, was
decreased in this study. This change may indirectly contribute to
the activity of another mutation (38–41).
Because the envelope (S) gene is completely overlapped by the RT
gene, the minor variant W196* may produce changes in the
overlapping RT region and amino acid substitution rtM204I. Although
we found no difference in the variant rtM204K/V between the two
groups, we suggest that this substitution should be monitored, even
in minor populations.
Two minor variations in the B cell epitope, E24D and
A40G, were more frequent in the ALD group. These mutations are
crucial for binding to neutralizing antibodies and to produce
immune escape mutants. At the C terminal of the X region, we found
that the frequencies of the variations I127N, K130M, and V131I
increased with severe disease progression. These variations
accumulated in the BCP region and can cause a substantial decrease
in HBeAg expression, interfere with DNA repair, and enhance viral
genome replication, which may contribute to liver disease
progression via increased viral invasion and inflammation (20,42).
These variations may also contribute to hepatocarcinogenesis via
downregulated p21, a tumor suppressor protein, leading to rapid and
uncontrolled cell proliferation (20,39).
Similarly, a previous study reported that the variants K130M and
V131I in genotype C were associated with the progression of ALD
(10,21,43).
The double mutation K130M/V131I was associated with severe clinical
outcomes and progression of liver disease in chronic HBV patients
in India (44).
A limitation of this study is the small number of
patients and samples. A larger number of patients is needed to
obtain more reproducible data.
In this study, we found that the accumulation of
quasispecies variations in the S and X regions was predominant in
patients with ALD. By using high-throughput sequencing, we found
that quasispecies variants accumulated in the small S gene, RT box
G, RNAseH, BCP, and B cell epitope genes, presumably in relation to
increased disease severity. We suggest that viral quasispecies
change dynamically during disease progression and might be related
to progression of severe liver disease. Despite the limited number
of samples, our results are clinically significant and provide new
insight into the quasispecies variations of HBV isolated from
Indonesian patients. Larger cohorts studies are needed to clarify
the dynamics of viral quasispecies as well as their clinical
implications.
Acknowledgements
Not applicable.
Funding
This study was supported by a Grant-in-Aid from the
Ministry of Education, Culture, Sports, Science and Technology,
Japan (grant no. 16H05826), and a Grant-in-Aid from the Japan
Initiative for Global Research Network on Infectious Disease
(J-GRID) supported by The Ministry of Education, Culture, Sports,
Science and Technology.
Availability of data and materials
The datasets analyzed during the current study are
available in the DDBJ under the following accession numbers: CHB
group LC349872, LC349873, LC349875, and LC349879; ALD group
LC349868, LC349869, LC349870, LC349871, LC349874, LC349876,
LC349877, and LC349878.
Authors' contributions
WAP, YY, LNY, S, MIL and YH conceived and designed
the study. WAP, YL and YM performed the experiments. WAP analyzed
the data and wrote the study. YY and TU performed some of the
experimental methods and techniques. YY, S, MIL and YH revised the
manuscript and supervised the study.
Ethics approval and consent to
participate
The research protocols were approved by the
Institutional Review Board of the Institute of Tropical Disease,
Airlangga University (Surabaya, Indonesia), and conformed to the
Declaration of Helsinki (1975). Informed consent was obtained from
all of the subjects involved in the study.
Patient consent for publication
Informed consent was obtained from all of the
subjects involved in the study.
Competing interests
The authors declare that they have no conflicts of
interest.
Glossary
Abbreviations
Abbreviations:
|
ALD
|
advanced liver disease
|
|
ALT
|
alanine transaminase
|
|
AST
|
aspartate transaminase
|
|
BCP
|
basal core promoter
|
|
CHB
|
chronic Hepatitis B
|
|
CT
|
computed tomography
|
|
HBeAg
|
Hepatitis B e antigen
|
|
HBsAg
|
Hepatitis B s antigen
|
|
HBV
|
Hepatitis B virus
|
|
HBx
|
Hepatitis B × protein
|
|
HCC
|
hepatocellular carcinoma
|
|
HIF-1α
|
Hypoxia-inducible factor-1α
|
|
LC
|
liver cirrhosis
|
|
MFI
|
mutation frequency index
|
|
MRI
|
magnetic resonance imaging
|
|
NF-κB
|
nuclear factor-κB
|
|
nt
|
nucleotide
|
|
ORF
|
open reading frame
|
|
ORP
|
overlapping read pairs
|
|
PCR
|
polymerase chain reaction
|
|
pgRNA
|
pregenomic RNA
|
|
RNAseH
|
ribonuclease H
|
|
RT
|
reverse transcription
|
References
|
1
|
Chen GF, Wang C and Lau G: Treatment of
chronic hepatitis B infection-2017. Liver Int. 37 (Suppl 1):59–66.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Locarnini SA: Hepatitis B virus surface
antigen and polymerase gene variants: potential virological and
clinical significance. Hepatology. 27:294–297. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heriyanto DS, Yano Y, Utsumi T,
Anggorowati N, Rinonce HT, Lusida MI, Soetjipto, Triwikatmani C,
Ratnasari N, Maduseno S, et al: Mutations within enhancer II and
BCP regions of hepatitis B virus in relation to advanced liver
diseases in patients infected with subgenotype B3 in Indonesia. J
Med Virol. 84:44–51. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu HL, Li X, Li J and Zhang ZH: Genetic
variation of occult hepatitis B virus infection. World J
Gastroenterol. 22:3531–3546. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kendrick S and Day C: Natural history and
factors influencing the course of alcohol-related liver disease.
Clin Liver Dis. 2:61–63. 2013. View
Article : Google Scholar
|
|
6
|
Luangsay S and Zoulim F: Structure and
molecular virology. In: Viral hepatitis. 4th edition.
Wiley-Blackwell; Hoboken, NJ: pp. 63–80. 2013
|
|
7
|
Gao S, Duan ZP and Coffin CS: Clinical
relevance of hepatitis B virus variants. World J Hepatol.
7:1086–1096. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rehermann B and Nascimbeni M: Immunology
of hepatitis B virus and hepatitis C virus infection. Nat Rev
Immunol. 5:215–229. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamani LN, Yano Y, Utsumi T, Juniastuti,
Wandono H, Widjanarko D, Triantanoe A, Wasityastuti W, Liang Y,
Okada R, et al: Ultradeep sequencing for detection of quasispecies
variants in the major hydrophilic region of hepatitis b virus in
Indonesian patients. J Clin Microbiol. 53:3165–3175. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liao Y, Hu X, Chen J, Cai B, Tang J, Ying
B, Wang H and Wang L: Precore mutation of hepatitis B virus may
contribute to hepatocellular carcinoma risk: evidence from an
updated meta-analysis. PLoS One. 7:e383942012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liang Y, Yano Y, Putri WA, Mardian Y,
Okada R, Tanahashi T, Murakami Y and Hayashi Y: Early changes in
quasispecies variant after antiviral therapy for chronic hepatitis
B. Mol Med Rep. 17:5528–5537. 2018.PubMed/NCBI
|
|
12
|
Kim BK, Revill PA and Ahn SH: HBV
genotypes: Relevance to natural history, pathogenesis and treatment
of chronic hepatitis B. Antivir Ther. 16:1169–1186. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang CH, Yuan Q, Chen PJ, Zhang YL, Chen
CR, Zheng QB, Yeh SH, Yu H, Xue Y, Chen YX, et al: Influence of
mutations in hepatitis B virus surface protein on viral
antigenicity and phenotype in occult HBV strains from blood donors.
J Hepatol. 57:720–729. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wilson JN, Nokes DJ and Carman WF: Current
status of HBV vaccine escape variants-a mathematical model of their
epidemiology. J Viral Hepat. 5 (Suppl 2):25–30. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wasityastuti W, Yano Y, Widasari DI,
Yamani LN, Ratnasari N, Heriyanto DS, Okada R, Tanahashi T,
Murakami Y, Azuma T, et al: Different variants in reverse
transcriptase domain determined by ultra-deep sequencing in
treatment-naïve and treated indonesian patients infected with
hepatitis B virus. Kobe J Med Sci. 62:E1–E8. 2016.PubMed/NCBI
|
|
16
|
Bobek V, Kolostova K, Pinterova D,
Kacprzak G, Adamiak J, Kolodziej J, Boubelik M, Kubecova M and
Hoffman RM: A clinically relevant, syngeneic model of spontaneous,
highly metastatic B16 mouse melanoma. Anticancer Res. 30:4799–4803.
2010.PubMed/NCBI
|
|
17
|
Yano Y, Azuma T and Hayashi Y: Variations
and mutations in the hepatitis B virus genome and their
associations with clinical characteristics. World J Hepatol.
7:583–592. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
An Introduction to Next-Generation
Sequencing Technology, . Illumina, Inc.; San Diego, CA: 2015,
https://www.illumina.com/documents/products/illumina_sequencing_introduction.pdf
|
|
19
|
Kim H, Lee SA, Do SY and Kim BJ:
Precore/core region mutations of hepatitis B virus related to
clinical severity. World J Gastroenterol. 22:4287–4296. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim H, Lee S and Kim BJ: X region
mutations of hepatitis B virus related to clinical severity. World
J Gastroenterol. 22:5467–5478. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Utama A, Purwantomo S, Siburian MD, Dhenni
R, Gani RA, Hasan I, Sanityoso A, Miskad UA, Akil F, Yusuf I, et
al: Hepatitis B virus subgenotypes and basal core promoter
mutations in Indonesia. World J Gastroenterol. 15:4028–4036. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Utsumi T, Yano Y, Lusida MI, Amin M,
Soetjipto, Hotta H and Hayashi Y: Serologic and molecular
characteristics of hepatitis B virus among school children in East
Java, Indonesia. Am J Trop Med Hyg. 83:189–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mulyanto TF, Tsuda F, Karossi AT,
Soewignjo S, Roestamsjah, Sumarsidi D, Trisnamurti RH,
SumardiSurayah, Udin LZ, et al: Distribution of the hepatitis B
surface antigen subtypes in Indonesia: Implications for ethnic
heterogeneity and infection control measures. Arch Virol.
142:2121–2129. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lamontagne RJ, Bagga S and Bouchard MJ:
Hepatitis B virus molecular biology and pathogenesis. Hepatoma Res.
2:163–186. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Trépo C, Chan HLY and Lok A: Hepatitis B
virus infection. Lancet. 384:2053–2063. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Capobianchi MR, Giombini E and Rozera G:
Next-generation sequencing technology in clinical virology. Clin
Microbiol Infect. 19:15–22. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gong L, Han Y, Chen L, Liu F, Hao P, Sheng
J, Li XH, Yu DM, Gong QM, Tian F, et al: Comparison of
next-generation sequencing and clone-based sequencing in analysis
of hepatitis B virus reverse transcriptase. J Clin Microbiol.
51:4087–4094. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han Y, Gong L, Sheng J, Liu F, Li XH, Chen
L, Yu DM, Gong QM, Hao P and Zhang XX: Prediction of virological
response by pretreatment hepatitis B virus reverse transcriptase
quasispecies heterogeneity: the advantage of using next-generation
sequencing. Clin Microbiol Infect. 21:797.e1–8. 2015. View Article : Google Scholar
|
|
29
|
Solmone M, Vincenti D, Prosperi MCF,
Bruselles A, Ippolito G and Capobianchi MR: Use of massively
parallel ultradeep pyrosequencing to characterize the genetic
diversity of hepatitis B virus in drug-resistant and drug-naive
patients and to detect minor variants in reverse transcriptase and
hepatitis B S antigen. J Virol. 83:1718–1726. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Du Y, Chi X, Wang C, Jiang J, Kong F, Yan
H, Wang X, Li J, Wu NC, Dai L, et al: Quantifying perinatal
transmission of Hepatitis B viral quasispecies by tag linkage deep
sequencing. Sci Rep. 7:101682017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nishijima N, Marusawa H, Ueda Y, Takahashi
K, Nasu A, Osaki Y, Kou T, Yazumi S, Fujiwara T, Tsuchiya S, et al:
Dynamics of hepatitis B virus quasispecies in association with
nucleos(t)ide analogue treatment determined by ultra-deep
sequencing. PLoS One. 7:e350522012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fan C, Yang J and Engelhardt JF: Temporal
pattern of NFkappaB activation influences apoptotic cell fate in a
stimuli-dependent fashion. J Cell Sci. 115:4843–4853. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mukherjee A, Choudhury M, Peruani F,
Ganguly N and Mitra B: Dynamics on and of complex networks. 2
Birkhäuser; Basel: 2013
|
|
34
|
Shishodia S and Aggarwal BB: Nuclear
factor-kappaB: A friend or a foe in cancer? Biochem Pharmacol.
68:1071–1080. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang X and Ding H-G: Key role of
hepatitis B virus mutation in chronic hepatitis B development to
hepatocellular carcinoma. World J Hepatol. 7:1282–1286. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tarocchi M, Polvani S, Marroncini G and
Galli A: Molecular mechanism of hepatitis B virus-induced
hepatocarcinogenesis. World J Gastroenterol. 20:11630–11640. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Badtke MP, Khan I, Cao F, Hu J and Tavis
JE: An interdomain RNA binding site on the hepadnaviral polymerase
that is essential for reverse transcription. Virology. 390:130–138.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhu B, Wang T, Wei X, Zhuo Y, Liu A and
Zhang G: Accumulation of mutations in reverse transcriptase of
hepatitis B virus is associated with liver disease severity in
treatment-naïve Chinese patients with chronic hepatitis B. Adv Clin
Exp Med. 26:1123–1129. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tong S and Revill P: Overview of hepatitis
B viral replication and genetic variability. J Hepatol. 64 (Suppl
1):S4–S16. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rodriguez-Frias F, Buti M, Tabernero D and
Homs M: Quasispecies structure, cornerstone of hepatitis B virus
infection: Mass sequencing approach. World J Gastroenterol.
19:6995–7023. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pacheco SR, Dos Santos MIMA, Stocker A,
Zarife MAS, Schinoni MI, Paraná R, Dos Reis MG and Silva LK:
Genotyping of HBV and tracking of resistance mutations in
treatment-naïve patients with chronic hepatitis B. Infect Drug
Resist. 10:201–207. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shi YH and Shi CH: Molecular
characteristics and stages of chronic hepatitis B virus infection.
World J Gastroenterol. 15:3099–3105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Khan A, Al Balwi MA, Tanaka Y, Hajeer A,
Sanai FM, Al Abdulkarim I, Al Ayyar L, Badri M, Saudi D, Tamimi W,
et al: Novel point mutations and mutational complexes in the
enhancer II, core promoter and precore regions of hepatitis B virus
genotype D1 associated with hepatocellular carcinoma in Saudi
Arabia. Int J Cancer. 133:2864–2871. 2013.PubMed/NCBI
|
|
44
|
Asim M, Malik A, Sarma MP, Polipalli SK,
Begum N, Ahmad I, Khan LA, Husain SA, Akhtar N, Husain S, et al:
Hepatitis B virus BCP, Precore/core, X gene mutations/genotypes and
the risk of hepatocellular carcinoma in India. J Med Virol.
82:1115–1125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Clark DN and Hu J: Unveiling the roles of
HBV polymerase for new antiviral strategies. Future Virol.
10:283–295. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jones SA and Hu J: Hepatitis B virus
reverse transcriptase: diverse functions as classical and emerging
targets for antiviral intervention. Emerg Microbes Infect.
2:e562013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jones SA, Clark DN, Cao F, Tavis JE and Hu
J: Comparative analysis of hepatitis B virus polymerase sequences
required for viral RNA binding, RNA packaging, and protein priming.
J Virol. 88:1564–1572. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Saha D, Pal A, Biswas A, Panigrahi R,
Sarkar N, Das D, Sarkar J, Guha SK, Saha B, Chakrabarti S and
Chakravarty R: Molecular characterization of HBV strains
circulating among the treatment-naive HIV/HBV co-infected patients
of eastern India. PLoS One. 9:e904322014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shi Y, Wei F, Hu D Li Q, Smith D, Li N and
Chen D: Mutations in the major hydrophilic region (MHR) of
hepatitis B virus genotype C in North China. J Med Virol.
84:1901–1906. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Juniastuti UT, Utsumi T, Aksono EB, Yano
Y, Soetjipto, Hayashi Y, Hotta H, Rantam FA, Kusumobroto HO and
Lusida MI: Predominance of precore mutations and clinical
significance of basal core promoter mutations in chronic hepatitis
B virus infection in Indonesia. Biomed Rep. 1:522–528. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yang Z, Zhuang L, Lu Y, Xu Q, Tang B and
Chen X: Naturally occurring basal core promoter A1762T/G1764A dual
mutations increase the risk of HBV-related hepatocellular
carcinoma: a meta-analysis. Oncotarget. 7:12525–12536.
2016.PubMed/NCBI
|