Introduction
Parkinson's disease (PD) is one of the most common
neurodegenerative and motor disorders and principally affects
people over the age of 50, with an increased prevalence with
increasing age. The pathogenesis of PD has been proposed to be
related to mitochondrial disturbance, oxidative stress, the
inflammatory response of microglial cells or cell senescence
(1). PD is characterized
pathologically by the loss and/or dysfunction of dopaminergic (DA)
neurons in the substantia nigra and histologically by the presence
of Lewy body (LB) and Lewy neurite (LN) formation, comprised
primarily of abnormal aggregates of α-synuclein (α-syn) (2).
The SNCA gene encodes α-syn, a small (140
amino acid) protein that is mainly expressed in the brain, with a
primarily pre-synaptic localization (3). Physiologically, α-syn is an
indispensable anti-apoptosis factor in DA neurons in the
nigrostriatal pathway; however, pathologically, oligomeric and/or
fibrillary forms of α-syn lose their anti-apoptotic function and
are cytotoxic to DA neurons. These pathological forms can be caused
by certain mutations, such as a duplication or triplication of
SNCA, or by abnormal metabolism of α-syn (4–6).
Moreover, it appears that pathological α-syn aggregates can be
transferred via cell-to-cell transfer, which seeds further α-syn
aggregation in other neurons in a ‘prion-like’ manner (7–10).
The autophagy pathway is one of the degradation
systems for cytosolic proteins in the central nervous system. It
has been confirmed that neuronal clearance of α-syn aggregates
relies on macroautophagy (a type of autophagy), because the
aggregates cannot pass through the narrow proteasomal core for
degradation (11). In addition, to
halt PD development, autophagy may prevent the early events in
α-syn exosomal release and uptake by neurons (12). Conversely, α-syn aggregates can
impair autophagy in DA neurons by decreasing the clearance of
autophagosomes, leading to neuronal death (13). Therefore, there seems to be a
strong relationship between α-syn aggregates and impaired autophagy
in DA neurons during PD development, and the protection of neuronal
autophagic function could be an effective therapeutic strategy
against PD.
Salidroside (SAL) is an ingredient extracted from
the root of Rhodiola rosea, and is reported to have various
pharmacological activities, including antioxidant, anti-apoptosis,
anti-tumor, cardioprotective and hepatoprotective functions
(14). A previous study showed
that SAL could inhibit cell senescence by regulating p53, p21 and
p16 expression in oxidant-impaired cells (15). Furthermore, SAL has been reported
to play a role in the downregulation of reactive oxygen species
(ROS) and amyloid-β aggregation in damaged neurons (16,17)
and in the protection of myocardial cells via PI3K/Akt/mTOR
signaling, which is related to autophagy repair (18).
The present study investigated the therapeutic
potential of SAL in a PD cell model [overexpression of wild-type
(WT) α-syn or A53T mutation of α-syn in SH-SY5Y cells] and explored
the underlying mechanism of its autophagy promotion via the
mTOR/p70S6K and PI3K/Akt signaling pathways.
Materials and methods
Materials
SAL (cat. no. S101157) and EDTA (cat. no. 431788)
were purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd.
Rapamycin (Rap; cat. no. HY-10219) and 3-methyladenine (3-MA; cat.
no. HY-19312) were purchased from MedChemExpress. FBS (cat. no.
16000-044) and OPTI-MEM (cat. no. 51985-034) were purchased from
Gibco (Thermo Fisher Scientific, Inc.). DMEM-F12 (cat. no.
SH30023.01B) and MEM (cat. no. SH30024.01B) were purchased from
HyClone (GE Healthcare Life Sciences). L-glutamine (cat. no.
1294808), sodium pyruvate (cat. no. 792500), non-essential amino
acid (NEAA) solution (100X; cat. no. M7145) and MTT kit (cat. no.
M5655) were purchased from Sigma-Aldrich (Merck KGaA).
Penicillin-streptomycin (100X; cat. no. P1400-100), trypsin-EDTA
(0.25%; cat. no. T1300-100) and agarose gel DNA extraction (cat.
no. D1200) were purchased from Beijing Solarbio Science &
Technology Co., Ltd. DMSO (cat. no. 302A0316) was purchased from
Ameresco, Inc. Tween-20 (cat. no. BYL40713) and Tris-HCl (pH 7.5)
(cat. no. BYL40909) were purchased from JRDUN Biotechnology Co.,
Ltd. The Hoechst Staining kit (cat. no. C003), DAPI (cat. no.
C1002) and 3% bovine serum albumin (BSA; cat. no. ST023) were
purchased from Beyotime Institute of Biotechnology. LA
Taq® (cat. no. RR02MA) and DNA marker (cat. no. 3590Q)
were purchased from Takara Biotechnology Co., Ltd. T4 DNA Ligase
(cat. no. 15224017), Anza™ 10-Pack restriction enzyme starter kit
(for the digestion of HindIII-XhoI sites; cat. no.
IVGN3006) and Lipofectamine® 2000 (cat. no. 11668-019)
were purchased from Invitrogen (Thermo Fisher Scientific, Inc.).
High Pure dNTPs (cat. no. AD101) and competent DH5α cells (cat. no.
CD521) were purchased from Transgene SA. The plasmid extraction kit
was purchased from Omega Bio-Tek, Inc. (cat. no. D6940), and
pCDNA3.1(+) was purchased from Addgene, Inc. (cat. no. V790-20).
The following antibodies were all purchased from Abcam with human
species reactivity: pSer129-α-syn (cat. no. ab51253), PI3K (cat.
no. ab86714), phosphorylated (p)-PI3K (cat. no. ab151549), mTOR
(cat. no. ab2732), p-mTOR (cat. no. ab109268), p70S6K (cat. no.
ab32529), p-p70S6K (cat. no. ab5231) and microtubule-associated
proteins 1A/1B light chain 3B (LC3B; cat. no. ab48394). The
following antibodies were all purchased from CST with human species
reactivity: Akt (cat. no. 4685), p-Akt (cat. no. 4060) and GAPDH
(cat. no. 5174). The following secondary antibodies were purchased
from Beyotime Institute of Biotechnology: Horseradish peroxidase
(HRP)-conjugated goat anti-rabbit IgG (cat. no. A0208),
HRP-conjugated donkey anti-goat IgG (cat. no. A0181),
HRP-conjugated goat anti-mouse IgG (cat. no. A0216), Alexa Fluor
555-conjugated donkey anti-rabbit IgG (H+L) (cat. no. A0453), Alexa
Fluor 488-conjugated goat anti-rabbit IgG (H+L) (cat. no. A0423)
and Alexa Fluor 488-conjugated goat anti-mouse IgG (H+L) (cat. no.
A0428).
Cell culture
SH-SY5Y cells (cat. no. BNCC100465; BeNa Culture
Collection) were cultured in medium containing 10% FBS, 1%
penicillin-streptomycin, 1% L-glutamine, 1% sodium pyruvate, 1%
NEAA, 42.5% MEM and 42.5% DMEM-F12 under 5% CO2 at 37°C.
Cells were seeded onto poly-L-lysine-coated plates and passaged
when they reached 60–70% confluence.
α-Syn plasmid transfection
Human WT/A53T-α-syn overexpression plasmids were
established by JRDUN Biotechnology. The PCR primers for the WT
α-syn in the vector plasmid pCDNA3.1(+) were as follows: Forward
primer,
5′-CAAGCTGGCTAGCGTTTAAACTTAAGCTTGCCGCCACCATGGATGTATTCATG-3′; and
reverse primer,
5′-GGGTTTAAACGGGCCCTCTAGACTCGAGTTAGGCTTCAGGTTCGTAGTC-3′. The PCR
primers for the A53T α-syn in the vector plasmid pCDNA3.1(+) were
as follows: Forward primer,
5′-GGGAGTGGTGCATGGTGTGACAACAGTGGCTGAGAAGACC-3′; and reverse primer,
5′-GGTCTTCTCAGCCACTGTTGTCACACCATGCACCACTCCC-3′. The two new
plasmids were verified by digestion with the restriction enzymes
HindIII and XhoI, according to the manufacturer's
guidelines, and were sent to General Biosystems, Ltd. for
sequencing. The sequencing results for full-length WT α-syn were
then aligned with a Homo sapiens SNCA mRNA sequence
(accession no. NM_001146055.1) from NCBI (https://www.ncbi.nlm.nih.gov/), while the mutation
A53T in α-syn was determined by aligning the sequence with
full-length WT α-syn using Cluster Omega Alignment Tools
(https://www.ebi.ac.uk/Tools/msa/clustalo/) (19). For the following experiments, cells
(5×105/well) were transfected with WT/A53T-α-syn plasmid
(1.6 µg) or the blank control pCDNA3.1(+) (1.6 µg) for 6 h, using
Lipofectamine® 2000, according to the manufacturer's
instructions.
Measurement of cell viability
Cell viability was determined using the MTT assay,
according to the manufacturer's instructions. First, SH-SY5Y cells
were seeded into a 96-well plate at a concentration of
3×103/well, with 100 µl medium as the blank. After an
overnight incubation, plates were first transfected with plasmids
[mock (blank control), WT and A53T] for 6 h before the transfected
solution was removed, and plates were then washed three times with
PBS. Cells were then treated with one of a range of solutions
(DMSO; 10 or 20 µM SAL; 10 µM SAL + 5 µM Rap; 10 µM SAL + 1 mM
3-MA; or serum-free medium as the positive control) for various
time periods (0, 24 or 48 h), according to the experimental
protocols. Subsequently, 20 µl MTT (5 mg/ml) was added to each well
and incubated at 37°C for 4 h. The medium was removed, and 150 µl
DMSO was added to each well. After shaking for 10 min, the
absorbance at 490 nm was measured in a microplate reader (Bio-Rad
Laboratories, Inc.).
Hoechst staining
SH-SY5Y cells (Mock, WT or A53T, according to the
aforementioned treatments) were plated in 6-well plates at a
density of 1×105 cells/well and treated with DMSO, or 10
or 20 µM SAL for 24 h. The cells were washed in PBS three times and
incubated in 0.5 ml Hoechst 33258 (Beyotime Institute of
Biotechnology; cat. no. C-0003) solution (5 µg/ml) at 4°C
overnight. Finally, fluorescence microscopy (magnification, ×200)
was performed to observe the nuclear changes in SH-SY5Y cells with
the various treatments. Each group was analyzed in triplicate.
Western blotting
Treated cells were harvested and lysed in lysis
buffer with complete protease inhibitors and phosphatase inhibitors
on ice. Proteins were extracted on ice for >1 h with occasional
gentle vortexing, and debris and insoluble materials were pelleted
by centrifugation at 14,000 × g for 10 min at 4°C. Pierce™ BCA
Protein Assays (Thermo Fisher Scientific, Inc.; cat. no. 23225)
were used to determine the concentration of protein, according to
the manufacturer's protocol. A total of 20 µg total proteins were
loaded for SDS-PAGE (10–15%), separated, and transferred onto a
nitrocellulose membrane. The immunoblots were incubated in 3%
bovine serum albumin (BSA), 10 mmol/l Tris-HCl (pH 7.5), 1 mmol/l
EDTA, and 0.1% Tween-20 at room temperature for 1 h before being
probed with primary (overnight at 4°C) and appropriate secondary
antibodies (1:1,000, for 1 h at 37°C). Primary antibody dilutions
were as follows: α-Syn, 1:6,000; pSer129-α-syn, 1:5,000; PI3K,
1:1,000; p-PI3K, 1:1,000; mTOR, 1:2,000; p70S6K, 1:5,000; p-p70S6K,
1:1,000; Akt, 1:1,000; p-Akt, 1:2,000; LC3B, 1:2,000; and GAPDH,
1:2,000. Protein bands were visualized using enhanced
chemiluminescence (cat. no. WBKLS0100; Merck KGaA), and protein
quantification was performed using Image J software (version 1.52;
National Institutes of Health).
Immunocytochemistry
SH-SY5Y cells treated with various solutions were
grown on poly-L-lysine-coated slides and fixed with 4%
paraformaldehyde for 15 min at room temperature, permeabilized with
0.1% Triton X-100 for 30 min, and then blocked in 2% BSA in PBS for
1 h at room temperature. Cells were washed three times with PBS and
incubated at 4°C overnight with primary antibodies against
pSer129-α-syn (1:500) or LC3B (1:500). The next day, the cells were
washed with PBS and labeled with Alexa Fluor 555-conjugated donkey
anti-rabbit IgG (H+L) and Alexa Fluor 488-conjugated goat
anti-rabbit IgG (H+L) (both 1:200) for 2 h at room temperature. The
nuclei were labeled with DAPI for 10 min at 4°C. The final images
were observed using a laser-scanning confocal microscope
(magnification, ×400).
Statistical analysis
All statistics were analyzed with SPSS software 14.0
(SPSS, Inc.). All experiments were performed three times, and the
data are presented as the mean ± SD. One-way ANOVA with Tukey's
post hoc test was used to assess multiple groups of data and the
Student's t-test was used to compare two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
SAL protects PD model neurons with
α-syn aggregation from apoptosis
SH-SY5Y cells were transfected with blank vector
(mock), WT α-syn plasmid, or the A53T mutation of α-syn (A53T
α-syn) plasmid, giving a total of three experimental groups:
Mock-SH, WT-SH, and Mut-SH, respectively. Firstly, it was indicated
that both WT and A53T-α-syn plasmid were successfully overexpressed
in cells transfected with the overexpression plasmids by western
blotting (Fig. 1A). Hence, W-SH
and M-SH were established as PD model neurons with α-syn
aggregation. Neurons from each group were treated with three
different SAL concentrations (0, 10 and 20 µM), and examined for
cell apoptosis at various time points using the MTT test and
Hoechst staining. Among the three groups with no SAL treatment
(Mock-SH-0, WT-SH-0 and Mut-SH-0), the cell proliferation indexes
of WT-SH-0 and Mut-SH-0 were both lower than that of Mock-SH-0,
especially at the 48 h time point. The result suggested that
increased α-syn expression in WT-SH and Mut-SH and potential
subsequent formation of α-syn aggregates affected cell
proliferation in a time-dependent manner; thus, a cell model of PD
was established (Fig. 1B). In the
SAL-treated groups, the cell proliferation index was increased in
both the WT-SH and Mut-SH groups in a dose-dependent manner
(Fig. 1C). On the other hands, the
results of the Hoechst staining indicated that SAL could protect
SH-SY5Y with α-syn aggregation (WT or A53T Mut) from apoptosis
(Fig. 1D and E).
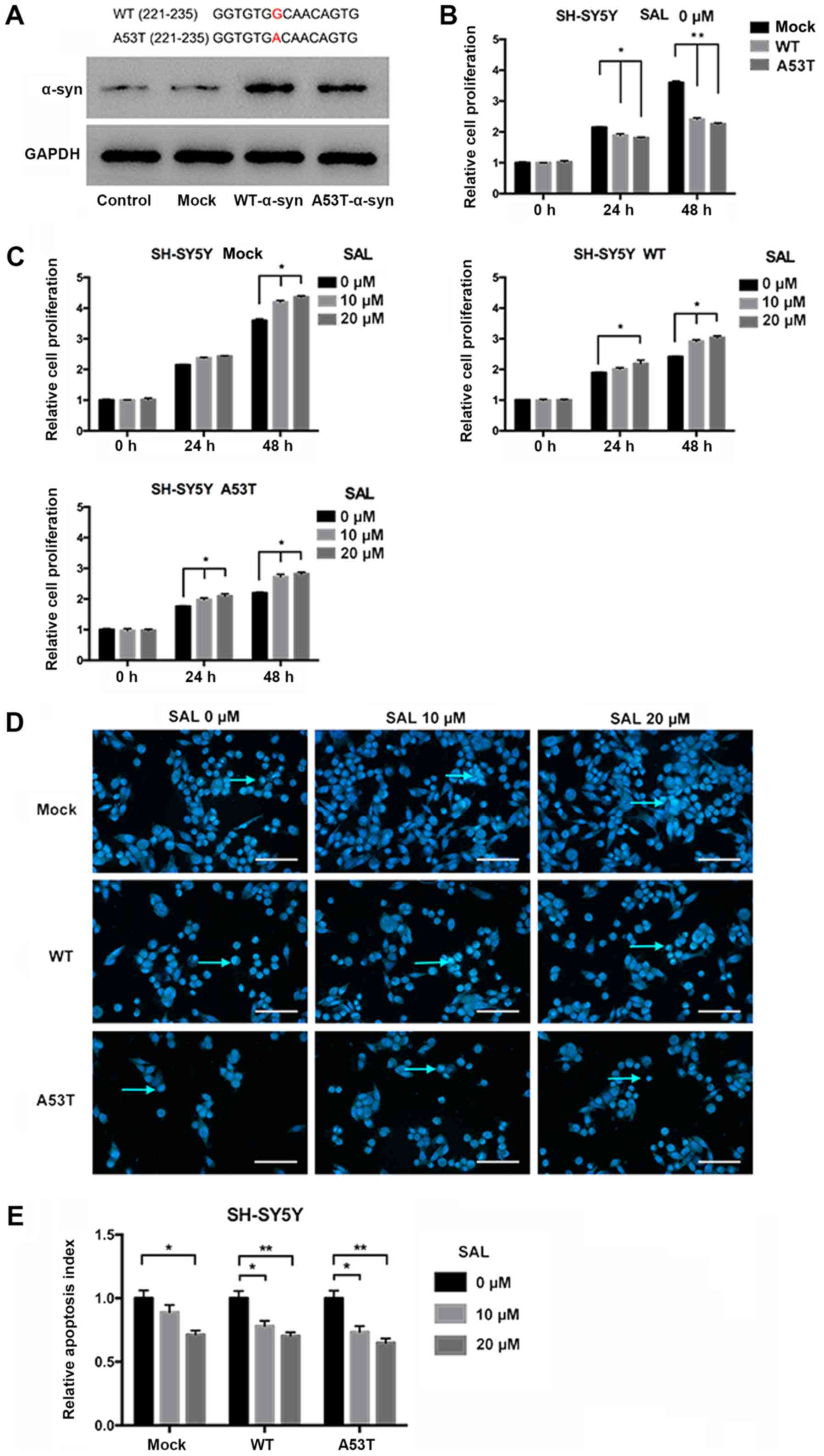 | Figure 1.SAL protects Parkinson's disease
model neurons with α-syn aggregation in a dose-dependent manner.
(A) SH-SY5Y cells transfected with WT- or A53T-α-syn overexpression
plasmids were assessed for overexpressed WT or A53T α-syn by
western blotting. The red letters above indicate the mutation site
in A53T α-syn. (B) Relative cell proliferation index comparison of
the three neuron groups, mock, WT and A53T, without SAL treatment
(0 µM SAL). The proliferation indexes of both the WT and A53T
groups were decreased compared to that of the mock group at 24 and
48 h. (C) Effects of SAL treatment on the viability of the three
groups of SH-SY5Y cells. The neurons of each group were treated
with various concentrations of SAL (0, 10 and 20 µM) for 24 h and
48 h. (D) Hoechst staining of each group of neurons with varying
concentrations of SAL treatment. Apoptotic neurons, with compact
and heavily stained nuclei, are indicated by the cyan arrows (scale
bar, 100 µm). (E) The relative apoptosis indexes of SH-SY5Y cells
were significantly lower in the SAL treatment (10 or 20 µM)
subgroups than in the no SAL treatment (0 µM) subgroup, in WT or
A53T SH-SY5Y cells. One-way ANOVA was used to assess multiple
groups of data. All data are expressed as the mean ± SD.
*P<0.05; **P<0.01. SAL, salidroside; A53T, A53T mutation; WT,
wild-type; α-syn, α-synuclein. |
SAL inhibits α-syn phosphorylation and
restores autophagy in PD model neurons
The phosphorylation of α-syn (at Ser129) is
considered a promoter of abnormal α-syn aggregation. SH-SY5Y cells
transfected with the three different plasmids (Mock-SH, W-SH and
Mut-SH) were treated with three different SAL concentrations (0, 10
and 20 µM), and cells cultured in serum-free medium were used as
the positive control (20). The
level of α-syn phosphorylation was decreased by SAL in all three
groups, and in a dose-dependent manner in both the WT-SH and Mut-SH
groups (Fig. 2A and B).
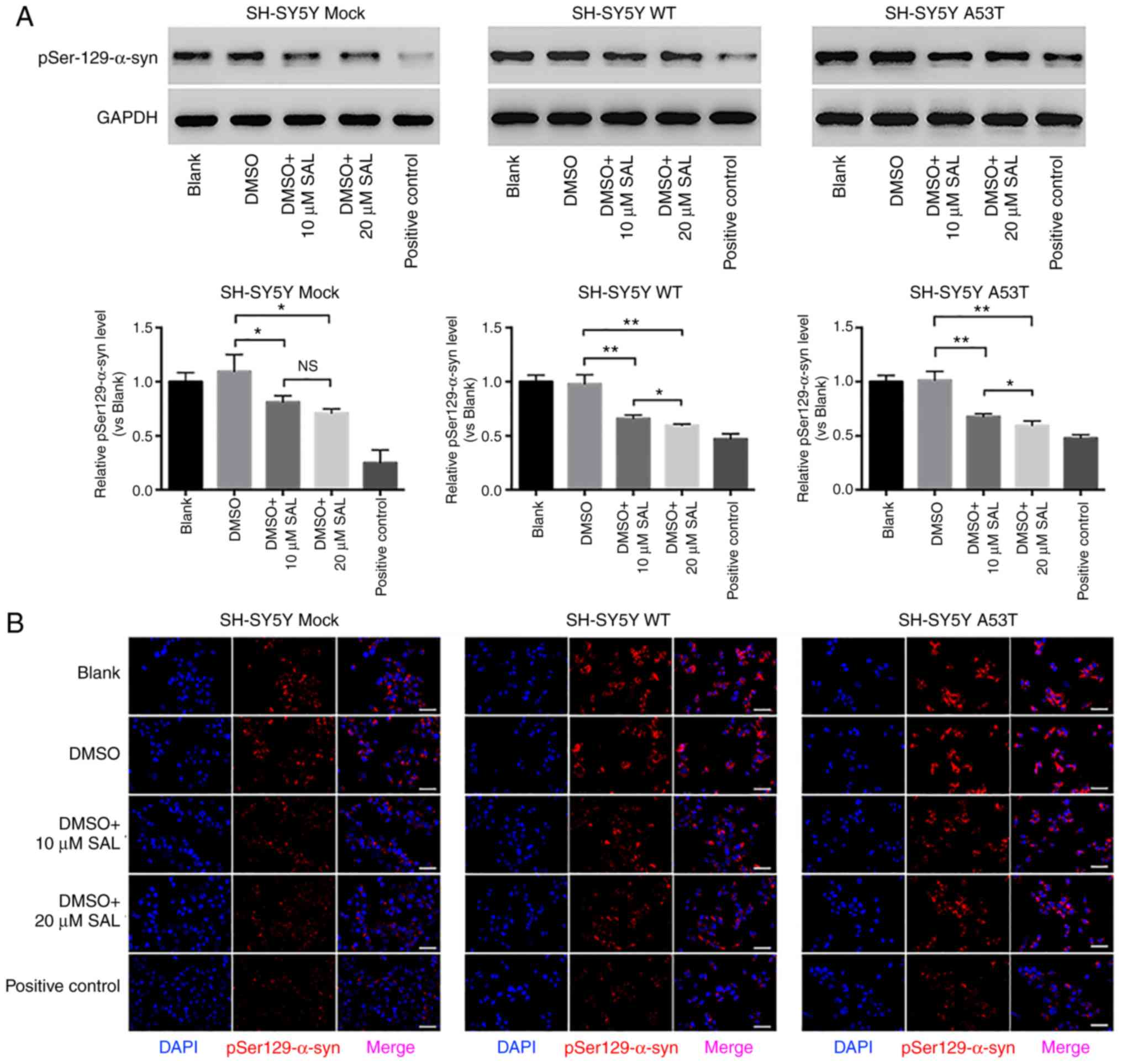 | Figure 2.SAL decreased the expression of
pSer129-α-syn and restored autophagy in SH-SY5Y Parkinson's disease
model neurons. (A) Western blot analysis of pSer129-α-syn in each
group of SH-SY5Y cells (mock, WT and A53T) with various treatments
(Blank, DMSO, 10 µM SAL, 20 µM SAL and positive control). (B)
Immunofluorescence staining of pSer129-α-syn (red) in each group of
SH-SY5Y cells (mock, WT and A53T) with various treatments at 24 h
(scale bar, 50 µm). (C) Western blot analysis of LC3-II/LC3-I in
each group of SH-SY5Y cells (mock, WT and A53T) with various
treatments (blank, DMSO, 10 µM SAL, 20 µM SAL and positive
control). (D) Immunofluorescence staining of LC3-II (green) in each
group of SH-SY5Y cells (mock, WT, and A53T) with various treatments
at 24 h (scale bar, 50 µm). *P<0.05; **P<0.01; ***P<0.001.
NS, not significant; SAL, salidroside; A53T, A53T mutation; WT,
wild-type; α-syn, α-synuclein; DMSO, dimethyl sulfoxide;
pSer129-α-syn, phosphorylated α-syn; LC3, microtubule-associated
proteins 1A/1B light chain 3. |
LC3 is currently considered to be a marker of
autophagy. LC3-I is cytosolic, and LC3-II is membrane bound, and
this was the most fully characterized mammalian protein identified
to be specifically and essentially associated with autophagosome
membranes (21).
Immunofluorescence results demonstrated that the expression of
LC3-II in W-SH-0 and M-SH-0 was lower than that in B-SH-0,
indicating that the autophagic function of both W-SH and M-SH was
impaired. The levels of LC3-II in all three cell models (B-SH, W-SH
and M-SH) were increased by SAL, and the results of the western
blot experiments indicated that the ratio of LC3-II:LC3-I was
increased by SAL treatment, also in a dose-dependent manner
(Fig. 2C and D). These results
implied that SAL could repair autophagic function in SH-SY5Y cells
that have α-syn aggregation.
Protective effect of SAL against α-syn
aggregation in PD model neurons is affected by Rap and 3-MA
The M-SH model was used in this experiment, and
there were five treatment groups for this model: DMSO, 10 µM SAL,
10 µM SAL + 5 µM Rap (an mTOR inhibitor, used to induce mild
autophagy in neurons), 10 µM SAL + 1 mM 3-MA (an autophagy
inhibitor), and cells cultured in serum-free medium (as the
positive control). The MTT results indicated that the relative cell
proliferation value of the 10 µM SAL + 5 µM Rap group was higher
than that of the 10 µM SAL group, while the value of the 10 µM SAL
+ 1 mM 3-MA group was much lower than that of the 10 µM SAL group.
These results indicated that the protective effect of SAL on the PD
model neurons (Fig. 1) could be
enhanced by Rap, but was inhibited by 3-MA, especially at the 48-h
time point (Fig. 3A and B). In
addition, Rap intensified the inhibitory effect of SAL on α-syn
phosphorylation, but 3-MA appeared to attenuate the effect of SAL
on α-syn phosphorylation (Fig. 3C and
D), indicating that SAL may protect SH-SY5Y PD model neurons
against α-syn aggregation through autophagy via PI3K/mTOR
signaling.
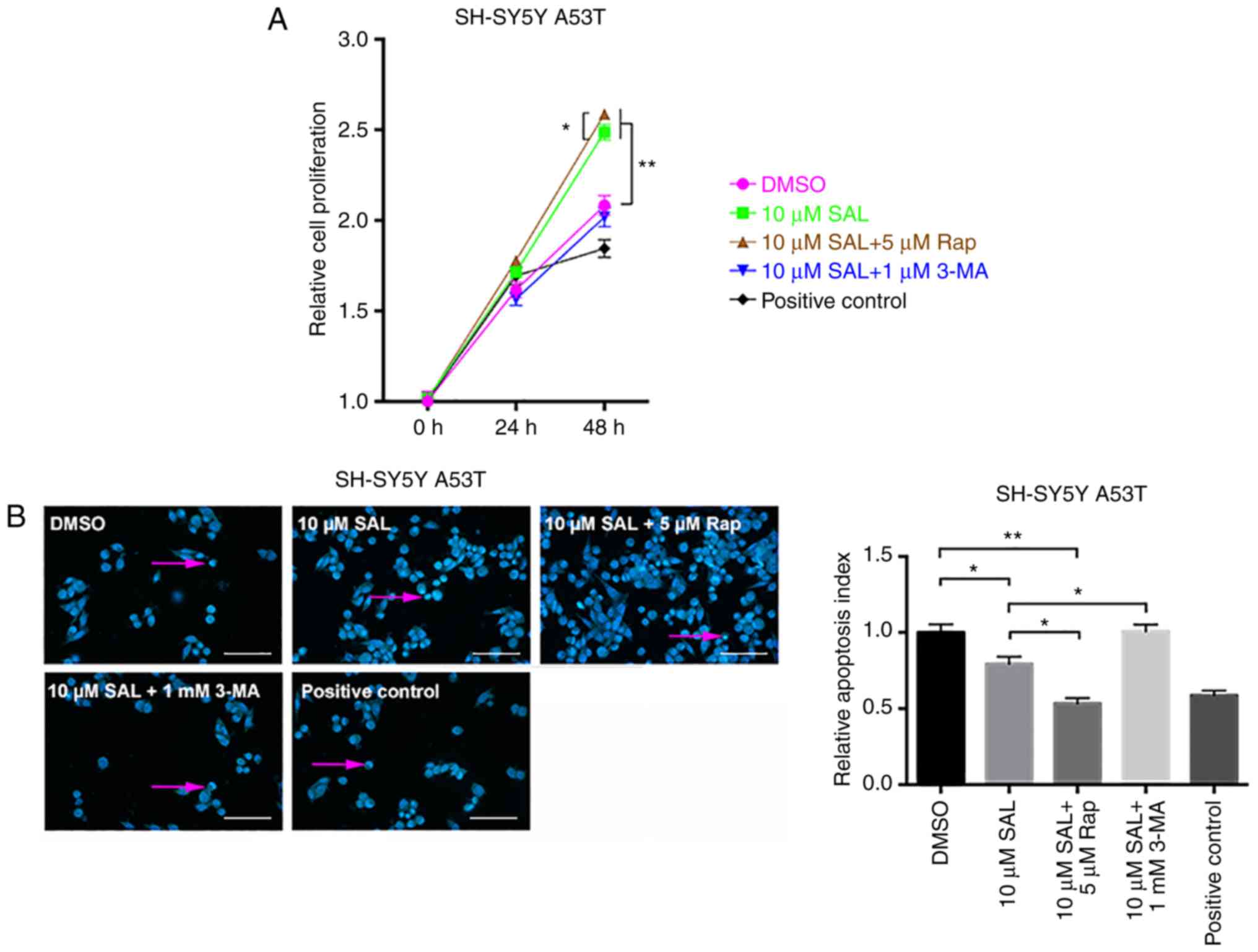 | Figure 3.Rapamycin and 3-methyladenine
influenced the protective effect of SAL on SH-SY5Y Parkinson's
disease model neurons with α-syn aggregation. (A) Relative cell
proliferation index comparison of SH-SY5Y PD model neurons with
various treatments (DMSO, 10 µM SAL, 10 µM SAL + 5 µM Rap, 10 µM
SAL + 1 mM 3-MA and positive control) at different time points (0,
24 and 48 h). (B) Hoechst staining of A53T SH-SY5Y cells with
various treatments (DMSO, 10 µM SAL, 10 µM SAL + 5 µM Rap, 10 µM
SAL + 1 mM 3-MA and positive control). Apoptotic neurons, with
compact and heavily stained nuclei, are indicated by pink arrows
(scale bar, 100 µm). (C) Western blot analyses of A53T SH-SY5Y
cells with various treatments (DMSO, 10 µM SAL, 10 µM SAL + 5 µM
Rap, 10 µM SAL + 1 mM 3-MA and positive control) at 24 h for the
pSer129-α-syn expression level. (D) Immunofluorescence staining of
pSer129-α-syn (red) of each group of A53T SH-SY5Y cells with
various treatments at 24 h (scale bar, 50 µm). One-way ANOVA was
used to assess multiple groups. All data are presented as the mean
± SD. *P<0.05; **P<0.01. SAL, salidroside; A53T, A53T
mutation; Rap, rapamycin; 3-MA, 3-methyladenine; α-syn,
α-synuclein; DMSO, dimethyl sulfoxide; pSer129-α-syn,
phosphorylated α-syn. |
SAL preserves the autophagic function
of M-SH cells via the inhibition of mTOR/p70S6K, independent of
PI3K/Akt signaling
Additionally, in Mut-SH neurons, SAL (10 µM)
treatment decreased the expression of phosphorylated mTOR and
p70S6K, but increased the ratio of LC3-II to LC3-I. The impact of
SAL treatment was promoted by Rap (5 µM) and reversed by 3-MA (1
mM). Conversely, the levels of phosphorylated PI3K and Akt, two
upstream proteins of PI3K/Akt/mTOR signaling, were both increased
by SAL, and treatment with Rap further elevated the levels of both
phosphorylated proteins, while 3-MA treatment reduced their levels
(Fig. 4). These results indicated
that SAL may preserve the autophagic function of SH-SY5Y cells with
α-syn aggregation by inhibiting mTOR/p70S6K, but that this effect
is independent of PI3K/Akt signaling.
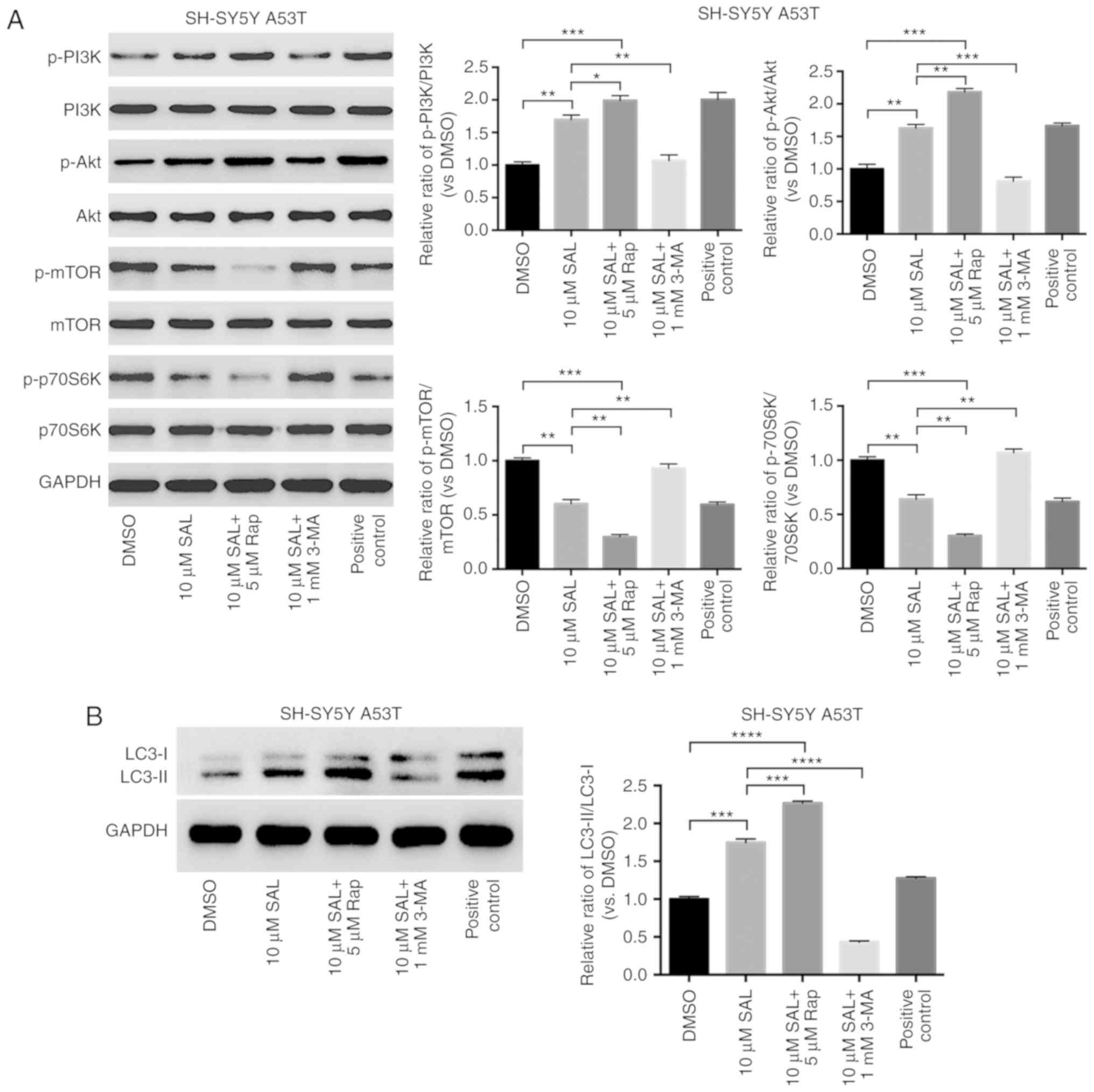 | Figure 4.SAL preserved the autophagy of A53T
SH-SY5Y cells by inhibiting the phosphorylation of mTOR/p70S6K.
Western blot analyses of A53T SH-SY5Y cells with various treatments
(DMSO, 10 µM SAL, 10 µM SAL + 5 µM Rap, 10 µM SAL + 1 mM 3-MA, and
the positive control) at 24 h for the expression level of (A) PI3K,
Akt, mTOR, p70S6K, p-PI3K, p-Akt, p-mTOR, p-p70S6K, and (B)
LC3-II/LC3-I. The ratios of LC3-II to LC3-I, as well as those of
the phosphorylated proteins to the total relevant proteins in
PI3K/Akt/mTOR/p70S6K signaling, were compared between the groups
above. (C) Immunofluorescence staining of LC-II (green) in each
group of A53T SH-SY5Y cells with various treatments at 24 h (scale
bar, 50 µm). *P<0.05; **P<0.01; ***P<0.001;
****P<0.0001. SAL, salidroside; A53T, A53T mutation; Rap,
rapamycin; 3-MA, 3-methyladenine; DMSO, dimethyl sulfoxide; LC3,
microtubule-associated proteins 1A/1B light chain 3; p,
phosphorylated. |
Discussion
Accumulation of misfolded α-syn into aggregates is
considered part of the pathogenesis of PD. During the pathogenic
process, the misfolded α-syn forms insoluble protein amyloid
fibrils known as Lewy bodies, the pathological hallmark of this
disease (22). Aside from point
mutations (e.g. p.A53T, p.A30P, and p.E46K) and single nucleotide
polymorphisms of SNCA that alter the α-syn protein
structure, gene multiplications or normal aging can also lead to
significantly increased cytoplasmic levels of soluble α-syn, and
are also associated with PD (23).
This suggests that simply decreasing the cellular levels of α-syn
protein, which is the source of the misfolded α-syn or α-syn
aggregates, is a possible therapeutic strategy against PD,
especially in elderly patients with sporadic disease. A PD model
was established by transfecting WT or p.A53T mutant (to increase
the propensity for aggregate formation) (24) α-syn-overexpressing plasmids into
SH-SY5Y cells, which is a common cell model used in PD
research.
It is known that the α-syn protein undergoes
extensive post-translational modifications, such as
phosphorylation, nitration and dopamine modification, which all
tend toward the oligomerization of α-syn (25). Phosphorylation at Ser129 (pSer129)
of α-syn is the most prevalent modification in PD brains (26). More importantly, the
phosphorylation of α-syn at Ser129 can promote the accumulation of
oligomeric α-syn in SH-SY5Y cells (27), cause neuronal loss in transgenic
mice overexpressing α-syn (28),
and affect α-syn solubility and subcellular distribution (29). Therefore, phosphorylation at Ser129
is implicated in the PD process, and was used as a disease
indicator in the present study.
Autophagy has been demonstrated to be cytoprotective
during brain aging and neurodegeneration (30). In particular, α-syn can be degraded
either by autophagy or by proteasomes, but the degradation of toxic
oligomeric α-syn can only be initiated by the autophagy-lysosome
pathway, rather than by proteasomes (31). Furthermore, mutant α-syn actually
inhibits chaperone-mediated autophagy (32), while WT α-syn inhibits
macroautophagy (33). It thus
seems reasonable that breaking the interaction between α-syn and
the autophagy pathway may be an effective therapy for PD, although
the mechanism is still not well understood.
SAL is the main bioactive component in Rhodiola
rosea L., a botanical medicine that has historically been used
widely in Asia, Europe and North America to prevent and treat a
large variety of diseases (34).
It has been reported that the pharmacological effects of SAL are
effective against Alzheimer's disease, PD, stroke, depression,
cancer, and diabetes, and provide organ protection and
neurofunctional improvement. Although some studies have
investigated the neuroprotective role of SAL, the exact mechanisms
are not clear (35). The present
study provides the first evidence, to the best of our knowledge,
that SAL protects neurons in a PD model (SH-SY5Y with pathogenic
α-syn) from misfolded α-syn aggregate toxicity, by stimulating
autophagy and thereby reducing α-syn aggregation. Furthermore, this
study demonstrated that mTOR/p70S6K signaling mediated the effects
of SAL on PD by inducing autophagy.
Previous studies have confirmed that SAL can
efficiently protect PD model cells via various mechanisms. It has
been demonstrated that SAL has protective effects on MPTP/MPP+
models of PD by reducing α-syn aggregation through modulation of
the ROS-NO-related mitochondrial pathway, both in vitro and
in vivo (36). Moreover,
SAL was also reported to prevent 1-methyl-4-phenylpyridinium
(MPP+)-induced apoptosis in PC12 cells, partly through activation
of the PI3K/Akt pathway (37).
MPP+, the active metabolite of MPTP, acts as a selective toxin for
DA neurons, finally leading to oxidative stress in the neurons via
the ROS/NO-related mitochondrial pathway that promotes α-syn
aggregation. These factors therefore interact in the
pathophysiology of PD (38,39).
In the present study, SH-SY5Y cells overexpressing WT/A53T-α-syn
were the PD cell model, and numerous misfolded α-syn aggregates
were the direct pathogenic factor. More importantly, it was
verified that SAL could decrease the level of phosphorylated α-syn,
which is the source of α-syn aggregation, and clear α-syn
aggregation by autophagy through mTOR/p70S6K activation,
independently of PI3K/Akt. However, it remains possible that SAL
may repair the abnormal ROS/NO mitochondrial pathway that was
secondarily impaired by α-syn aggregation in the present study.
Additionally, the PI3K/Akt pathway is involved in survival and the
inhibition of apoptosis in different cells via PI3K activity and
the phosphorylation of Akt at serine residue 473 (Ser473), leading
to the inhibition of apoptotic machinery molecules such as Bcl-2,
BAD, Caspase 9 and Fas ligand (40). Therefore, the results from the
present study and a previously published study (33) are not paradoxical; that is, SAL may
rescue PD model cells not only via autophagy, induced by the
inhibition of mTOR/p70S6K, but also via the promotion of survival
through the direct activation of PI3K/Akt. Moreover, mTOR
inhibition could stimulate the PI3K/Akt pathway via a negative
feedback mechanism, so the anti-apoptosis effect of PI3K/Akt may be
indirectly augmented. Lastly, it is not clear how mTOR may be
inhibited by SAL, and this is a question for future research.
Besides autophagy, the ubiquitin-proteasome system
(UPS) is the major degradation pathway in eukaryotic cells. The two
systems share several common characteristics, and constrain each
other (41). Previously, Li et
al (42) used
6-hydroxydopamine and overexpression of WT-/A30P-α-syn to establish
a PD cell model in SH-SY5Y cells; it was found that SAL could
promote α-syn clearance and protect SH-SY5Y cells by restoring UPS
activity, with the inhibition of autophagy. Therefore, although the
present study demonstrated that SAL promoted autophagy via the
inhibition of mTOR/p70S6K signaling to protect SH-SY5Y cells, it is
also possible that SAL may impact signaling to maintain UPS
function. In future research, it will be worthwhile to investigate
the relationship between these two systems in PD pathogenesis, in
order to develop new and improved pharmacological solutions for PD
treatment.
In conclusion, the present study demonstrated for
the first time, to the best of our knowledge, that SAL could
inhibit the phosphorylation of α-syn at Ser129, as well as the
formation of WT/A53T-α-syn aggregates in SH-SY5Y cells, by inducing
autophagy via the inhibition of mTOR/p70S6K, independent of the
PI3K/Akt pathway. These results suggest that SAL acts as a
potential protective agent for DA neurons by restoring autophagic
function and eliminating pathogenic α-syn aggregation. However, it
is necessary to further investigate the mechanisms underlying the
protective effect of SAL on PD model neurons, to better understand
the potential clinical implications for PD and other
neurodegenerative diseases in the future.
Acknowledgements
The authors would like to thank Dr Bronwen Gardner
for editing the English text of a draft of this manuscript.
Funding
This work was supported by the National Natural
Science Foundation of China (grant nos. 81501113, 81502321 and
81771520), and funds from the Medical and Health Research Project
of Zhejiang Province (grant nos. 2015KYA001, 2017KY188, 2014KYA089
and 2014KYA093).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SC, JY and GM designed the study. SC, FC and JW
performed the major experiments. SC, FC, GM, JY, ZY, GW and CG
analyzed the data and discussed the results. SC and FC wrote the
manuscript. SC, FC and CG revised the manuscript and figures. All
authors approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PD
|
Parkinson's disease
|
|
α-syn
|
α-synuclein
|
|
SAL
|
salidroside
|
References
|
1
|
Jenner P, Morris HR, Robbins TW, Goedert
M, Hardy J, Ben-Shlomo Y, Bolam P, Burn D, Hindle JV and Brooks D:
Parkinson's disease-the debate on the clinical phenomenology,
aetiology, pathology and pathogenesis. J Parkinsons Dis. 3:1–11.
2013.PubMed/NCBI
|
|
2
|
Spillantini MG, Schmidt ML, Lee VM,
Trojanowski JQ, Jakes R and Goedert M: Alpha-synuclein in Lewy
bodies. Nature. 388:839–840. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jakes R, Spillantini MG and Goedert M:
Identification of two distinct synucleins from human brain. FEBS
Lett. 345:27–32. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Alves da Costa C, Dunys J, Brau F, Wilk S,
Cappai R and Checler F: 6-Hydroxydopamine but not
1-methyl-4-phenylpyridinium abolishes alpha-synuclein
anti-apoptotic phenotype by inhibiting its proteasomal degradation
and by promoting its aggregation. J Biol Chem. 281:9824–9831. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Del Tredici K and Braak H: Review:
Sporadic Parkinson's disease: Development and distribution of
alpha-synuclein pathology. Neuropathol Appl Neurobiol. 42:33–50.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Volpicelli-Daley LA, Luk KC, Patel TP,
Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ and Lee
VM: Exogenous α-synuclein fibrils induce Lewy body pathology
leading to synaptic dysfunction and neuron death. Neuron. 72:57–71.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Desplats P, Lee HJ, Bae EJ, Patrick C,
Rockenstein E, Crews L, Spencer B, Masliah E and Lee SJ: Inclusion
formation and neuronal cell death through neuron-to-neuron
transmission of alpha-synuclein. Proc Natl Acad Sci USA.
106:13010–13015. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hansen C, Angot E, Bergstrom AL, Steiner
JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, et al:
α-Synuclein propagates from mouse brain to grafted dopaminergic
neurons and seeds aggregation in cultured human cells. J Clin
Invest. 121:715–725. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luk KC, Kehm V, Carroll J, Zhang B,
O'Brien P, Trojanowski JQ and Lee VM: Pathological alpha-synuclein
transmission initiates Parkinson-like neurodegeneration in
nontransgenic mice. Science. 338:949–953. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mougenot AL, Nicot S, Bencsik A, Morignat
E, Verchère J, Lakhdar L, Legastelois S and Baron T: Prion-like
acceleration of a synucleinopathy in a transgenic mouse model.
Neurobiol Aging. 33:2225–2228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tyedmers J, Mogk A and Bukau B: Cellular
strategies for controlling protein aggregation. Nat Rev Mol Cell
Biol. 11:777–788. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Danzer KM, Kranich LR, Ruf WP,
Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR and McLean PJ:
Exosomal cell-to-cell transmission of alpha synuclein oligomers.
Mol Neurodegener. 7:422012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tanik SA, Schultheiss CE, Volpicelli-Daley
LA, Brunden KR and Lee VM: Lewy body-like alpha-synuclein
aggregates resist degradation and impair macroautophagy. J Biol
Chem. 288:15194–15210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Grech-Baran M, Syklowska-Baranek K and
Pietrosiuk A: Biotechnological approaches to enhance salidroside,
rosin and its derivatives production in selected Rhodiola spp. in
vitro cultures. Phytochem Rev. 14:657–674. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mao GX, Wang Y, Qiu Q, Deng HB, Yuan LG,
Li RG, Song DQ, Li YY, Li DD and Wang Z: Salidroside protects human
fibroblast cells from premature senescence induced by H(2)O(2)
partly through modulating oxidative status. Mech Ageing Dev.
131:723–731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li X, Ye X, Li X, Sun X, Liang Q, Tao L,
Kang X and Chen J: Salidroside protects against MPP(+)-induced
apoptosis in PC12 cells by inhibiting the NO pathway. Brain Res.
1382:9–18. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang L, Yu H, Zhao X, Lin X, Tan C, Cao G
and Wang Z: Neuroprotective effects of salidroside against
beta-amyloid-induced oxidative stress in SH-SY5Y human
neuroblastoma cells. Neurochem Int. 57:547–555. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu Y, Shi YP, Wu D, Ji YJ, Wang X, Chen
HL, Wu SS, Huang DJ and Jiang W: Salidroside protects against
hydrogen peroxide-induced injury in cardiac H9c2 cells via PI3K-Akt
dependent pathway. DNA Cell Biol. 30:809–819. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chojnacki S, Cowley A, Lee J, Foix A and
Lopez R: Programmatic access to bioinformatics tools from EMBL-EBI
update: 2017. Nucleic Acids Res. 45:W550–W553. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mohan N, Banik NL and Ray SK: Combination
of N-(4-hydroxyphenyl) retinamide and apigenin suppressed
starvation-induced autophagy and promoted apoptosis in malignant
neuroblastoma cells. Neurosci Lett. 502:24–29. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schaaf MB, Keulers TG, Vooijs MA and
Rouschop KM: LC3/GABARAP family proteins: Autophagy-(un)related
functions. FASEB J. 30:3961–3978. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lashuel HA, Overk CR, Oueslati A and
Masliah E: The many faces of alpha-synuclein: From structure and
toxicity to therapeutic target. Nat Rev Neurosci. 14:38–48. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chu Y and Kordower JH: Age-associated
increases of alpha-synuclein in monkeys and humans are associated
with nigrostriatal dopamine depletion: Is this the target for
Parkinson's disease? Neurobiol Dis. 25:134–149. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee VM, Giasson BI and Trojanowski JQ:
More than just two peas in a pod: Common amyloidogenic properties
of tau and alpha-synuclein in neurodegenerative diseases. Trends
Neurosci. 27:129–134. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barrett PJ and Timothy Greenamyre J:
Post-translational modification of alpha-synuclein in Parkinson's
disease. Brain Res. 1628:247–253. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fujiwara H, Hasegawa M, Dohmae N,
Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K and Iwatsubo
T: Alpha-Synuclein is phosphorylated in synucleinopathy lesions.
Nat Cell Biol. 4:160–164. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Smith WW, Margolis RL, Li X, Troncoso JC,
Lee MK, Dawson VL, Dawson TM, Iwatsubo T and Ross CA:
Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic
inclusion formation in SH-SY5Y cells. J Neurosci. 25:5544–5552.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schell H, Hasegawa T, Neumann M and Kahle
PJ: Nuclear and neuritic distribution of serine-129 phosphorylated
alpha- synuclein in transgenic mice. Neuroscience. 160:796–804.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou J, Broe M, Huang Y, Anderson JP, Gai
WP, Milward EA, Porritt M, Howells D, Hughes AJ, Wang X and
Halliday GM: Changes in the solubility and phosphorylation of
α-synuclein over the course of Parkinson's disease. Acta
Neuropathol. 121:695–704. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rubinsztein DC, Gestwicki JE, Murphy LO
and Klionsky DJ: Potential therapeutic applications of autophagy.
Nat Rev Drug Discov. 6:304–312. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bennett MC, Bishop JF, Leng Y, Chock PB,
Chase TN and Mouradian MM: Degradation of alpha-synuclein by
proteasome. J Biol Chem. 274:33855–33858. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cuervo AM, Stefanis L, Fredenburg R,
Lansbury PT and Sulzer D: Impaired degradation of mutant
alpha-synuclein by chaperone-mediated autophagy. Science.
305:1292–1295. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Winslow AR, Chen CW, Corrochano S,
Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM,
Ravikumar B, Imarisio S, et al: α-Synuclein impairs macroautophagy:
Implications for Parkinson's disease. J Cell Biol. 190:1023–1037.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Panossian A, Wikman G and Sarris J:
Rosenroot (Rhodiola rosea): Traditional use, chemical composition,
pharmacology and clinical efficacy. Phytomedicine. 17:481–493.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhong Z, Han J, Zhang J, Xiao Q, Hu J and
Chen L: Pharmacological activities, mechanisms of action, and
safety of salidroside in the central nervous system. Drug Des Devel
Ther. 12:1479–1489. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang S, He H, Chen L, Zhang W, Zhang X and
Chen J: Protective effects of salidroside in the
MPTP/MPP(+)-induced model of Parkinson's disease through
ROS-NO-related mitochondrion pathway. Mol Neurobiol. 51:718–728.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang L, Ding W, Sun H, Zhou Q, Huang J,
Li X, Xie Y and Chen J: Salidroside protects PC12 cells from
MPP+-induced apoptosis via activation of the PI3K/Akt
pathway. Food Chem Toxicol. 50:2591–2597. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Parihar MS, Parihar A, Fujita M, Hashimoto
M and Ghafourifar P: Alpha-synuclein overexpression and aggregation
exacerbates impairment of mitochondrial functions by augmenting
oxidative stress in human neuroblastoma cells. Int J Biochem Cell
Biol. 41:2015–2024. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tsang AH and Chung KK: Oxidative and
nitrosative stress in parkinson's disease. Biochim Biophys Acta.
1792:643–650. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Heras-Sandoval D, Perez-Rojas JM,
Hernandez-Damian J and Pedraza-Chaverri J: The role of
PI3K/AKT/mTOR pathway in the modulation of autophagy and the
clearance of protein aggregates in neurodegeneration. Cell Signal.
26:2694–2701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kocaturk NM and Gozuacik D: Crosstalk
between mammalian autophagy and the ubiquitin-proteasome system.
Front Cell Dev Biol. 6:1282018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li T, Feng Y, Yang R, Wu L, Li R, Huang L,
Yang Q and Chen J: Salidroside promotes the pathological
alpha-synuclein clearance through ubiquitin-proteasome system in
SH-SY5Y Cells. Front Pharmacol. 9:3772018. View Article : Google Scholar : PubMed/NCBI
|














