Introduction
Asthma is a chronic inflammatory disease of the
airways, characterized by airway hyperresponsiveness, chronic lung
inflammation and airway remodeling (1,2).
Asthma affects an estimated 358 million people worldwide, leading
to a mortality rate of 0.4 million in 2015. Inhaled corticosteroids
are the main method of pharmaceutical treatment; however, their
effects are not satisfactory. Smoking and occupational asthmagens
are the primary risk factors for asthma (3). Asthma affects people of all ages and
there are no effective clinical treatments due to the complex
pathophysiology of the condition (4). Therefore, understanding the molecular
basis underlying the pathophysiology of asthma, and the screening
of molecular markers for the development of therapeutic targets
remains critical.
Long non-coding RNAs (lncRNAs) are RNA transcripts
>200 nucleotides in length (5).
The various functions of lncRNAs have been reported with advances
in sequencing and bioinformatics analysis (6). Previous studies have suggested that
lncRNAs regulate gene expression by interacting with DNA, RNA or
proteins (5,6). For example, the lncRNA MAR1 acts as a
sponge for miR-487b, promoting skeletal muscle differentiation and
regeneration (7). Thus, these
transcripts may be considered as potential biomarkers and
therapeutic targets (8–12); however, the expression profiles of
lncRNAs and mRNAs in asthma remain unclear.
In the present study, microarray data was downloaded
from the National Center of Biotechnology Information (NCBI) Gene
Expression Omnibus (GEO) database for the analysis of
differentially expressed mRNA and lncRNA profiles in asthma. In
addition, the database was used to investigate asthma-associated
mRNAs and lncRNAs in airway epithelial brushings. Gene Ontology
(GO), Kyoto Encyclopedia of Genes and Genomes (KEGG),
protein-protein interaction (PPI) network and weighted correlation
network analyses (WGCNAs) were applied to investigate the role of
the differentially expressed lncRNAs and mRNAs in asthma. To the
best of our knowledge, the present study is the first to
comprehensively analyze mRNAs and lncRNAs in asthma. The findings
may provide in-depth molecular insight into the pathophysiology of
this condition.
Materials and methods
Tissue samples and data
acquisition
The gene expression data of the GSE67472 dataset was
downloaded from the NCBI GEO (www.ncbi.nlm.nih.gov/geo). The GEO is the largest
database for high-throughput molecular data, primarily gene
expression data (13), and
contains links to ~20,000 published studies comprising 800,000
samples derived from >1,600 organisms (14). Following screening, the GSE67472
dataset with a large sample size was selected for analysis to
ensure the stability and reliability of the data (15). Analysis was conducted via the
GPL1355 platform of Affymetrix Human Genome U133 Plus 2.0 (Thermo
Fisher Scientific, Inc.). The GSE67472 dataset containing the data
of airway epithelial gene expression in 62 patients with asthma and
43 healthy control samples from airway epithelial brushings was
included in the microarray analysis (16).
GeneChip probe re-annotation
Numerous lncRNAs were identified via the Affymetrix
microarray according to the lncRNA classification pipeline
developed in a previous study (17). The latest version of NetAffx
Annotation File (release 36; HG-U133_Plus_2 Annotations; CSV
format; 36 MB; accessed on 7th December 2016) was obtained from
Affymetrix (Thermo Fisher Scientific, Inc.). This annotation file
was mapped to the identifications (IDs) of the HG-U133_Plus_2 probe
sets. For the probe sets from the RefSeq database (RefSeq Release
89; www.ncbi.nlm.nih.gov/refseq/), probes labeled with
protein mixed (NP) were removed, but those with an ID beginning
with non-coding RNAs (NR) were included. For the probe sets from
the Ensembl database (ensemble 96; www.ensembl.org/index.html), the online software
BioMart (GRCh38.p11; http://asia.ensembl.org/biomart/martview/f0b2ccb5ee23510bf3f1e71d87ba7122)
was applied to convert Affymetrix microarray IDs to Ensembl IDs
with the corresponding gene type. Probe sets from NONCODE were
retained. Furthermore, genes annotated as ‘lincRNA’,
‘sense_intronic’, ‘processed_transcript’, ‘antisense’,
‘sense_overlapping’, ‘3prime_overlapping_ncRNA’ or ‘misc_RNA’ were
retained. Finally, probe set IDs annotated as ‘microRNA’ or
‘snoRNAs’, and other small RNAs were removed.
Data preprocessing
Affymetrix Expression Console (v1.4; http://www.affymetrix.com/support/technical/byproduct.affx?product=expressionconsole)
with Ro-bust microarray was applied to normalize the raw files.
Limma package (http://bioinf.wehi.edu.au/limma) in R (version 3.5.1;
http://cran.r-project.org/) was used to
identify differentially expressed lncRNAs and mRNAs among the
asthma and control groups via a t-test (18). Fold change (FC)>1.2 and
P<0.05 were set as the criteria for differential expression
(19). Hierarchical clustering was
conducted in R scripts.
GO and pathway enrichment
analysis
GO (geneontology.org) analysis, frequently used in
functional enrichment studies of large-scale genes (20), and KEGG (www.genome.jp/kegg) enrichment analysis were performed
to investigate the biological pathways that involve differentially
expressed mRNAs. In the present study, clusterProfiler (v3.12.0;
guangchuangyu.github.io/software/clusterProfiler) and Database for
Annotation, Visualization and Integrated Discovery tools (v6.8;
http://david.ncifcrf.gov/) were used to
analyze the functional enrichment conditions for dysregulated mRNAs
(21–23). The false discovery rate (FDR) was
calculated to correct the P-value and FDR<0.05 was selected as
the threshold for a statistically significant difference.
PPI network construction
Protein interactions were analyzed using the online
Search Tool for the Retrieval of Interacting Genes (v 10.5;
string-db.org) tool and a combined score of
>0.7 was used as the cut-off criterion (24–26).
PPI networks were subsequently generated using Cytoscape software
(v3.2.8; cytoscapeweb.cytoscape.org) (27,28).
Construction of the lncRNA-mRNA
WGCNA
The WGCNA package (29) in R was used to construct the
co-expression network of lncRNAs and mRNAs as follows: i) Network
construction, the weighted co-expression network of lncRNAs and
mRNAs was specified in its adjacency matrix (amn), which
encodes the network connection strength between nodes m and n. In
order to calculate the amn, the default approach was
employed, which defines the co-expression similarity
(Smn) as the absolute value of the correlation
coefficient between the nodes of m and n, and Smn=|cor
(am, an)|. The weighted adjacency
amn between two genes is proportional to their
similarity on a logarithmic scale with absolute value of the
correlation to a power β≥0.8, log (amn)=β × log
(Smn). Adjacency functions were obtained by using the
approximate scale-free topology criterion (30). The amn was converted
into a topology matrix; and ii) module detection: The Dynamic Tree
Cut and Static Tree Cut methods were applied to detect modules with
≥30 lncRNAs/genes (31). The
cluster dendrogram was visualization using the WGCNA package.
Results
Identification of differentially
expressed lncRNAs and mRNAs
Based on the screening criteria, FC>1.2 and
P<0.05, differentially expressed lncRNAs and mRNAs in the
GSE67472 dataset were identified. A total of 159 differentially
expressed lncRNAs and 1,261 mRNAs were identified. In total, 48
lncRNAs were found to be upregulated and 111 lncRNAs were found to
be downregulated while 744 mRNAs were found to be upregulated and
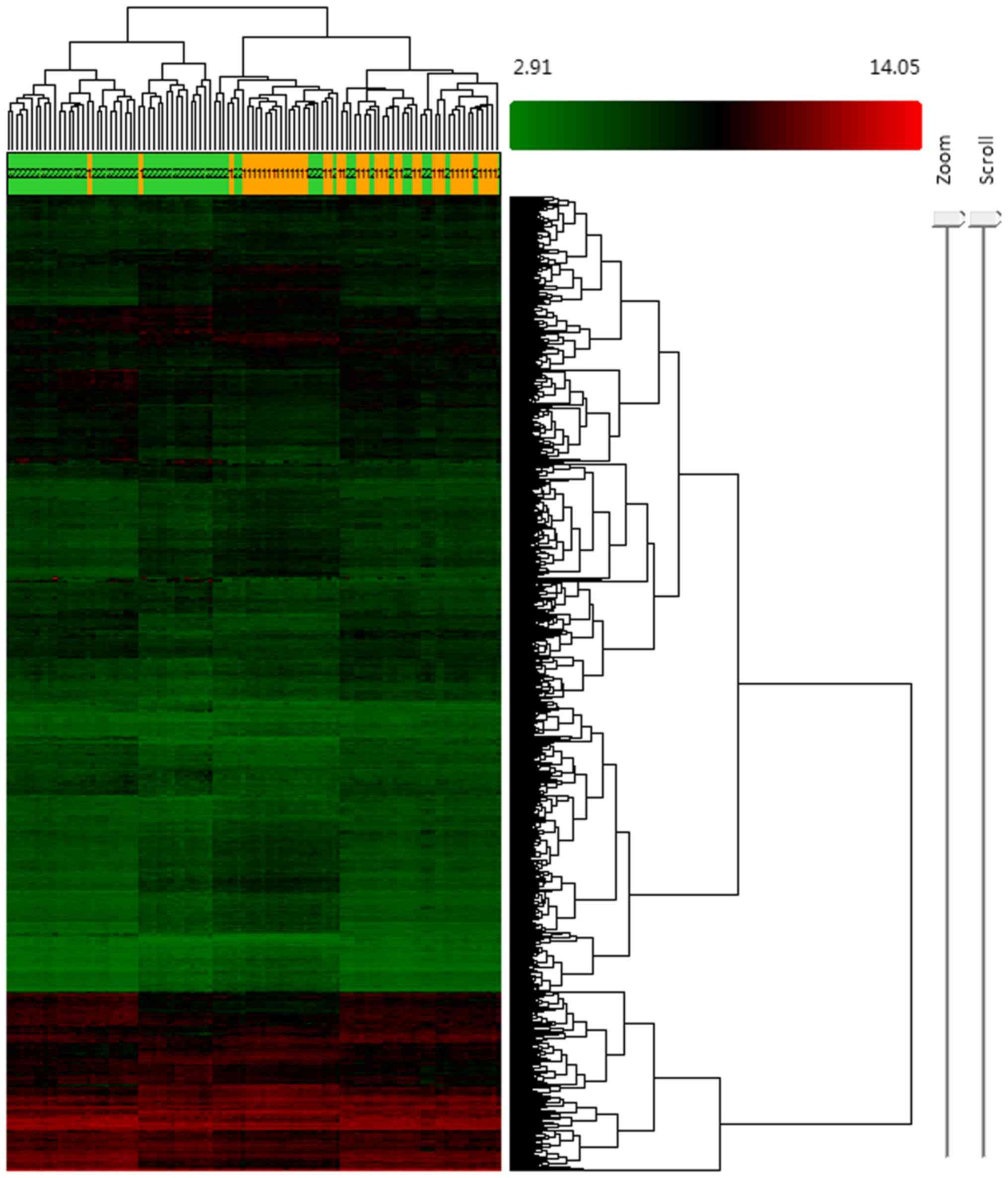
517 mRNAs were found to be downregulated. Hierarchical clustering
was conducted to evaluate the altered expression of lncRNAs and
mRNAs in the samples (Fig. 1).
Functional annotation and pathway
analysis for differentially expressed mRNAs
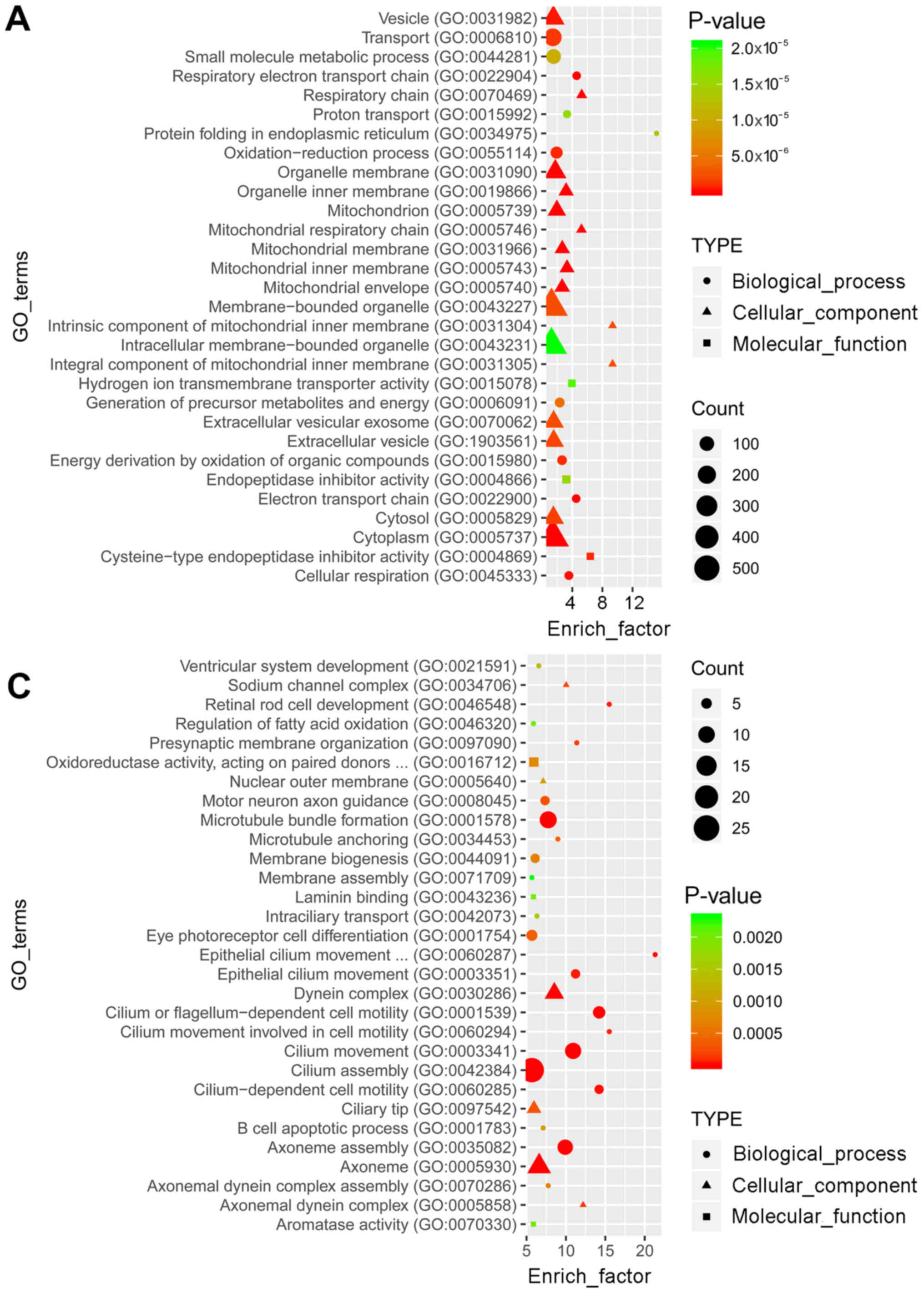
GO and KEGG pathway enrichment analyses were
performed to determine the functions of the identified
differentially expressed mRNAs. GO analysis revealed that the top
30 terms of the 2,184 and 1,605 GO terms were enriched in the
upregulated (Fig. 2A) and
downregulated (Fig. 2C) genes,
respectively. In the pathway analysis, the top 30 of the 245 and
219 pathways were enriched in the upregulated (Fig. 2B) and downregulated (Fig. 2D) genes, respectively.
PPI network
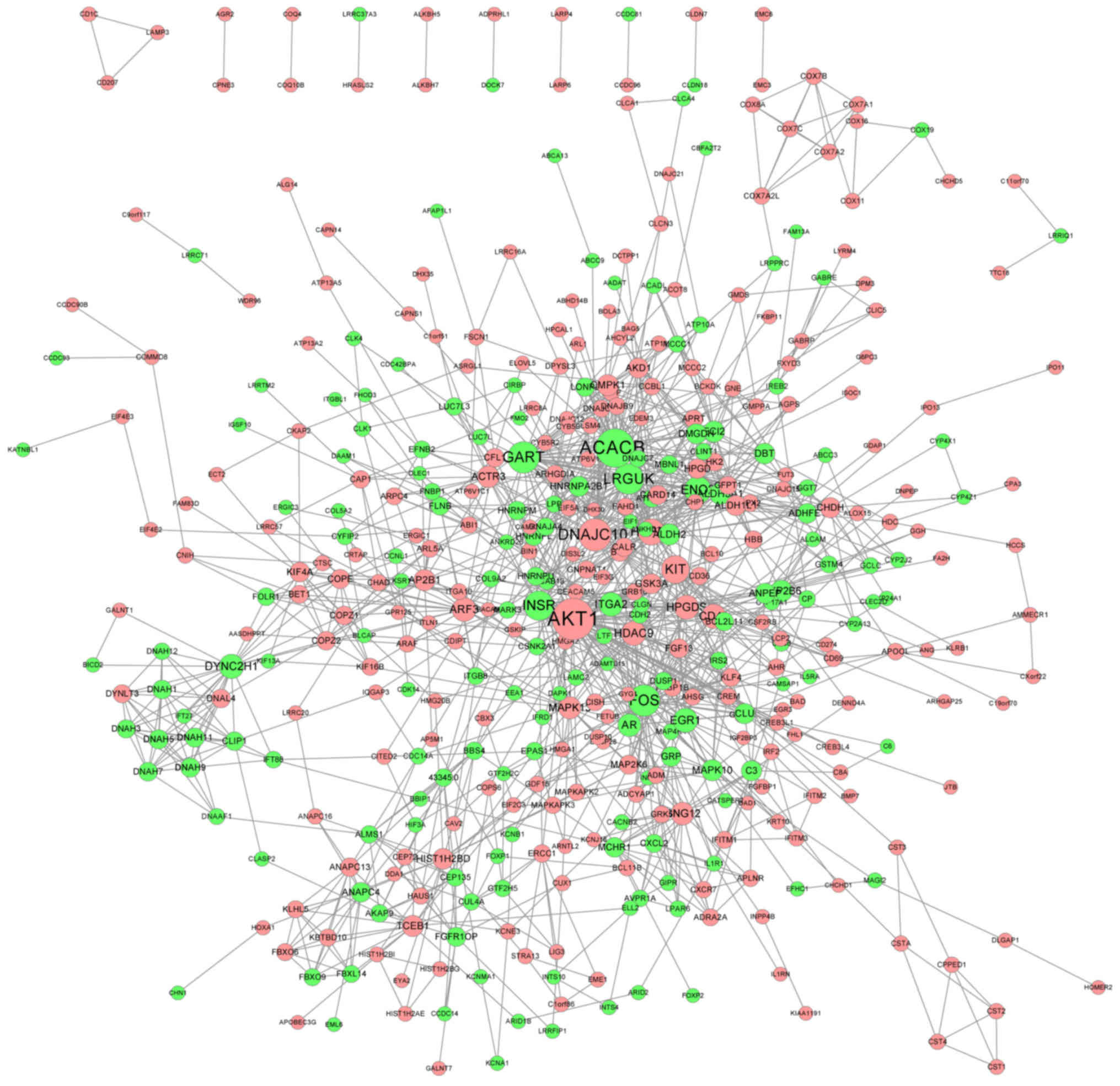
The PPI network of genes with significantly altered
expression (FC>1.2 and P<0.05) was delineated using the
STRING database. The PPI network contained 421 nodes and 1,232
edges (Fig. 3). Genes with the top
15 highest degrees are presented in Table I. Genes with a high degree may be
potential targets for clinical treatment (32).
 | Table I.Top 15 genes with the connectivity in
the protein-protein interaction network. |
Table I.
Top 15 genes with the connectivity in
the protein-protein interaction network.
| Gene | Node degree | Fold change
(asthma/control) | P-value | Trend |
|---|
| AKT1 | 55 | 1.24 |
3.13×10−4 | Up |
| ACACB | 49 | 1.27 |
1.71×10−3 | Down |
| DNAJC10 | 37 | 1.36 |
2.09×10−2 | Up |
| FOS | 35 | 1.37 |
6.04×10−3 | Down |
| GART | 35 | 1.43 |
3.91×10−4 | Down |
| LRGUK | 33 | 1.25 |
3.20×10−3 | Down |
| INSR | 31 | 1.44 |
1.00×10−5 | Down |
| KIT | 28 | 1.62 |
2.25×10−7 | Up |
| ENO2 | 26 | 1.22 |
1.75×10−3 | Down |
| HSPA5 | 25 | 1.33 |
1.33×10−2 | Up |
| CD44 | 21 | 1.62 |
4.30×10−5 | Up |
| DYNC2H1 | 21 | 1.33 |
4.00×10−5 | Down |
| EGR1 | 21 | 1.31 |
4.19×10−2 | Down |
| HDAC9 | 21 | 1.66 |
1.59×10−7 | Up |
| HPGDS | 21 | 1.48 |
2.00×10−5 | Up |
Construction of the lncRNA-mRNA
WGCNA
The lncRNA-mRNA co-expression network was
established to investigate the association between differentially
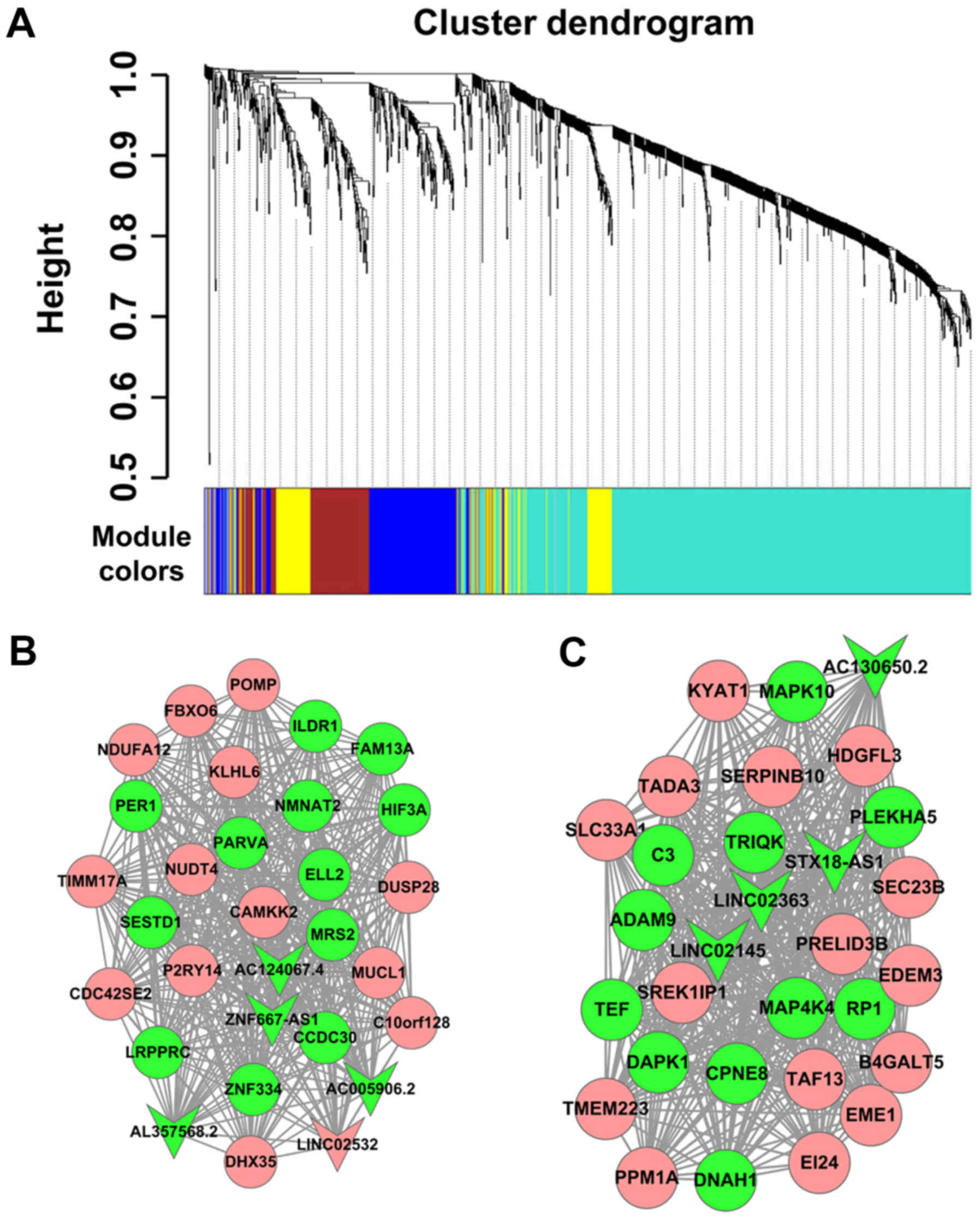
expressed lncRNAs and mRNAs. In the present study, a cluster
dendrogram was obtained using the WGCNA package in R (Fig. 4A); two weighted co-expression
subnetworks were identified. lncRNA-mRNA weighted co-expression
networks were generated based on genes with the top 30 degrees in
the two modules, which comprised 8 lncRNAs and 52 mRNAs. The two
modules included a number of lncRNA-mRNA co-expression
interactions, which indicated the mRNAs that could be regulated by
certain lncRNAs. The lncRNAs in the blue and turquoise modules are
listed in Table II. As presented
in Fig. 4B and C, the round and
arrow-shaped nodes represented mRNAs and lncRNAs, respectively; red
indicated upregulation, while green indicated downregulation
expression.
| Table II.lncRNAs in the blue module and the
turquoise module. |
Table II.
lncRNAs in the blue module and the
turquoise module.
| lncRNA | Module | Trend | P-value | Fold change |
|---|
| AC124067.4 | Blue | Down |
1.99×10−2 | 1.31 |
| ZNF667-AS1 | Blue | Down |
4.80×10−2 | 1.24 |
| AC005906.2 | Blue | Down |
2.28×10−2 | 1.26 |
| AL357568.2 | Blue | Down |
6.92×10−4 | 1.22 |
| AC130650.2 | Turquoise | Down |
9.64×10−7 | 1.31 |
| STX18-AS1 | Turquoise | Down |
8.91×10−4 | 1.24 |
| LINC02363 | Turquoise | Down |
1.34×10−2 | 1.22 |
| LINC02145 | Turquoise | Down |
2.00×10−6 | 1.37 |
Functional annotation and pathway
analysis for the differentially expressed mRNAs in the blue and
turquoise modules
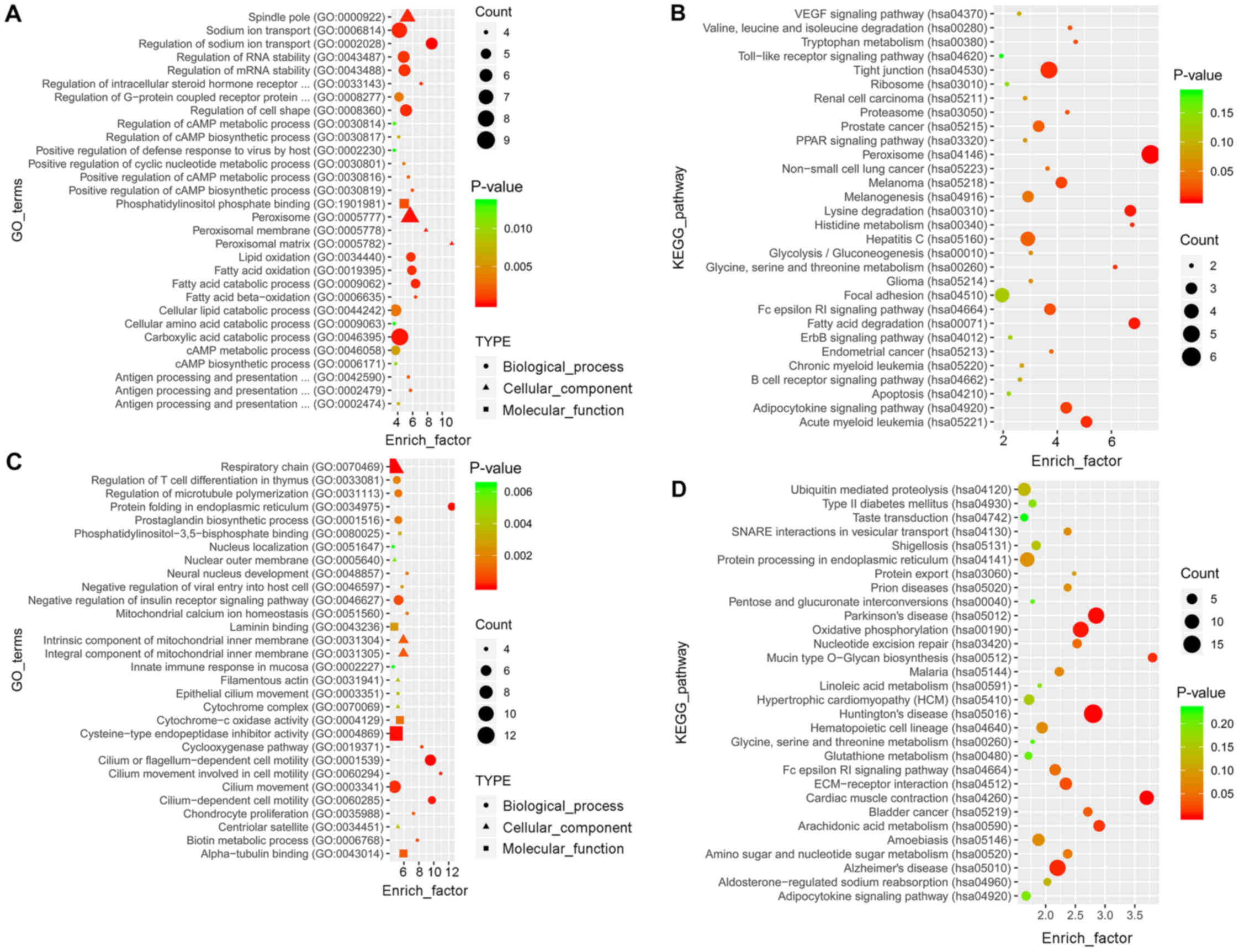
GO analysis revealed a total of 756 and 1,882
enriched terms in the blue and the turquoise modules, respectively.
The top 30 terms are presented in Fig.
5A and C. KEGG analysis revealed that 116 and 164 pathways were
enriched in the two modules, respectively. The top 30 pathways are
presented in Fig. 5B and D.
Discussion
Asthma is a common health issue, which poses an
economic and social burden to patients (2); however, the pathogenesis of this
condition remains poorly understood. To investigate the
pathogenesis of asthma and to identify potential biomarkers for
clinical treatment in the present study, lncRNA and mRNA expression
data in the GSE67472 dataset were downloaded. A total of 159 and
1,261 dysregulated lncRNAs and mRNAs, respectively, were identified
in bronchial mucosa samples obtained from patients with asthma
compared with samples obtained from normal controls.
Genes and their protein products are the basic
components of cells, and can be assembled into functional modules.
Gene co-expression networks are used to investigate the
associations between gene transcripts (33). WGCNA is a biological application
for screening clusters (modules) of highly associated genes and
demonstrates the link between genes across microarray samples. This
analytic tool has been utilized in numerous studies (12,18,29,34).
Using WGCNA and bioinformatics analysis, the major biological
functions of asthma-associated lncRNAs, and the potential
underlying molecular mechanisms in which these lncRNAs may be
involved, were investigated in the present study. WGCNA analysis
identified 8 key lncRNAs and 52 genes in two modules; genes in the
blue module were enriched in the GOs of several metabolic and
catabolic processes. Previous studies have indicated that lncRNAs
are involved in asthma (35,36).
ZNF667 antisense RNA 1 (ZNF667-AS1) inhibits the inflammatory
response of spinal cord injury by suppressing the Janus kinase-STAT
pathway (37–41). The present study observed that the
level of lncRNA ZNF667-AS1 in asthma was downregulated in
comparison with healthy controls. Therefore, lncRNA ZNF667-AS1 may
play a role in the pathogenesis of asthma.
Airway remodeling plays an important role in the
progressive worsening of asthma and is irreversible (1,2).
Vascular endothelial growth factor (42,43),
oxidative stress (44,45), the Fc ε RI signaling pathway
(46,47), and the dysregulation of amino
acids, sugar and nucleotide metabolism (48,49)
have been implicated in asthma. Functional gene analysis of the 159
lncRNAs and 1,261 mRNAs identified in the present study
demonstrated that the aforementioned pathways are involved in the
pathogenesis of asthma. In order to investigate the role of the 8
key lncRNAs and 52 genes in two modules, functional analysis using
GO and KEGG was conducted. Certain pathways, including ‘Toll-like
receptors’ (50,51), ‘activation of PPAR signaling
pathway’ (52) and ‘eosinophil
apoptosis’ (53), are important
for elucidating the mechanisms underlying the pathogenesis of
asthma. Genes in the turquoise module were enriched in the
following GO terms: ‘respiratory chain’, ‘regulation of T cell
differentiation in thymus’ and ‘cilium movement involved in cell
motility’. KEGG analysis revealed that genes in the turquoise
module were enriched in the ‘Fc ε RI signaling pathway’ and
‘ECM-receptor interaction’. These results indicated that genes in
the two modules may serve key roles in the initiation and
development of asthma.
In the present study, upregulated protein kinase B
(Akt) exhibited the highest degree in the PPI network and was
significantly enriched in the ‘VEGF signaling pathway’ and ‘Fc ε RI
signaling pathway’. Akt is also involved in the phosphoinositide
3-kinase/Akt signaling pathway, which served a key role in lung
inflammation and airway remodeling in a rat model of ovalbumin
(OVA)-induced asthma (54). The
expression of phosphorylated-Akt was increased in the lung tissues
of rats with OVA-induced asthma compared with controls.
lncRNAs regulate the transcription and expression of
mRNAs, and participate in the initiation and development of various
diseases (7,55). Therefore, the lncRNAs identified in
the present study and their interacting genes may serve important
roles in the onset and development of asthma. There are some
limitations to the present study. The data was only extracted from
one dataset that was not confirmed in independent studies.
Additionally, cell, animal and clinical experiments are required to
corroborate the results obtained in the present study as the mRNA
expression level may not always be consistent with the protein
level (56). Furthermore, the
interactions between the identified lncRNA and mRNA requires
experimental verification. Such future research may elucidate the
mechanisms underlying lncRNAs and mRNAs in the development of
asthma.
Acknowledgements
The authors would like to thank Mr Qiang Fan (Ao Ji
Bio-tech Co., Ltd.) for assisting with the data analysis.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81804043).
Availability of data and materials
The datasets analyzed during the present study are
available from the Gene Expression Omnibus repository, www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67472.
Authors' contributions
JG conceived and designed the study. XL and YZ
acquired, analyzed and interpreted data and wrote the manuscript.
NJ and HJ analyzed data and critically revised the manuscript. JG
generally supervised the research group and gave final approval.
All authors have read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Abreu SC, Lopes-pacheco M, da Silva AL,
Xisto DG, de Oliveira TB, Kitoko JZ, de Castro LL, Amorim NR,
Martins V, Silva LHA, et al: Eicosapentaenoic acid enhances the
effects of mesenchymal stromal cell therapy in experimental
allergic asthma. Front Immunol. 9:11472018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mejias SG and Ramphul K: Prevalence and
associated risk factors of bronchial asthma in children in santo
domingo, dominican republic. Cureus. 10:e22112018.PubMed/NCBI
|
|
3
|
Soriano JB, Abajobir AA, Abate KH, Abera
SF, Agrawal A, Ahmed MB, Aichour AN, Aichour I, Aichour MT, Alam K,
et al: Global, regional, and national deaths, prevalence,
disability-adjusted life years, and years lived with disability for
chronic obstructive pulmonary disease and asthma, 1990–2015: A
systematic analysis for the Global Burden of Disease Study 2015.
Lancet Respir Med. 5:691–706. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
de Castro LL, Xisto DG, Kitoko JZ, Cruz
FF, Olsen PC, Redondo PAG, Ferreira TPT, Weiss DJ, Martins MA,
Morales MM and Rocco PRM: Human adipose tissue mesenchymal stromal
cells and their extracellular vesicles act differentially on lung
mechanics and inflammation in experimental allergic asthma. Stem
Cell Res Ther. 8:151–162. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang ZK, Li J, Guan D, Liang C, Zhuo Z,
Liu J, Lu A, Zhang G and Zhang BT: A newly identified lncRNA MAR1
acts as a miR-487b sponge to promote skeletal muscle
differentiation and regeneration. J Cachexia Sarcopenia Muscle.
9:613–626. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li XQ, Ren ZX, Li K, Huang JJ, Huang ZT,
Zhou TR, Cao HY, Zhang FX and Tan B: Key Anti-fibrosis associated
long noncoding RNAs identified in human hepatic stellate cell via
transcriptome sequencing analysis. Int J Mol Sci. 19:E6752018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yan B, Liu JY, Yao J, Li XM, Wang XQ, Li
YJ, Tao ZF, Song YC, Chen Q and Jiang Q: lncRNA-MIAT regulates
microvascular dysfunction by functioning as a competing endogenous
RNA. Circ Res. 116:1143–1156. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang K, Liu CY, Zhou LY, Wang JX, Wang M,
Zhao B, Zhao WK, Xu SJ, Fan LH, Zhang XJ, et al: APF lncRNA
regulates autophagy and myocardial infarction by targeting
miR-188-3p. Nat Commun. 6:67792015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xing Z, Park PK, Lin C and Yang L: LncRNA
BCAR4 wires up signaling transduction in breast cancer. RNA Biol.
12:681–689. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gao JR, Qin XJ, Jiang H, Gao YC, Guo MF
and Jiang NN: Potential role of lncRNAs in contributing to
pathogenesis of chronic glomerulonephritis based on microarray
data. Gene. 643:46–54. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res 41 (Database).
D991–D995. 2013.
|
|
14
|
Sharma M, Batra J, Mabalirajan U, Sharma
S, Nagarkatti R, Aich J, Sharma SK, Niphadkar PV and Ghosh B: A
genetic variation in inositol polyphosphate 4 phosphatase a
enhances susceptibility to asthma. Am J Respir Crit Care Med.
177:712–719. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Christenson SA, Steiling K, van den Berge
M, Hijazi K, Hiemstra PS, Postma DS, Lenburg ME, Spira A and
Woodruff PG: Asthma-COPD overlap. Clinical relevance of genomic
signatures of type 2 inflammation in chronic obstructive pulmonary
disease. Am J Respir Crit Care Med. 191:758–766. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Christenson SA, Steiling K, van den Berge
M, Hijazi K, Hiemstra PS, Postma DS, Lenburg ME, Spira A and
Woodruff PG: Asthma-COPD Overlap: Clinical Relevance of Genomic
Signatures of Type 2 Inflammation in COPD. Am J Respir Crit Care
Med. 191:758–766. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang XQ, Sun S, Pu JK, Tsang AC, Lee D,
Man VO, Lui WM, Wong ST and Leung GK: Long non-coding RNA
expression profiles predict clinical phenotypes in glioma.
Neurobiol Dis. 48:1–8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu C, Ni HJ, Zhao YC, Chen K, Li M, Li C,
Zhu XD and Fu Q: Potential role of lncRNAs in contributing to
pathogenesis of intervertebral disc degeneration based on
microarray data. Med Sci Monit. 21:3449–3458. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Luchessi AD, Silbiger VN, Hirata RD,
Lima-Neto LG, Cavichioli D, Iñiguez A, Bravo M, Bastos G, Sousa AG,
Brión M, et al: Pharmacogenomics of anti-platelet therapy focused
on peripheral blood cells of coronary arterial disease patients.
Clin Chim Acta. 425:9–17. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xing ZH, Chu C, Chen L and Kong XY: The
use of Gene Ontology terms and KEGG pathways for analysis and
prediction of oncogenes. Biochim Biophys Acta. 1860:2725–2734.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maere S, Heymans K and Kuiper M: BiNGO: A
Cytoscape plugin to assess overrepresentation of Gene Ontology
categories in Biological Networks. Bioinformatics. 21:3448–3449.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang DW, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
von Mering C, Jensen LJ, Snel B, Hooper
SD, Krupp M, Foglierini M, Jouffre N, Huynen MA and Bork P: STRING:
Known and predicted protein-protein associations, integrated and
transferred across organisms. Nucleic Acids Res. 33:D433–D437.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cuesta-Astroz Y, Santos A, Oliveira G and
Jensen LJ: Analysis of predicted host-parasite interactomes reveals
commonalities and specificities related to parasitic lifestyle and
tissues tropism. Front Immunol. 10:2122019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Y, Ruan Z, Yu S, Tian T, Liang X,
Jing L, Li W, Wang X, Xiang L, Claret FX, et al: A four-methylated
mRNA signature-based risk score system predicts survival in
patients with hepatocellular carcinoma. Aging (Albany NY).
11:160–173. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Safari-Alighiarloo N, Taghizadeh M,
Tabatabaei SM, Shahsavari S, Namaki S, Khodakarim S and
Rezaei-Tavirani M: Identification of new key genes for type 1
diabetes through construction and analysis of protein–protein
interaction networks based on blood and pancreatic islet
transcriptomes. J Diabetes. 9:764–777. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Le DH and Pham VH: HGPEC: A Cytoscape app
for prediction of novel disease-gene and disease-disease
associations and evidence collection based on a random walk on
heterogeneous network. Bmc Syst Biol. 11:612017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. Bmc
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang B and Horvath S: A general framework
for weighted gene co-expression network analysis. Stat Appl Genet
Mol Biol. 4:Article 17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dudoit S and Fridlyand J: A
prediction-based resampling method for estimating the number of
clusters in a dataset. Genome Biol. 3:RESEARCH00362002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hsin KY, Ghosh S and Kitano H: Combining
machine learning systems and multiple docking simulation packages
to improve docking prediction reliability for network pharmacology.
PLoS One. 8:e839222013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Langfelder P and Horvath S: Eigengene
networks for studying the relationships between co-expression
modules. Bmc Syst Biol. 1:542007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao W, Langfelder P, Fuller T, Dong J, Li
A and Hovarth S: Weighted gene coexpression network analysis: State
of the art. J Biopharm Stat. 20:281–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang SY, Fan XL, Yu QN, Deng MX, Sun YQ,
Gao WX, Li CL, Shi JB and Fu QL: The lncRNAs involved in mouse
airway allergic inflammation following induced pluripotent stem
cell-mesenchymal stem cell treatment. Stem Cell Res Ther. 8:22017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang XY, Zhang LX, Tian CJ, Tang XY, Zhao
LM, Guo YL, Cheng DJ, Chen XL, Ma LJ and Chen ZC: LncRNAs BCYRN1
promoted the proliferation and migration of rat airway smooth
muscle cells in asthma via upregulating the expression of transient
receptor potential 1. Am J Transl Res. 8:3409–3418. 2016.PubMed/NCBI
|
|
37
|
Vrba L, Garbe JC, Stampfer MR and Futscher
BW: A lincRNA connected to cell mortality and
epigenetically-silenced in most common human cancers. Epigenetics.
10:1074–1083. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Meng W, Cui W, Zhao L, Chi W, Cao H and
Wang B: Aberrant methylation and downregulation of ZNF667-AS1 and
ZNF667 promote the malignant progression of laryngeal squamous cell
carcinoma. J Biomed Sci. 26:132019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li JW, Kuang Y, Chen L and Wang JF: LncRNA
ZNF667-AS1 inhibits inflammatory response and promotes recovery of
spinal cord injury via suppressing JAK-STAT pathway. Eur Rev Med
Pharmacol Sci. 22:7614–7620. 2018.PubMed/NCBI
|
|
40
|
Zhao LP, Li RH, Han DM, Zhang XQ, Nian GX,
Wu MX, Feng Y, Zhang L and Sun ZG: Independent prognostic Factor of
low-expressed LncRNA ZNF667-AS1 for cervical cancer and inhibitory
function on the proliferation of cervical cancer. Eur Rev Med
Pharmacol Sci. 21:5353–5360. 2017.PubMed/NCBI
|
|
41
|
Vrba L and Futscher BW: Epigenetic
silencing of lncRNA MORT in 16 TCGA cancer types. F1000Res.
7:2112018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Smith R: Is VEGF a potential therapeutic
target in asthma? Pneumologia. 63(194): 197–199. 2014.
|
|
43
|
Na HJ, Hwang JY, Lee KS, Choi YK, Choe J,
Kim JY, Moon HE, Kim KW, Koh GY, Lee H, et al: TRAIL negatively
regulates VEGF-induced angiogenesis via caspase-8-mediated
enzymatic and non-enzymatic functions. Angiogenesis. 17:179–194.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lan N, Luo G, Yang X, Cheng Y, Zhang Y,
Wang X, Wang X, Xie T, Li G, Liu Z and Zhong N: 25-Hydroxyvitamin
D3-deficiency enhances oxidative stress and corticosteroid
resistance in severe asthma exacerbation. PLoS One. 9:e1115992014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chung KF and Marwick JA: Molecular
mechanisms of oxidative stress in airways and lungs with reference
to asthma and chronic obstructive pulmonary disease. Ann N Y Acad
Sci. 1203:85–91. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu LC: Immunoglobulin E receptor signaling
and asthma. J Biol Chem. 286:32891–32897. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gounni AS, Wellemans V, Yang J, Bellesort
F, Kassiri K, Gangloff S, Guenounou M, Halayko AJ, Hamid Q and
Lamkhioued B: Human airway smooth muscle cells express the high
affinity receptor for IgE (Fc epsilon RI): A critical role of Fc
epsilon RI in human airway smooth muscle cell function. J Immunol.
175:2613–2621. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ho WE, Xu YJ, Xu F, Cheng C, Peh HY,
Tannenbaum SR, Wong WS and Ong CN: Metabolomics reveals altered
metabolic pathways in experimental asthma. Am J Resp Cell Mol.
48:204–211. 2013. View Article : Google Scholar
|
|
49
|
Xu W, Comhair SAA, Janocha AJ, Lara A,
Mavrakis LA, Bennett CD, Kalhan SC and Erzurum SC: Arginine
metabolic endotypes related to asthma severity. PLoS One.
12:e01830662017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xiao HT, Liao Z, Chen L and Tong RS: A
promising approach for asthma treatment by multiwayly modulating
toll-like receptors. Eur Rev Med Pharmacol Sci. 16:2088–2091.
2012.PubMed/NCBI
|
|
51
|
Bezemer GF, Sagar S, ven Bergenhenegouwen
J, Georgiou NA, Garssen J, Kraneveld AD and Folkerts G: Dual role
of Toll-like receptors in asthma and chronic obstructive pulmonary
disease. Pharmcol Rev. 64:337–358. 2012. View Article : Google Scholar
|
|
52
|
Xu J, Zhu YT, Wang GZ, Han D, Wu YY, Zhang
DX, Liu Y, Zhang YH, Xie XM, Li SJ, et al: The PPARγ agonist,
rosglitazone, attenuates airway inflammation and remodeling via
heme oxygenase-1 in murine model of asthma. Acta Pharmacol Sin.
36:171–178. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ilmarinen P and Kankaanranta H: Eosinophil
apoptosis as a therapeutic target in allergic asthma. Basic Clin
Pharmacol Toxicol. 114:109–117. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lin HY, Xu L, Xie SS, Yu F, Hu HY, Song XL
and Wang CH: Mesenchymal stem cells suppress lung inflammation and
airway remodeling in chronic asthma rat model via PI3K/Akt
signaling pathway. Int J Clin Exp Pathol. 8:8958–8967.
2015.PubMed/NCBI
|
|
55
|
Kung JT, Colognori D and Lee JT: Long
noncoding RNAs: Past, present, and future. Genetics. 193:651–669.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Huang KL, Li S, Mertins P, Cao S,
Gunawardena HP, Ruggles KV, Mani DR, Clauser KR, Tanioka M, Usary
J, et al: Proteogenomic integration reveals therapeutic targets in
breast cancer xenografts. Nat Commun. 8:148642017. View Article : Google Scholar : PubMed/NCBI
|