Introduction
Acute myocardial infarction (AMI) is a type of
severe ischemic heart disease, presenting as myocardial necrosis
induced by acute and continuous ischemia and hypoxia following the
acute occlusion of the coronary artery (1,2).
Ventricular remodeling and heart failure are two principal factors
that influence the cardiovascular event rate, long term survival
and quality of life following AMI (3). AMI is characterized by acute onset,
high mortality and disability, and is a major human health problem
(4). Therefore, the identification
of novel and effective AMI treatment strategies is required.
At present, numerous studies have reported that
inflammation and the apoptosis of cardiomyocytes are involved in
mediating impaired myocardial function and heart failure, thus
serving important roles in the pathogenesis of AMI (5,6).
Increasing evidence has indicated that a large quantity of
myocardial cell apoptosis is observed in the infarct area, with
partial irreversible apoptosis directly contributing to AMI-induced
cardiac injury (7). Following MI,
the developing apoptosis of cardiomyocytes, induced by ischemic
stress in the border zone, aggravates the ventricular remodeling of
the remaining active myocardium, leading to further heart failure
(8,9). Therefore, the prevention of
cardiomyocyte apoptosis is considered to be one strategy for
preventing cardiac remodeling and heart failure progression
following AMI, thereby improving the prognosis of patients. Further
investigation into the mechanisms underlying AMI-induced apoptosis
and antiapoptotic pathways in cardiomyocytes may aid the
identification of novel targets for intervention in the clinical
setting.
Previous studies have aimed to identify highly
sensitive and specific markers for the timely diagnosis, prevention
and control of the occurrence and development of AMI (10,11).
MicroRNAs (miRNAs/miRs) are a class of short non-coding RNAs (19–25
nucleotides in length) (12).
miRNAs regulate gene expression by binding to the 3′-untranslated
region (3′-UTR) of target mRNAs (12,13).
A large number of miRNAs have been identified in various species
(14). miRNAs serve important
roles in the regulation of various cellular functions, including
the differentiation, proliferation, metastasis and apoptosis of
cells (15,16). Numerous studies have demonstrated
that the abnormal expression of miRNAs is associated with
pathological processes in various cardiovascular diseases,
including AMI (17–21). A recent study reported that the
expression of miR-124 was abnormally upregulated in the blood of
patients with AMI (22),
suggesting that it may be involved in the occurrence and
development of AMI; however, the expression and roles of miR-124-3p
in AMI remain to be investigated. Therefore, the aims of the
present study were to investigate the role of miR-124-3p in the
development of AMI and to identify the underlying molecular
mechanisms.
Materials and methods
Clinical samples
A total of 30 blood samples were collected from 30
patients with AMI (18 males and 12 females, aged 27–57 years old)
who received percutaneous coronary intervention at the coronary
care unit of the Second Affiliated Hospital of Harbin Medical
University (Harbin, China) between May 2016 and May 2017. For
patients with AMI, the following exclusion criteria applied:
Previous history of myocardial infarction; having received
percutaneous coronary intervention treatment; having acute heart
failure upon admission; having myocardial disease, infectious
pericarditis or pericardial disease; having an infectious disease,
severe diabetes mellitus, malignant tumor, liver/kidney disease,
pulmonary fibrosis, bone metabolic disorder, systemic immune
disease or complications caused by malignant tumors; having cardiac
shock. A total of 30 blood samples from 30 healthy individuals (15
males and 15 females, aged 28–61 years) admitted for general
physical examination at the Second Affiliated Hospital of Harbin
Medical University (Harbin, China) between May 2016 and May 2017
were recruited as the control group. None of the healthy
individuals had a history of heart disease, vascular disease or
other major diseases. Blood samples (5 ml) were collected from
patients 6 h following the onset of AMI and frozen at −80°C prior
to the extraction of total RNA. The present study was approved by
the Ethical Committee of The Second Affiliated Hospital of Harbin
Medical University. All patients provided written informed
consent.
Cell culture
The rat cardiomyocyte cell line H9c2 was obtained
from the American Type Culture Collection (Manassas, VA, USA).
Cells were grown in Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10%
fetal bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.) and
1% penicillin-streptomycin mixed solution (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany). The cells were incubated to confluence
at 37°C with 5% CO2 and passaged every 2–3 days
(7).
Establishment of myocardial
hypoxic/ischemic injury
A cell model of myocardial hypoxia/ischemia injury
was established by exposing H9c2 cardiomyocytes to hypoxia for 48
h. In brief, H9c2 cells were cultured under standard conditions to
reach 80% confluence, and subsequently grown in a hypoxia chamber
(Thermo Fisher Scientific, Inc.) containing a gas mixture of 1%
O2, 5% CO2 and 94% N2 in a
humidified incubator (Thermo Fisher Scientific, Inc.) at 37°C for
48 h.
Luciferase reporter assay
TargetScan 7.1 bioinformatics software (www.targetscan.org/vert_71) was employed to
identify putative targets of miR-124-3p. It was revealed that
nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB)
repressing factor (NKRF) may be a target of miR-124-3p. To
investigate this prediction, the wild-type (NKRF-WT) and mutant
(NKRF-MUT) 3′-UTR of NKRF was cloned into a pmiR-RB-Report™ dual
luciferase reporter gene plasmid vector (Guangzhou RiboBio Co.,
Ltd., Guangzhou, China). To point-mutate the miR-124-3p binding
domain in the 3′-UTR of NKRF, a QuikChange Site-Directed
Mutagenesis kit (Stratagene; Agilent Technologies, Inc., Santa
Clara, CA, USA) was used, according to the manufacturer's
protocols. H9c2 cells were co-transfected with 100 ng NKRF-WT or
NKRF-MUT plasmid and 50 nM miR-124-3p mimic
(5′-UAAGGCACGCGGUGAAUGCC-3′; Shanghai GenePharma Co., Ltd.,
Shanghai, China) or mimic control (5′-UUCUCCATCGUGCCUCUAT-3′;
Shanghai GenePharma Co., Ltd.) using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocols. Luciferase activity was determined 48 h
following transfection at 37°C using the Dual-Luciferase Assay
system (Promega Corporation, Madison, WI, USA), according to the
manufacturer's protocols, and was normalized to Renilla
luciferase activity. Experiments were repeated in triplicate.
Cell transfection
H9c2 cells were seeded in 6-well plates
(4×105 cells/well). Subsequently, 100 nM negative
control of miR-124-3p inhibitor (NC; 5′-CCGUACUUCGCUAGAUCA-3′;
Shanghai GenePharma Co., Ltd.), 100 nM miR-124-3p inhibitor
(5′-UAAGGCACGCGGUGAAUGCC-3′; Shanghai GenePharma Co., Ltd.), 1 µM
control-small interfering RNA (siRNA; cat no. sc-36869; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), 1 µM NKRF-siRNA (cat no.
sc-72275; Santa Cruz Biotechnology, Inc.) or 100 nM miR-124-3p
inhibitor + 1 µM NKRF-siRNA was transfected into H9c2 cells using
Lipofectamine 3000, according to the manufacturer's protocols.
Cells were subjected to subsequent experiments 48 h following
transfection at 37°C. Transfection efficiency was determined via
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) analysis.
RT-qPCR
The expression of miR-124-3p and other genes was
determined via RT-qPCR analysis. Total RNA was extracted from blood
samples and cells using TRIzol® reagent (Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocols. cDNA
was synthesized using the Moloney Murine Leukemia Virus RT kit
(Takara Biotechnology Co., Ltd., Dalian, China), according to the
manufacturer's protocols. Reaction conditions for RT were: 50°C for
5 min and 80°C for 2 min. qPCR was performed on the synthesized
cDNAs with SYBR Green I (Applied Biosystems; Thermo Fisher
Scientific, Inc.) using a CFX Connect Real-Time system (Bio-Rad
Laboratories, Inc.) according to the manufacturers' protocols. The
primer sequences for qPCR were as follows: GAPDH, forward
5′-CTTTGGTATCGTGGAAGGACTC-3′, reverse 5′-GTAGAGGCAGGGATGATGTTCT-3′;
U6, forward 5′-GCTTCGGCAGCACATATACTAAAAT-3′, reverse
5′-CGCTTCACGAATTTGCGTGTCAT-3′; miR-124-3p, forward
5′-GCTAAGGCACGCGGTG-3′, reverse 5′-GTGCAGGGTCCGAGGT-3′; tumor
necrosis factor-α (TNF-α), forward 5′-GAACTGGCAGAAGAGGCACT-3′,
reverse 5′-GGTCTGGGCCATAGAACTGA-3′; interleukin (IL)-1β, forward
5′-TGTGAAATGCCACCTTTTGA-3′, reverse 5′-TGAGTGATACTGCCTGCCTG-3′;
IL-6, forward 5′-CCGGAGAGGAGACTTCACAG-3′, reverse
5′-CAGAATTGCCATTGCACA-3′; and NKRF, forward
5′-TATTGATATTGGGGAGATGCC-3′ and reverse
5′-GGATCTTCCTGTCTTTCATCT-3′. Thermocycling was conducted as
follows: 95°C for 5 min, followed by 40 cycles of denaturation at
95°C for 15 sec and annealing/elongation at 60°C for 30 sec. GAPDH
(for mRNA) and U6 (for miR-124-3p) were used as internal controls.
The 2−ΔΔCq method (23)
was used to determine the relative gene expression. Experiments
were repeated in triplicate.
Western blot analysis
To measure the protein levels of NKRF,
phosphorylated (p)-p65, B-cell lymphoma 2 (Bcl-2), and pro-caspases
and cleaved caspases 3 and 9, western blot analysis was performed.
Total protein was extracted from H9c2 cells using
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Shanghai, China), according to the manufacturer's
protocols. The concentration of protein was determined using a
Bicinchoninic Acid Protein Assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Lysate samples (25 µg/lane) were separated via
12% SDS-PAGE and transferred onto polyvinylidene difluoride
membranes prior to blocking with 5% skimmed milk at room
temperature for 1.5 h. The membranes were incubated overnight at
4°C with primary antibodies against: NKRF (1:1,000; cat. no.
ab168829; Abcam, Cambridge, MA, USA), p-p65 (1:1,000; cat no. 3033;
Cell Signaling Technology, Inc., Danvers, MA, USA), p65 (1:1,000;
cat no. 8242; Cell Signaling Technology, Inc.), Bcl-2 (1:1,000; cat
no. 4223; Cell Signaling Technology, Inc.), cleaved caspase 3
(1:1,000; cat no. 9664; Cell Signaling Technology, Inc.), cleaved
caspase 9 (1:1,000; cat no. 9505; Cell Signaling Technology, Inc.),
pro-caspase 3 (1:1,000; cat. no. ab32150; Abcam), pro-caspase 9
(1:1,000; cat. no. ab135544; Abcam) and β-actin (1:1,000; cat. no.
4970; Cell Signaling Technology, Inc.). Membranes were subsequently
incubated at room temperature for 4 h with the horseradish
peroxidase-conjugated anti-rabbit immunoglobulin G antibodies (cat
no. 7074; 1:2,000; Cell Signaling Technology, Inc.). Protein bands
were visualized using a SuperSignal West Dura Extended Duration
Substrate enhanced chemiluminescence detection system (Pierce;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocols. Protein expression was quantified using Gel-Pro Analyzer
Version 6.3 densitometry software (Media Cybernetics, Inc.,
Rockville, MD, USA). Experiments were repeated in triplicate.
MTT assay
The viability of cells was determined using an MTT
assay. Briefly, H9c2 cells were seeded into 96-well culture plates
and grown at 37°C for 24 h. Subsequently, 5 mg/ml MTT solution
(Amresco, LLC, Solon, OH, USA) was added to each culture well, and
cells were incubated for a further 4 h. Subsequently, 150 µl DMSO
was used to dissolve the purple formazan and the absorbance was
detected at 490 nm using a Synergy™ 2 Multi-Mode microplate reader
(BioTek Instruments, Inc., Winooski, VT, USA). Experiments were
repeated in triplicate.
Apoptosis assay
To determine the apoptosis of cells, an Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis
detection kit (cat no. 70-AP101-100; Hangzhou MultiSciences
Biotech, Co., Ltd., Hangzhou, China) was used. Briefly, H9c2 cells
were washed with PBS three times and fixed in 70% ethanol at 4°C
overnight, and stained with PI/Annexin V-FITC, according to the
manufacturer's protocols. A flow cytometer was used (BD
Biosciences, Franklin Lakes, NJ, USA) to analyze cell apoptosis,
and the apoptosis rate was calculated as the total percentage of
cells in the right-side quadrants (early + late apoptotic cells)
using FlowJo software (version 7.6.1; FlowJo LLC, Ashland, OR, USA)
was used for data analysis. Experiments were repeated in
triplicate.
ELISA
An ELISA was performed to determine the levels of
TNF-α, IL-1β and IL-6 in cell supernatants. Briefly, H9c2 cells
were transfected with NC, miR-124-3p inhibitor, or miR-124-3p
inhibitor + NKRF-siRNA for 2 h and then subjected to hypoxia for 48
h. Cell supernatants were harvested via centrifugation (1,000 × g,
15 min, 4°C). The expression levels of IL-1β (cat no. PI305;
Beyotime Institute of Biotechnology), TNF-α (cat no. PI518;
Beyotime Institute of Biotechnology), and IL-6 (cat no. PI330;
Beyotime Institute of Biotechnology) were detected using ELISA
kits, according to the manufacturer's protocols. Experiments were
repeated in triplicate.
Statistical analysis
Data are presented as the mean ± standard deviation
of at least three experiments. All data were analyzed using SPSS
version 17.0 software (SPSS, Inc., Chicago, IL, USA). Comparisons
between groups were performed using Student's t-tests or one-way
analyses of variance followed by a Tukey's test. P<0.05 was
considered to indicate a statistically significant difference.
Results
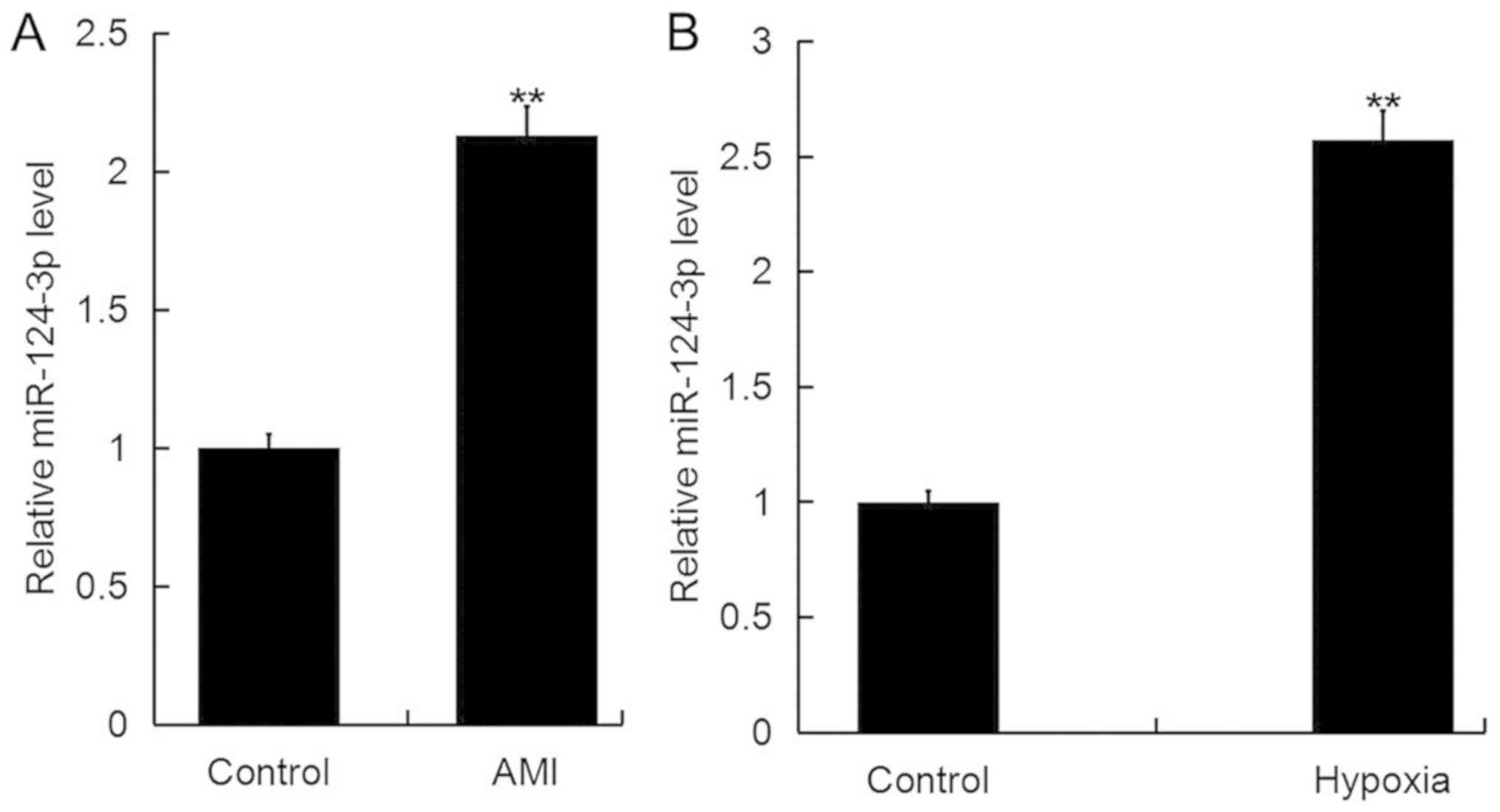
miR-124-3p is upregulated during
AMI
RT-qPCR analysis was performed to investigate the
expression of miR-124-3p in AMI. As presented in Fig. 1A, the expression levels of
miR-124-3p were significantly increased in the blood of patients
with AMI compared with healthy controls. Additionally, it was
revealed that the expression of miR-124-3p in the cardiomyocyte
cell line H9c2 was significantly upregulated following exposure to
hypoxic conditions for 48 h compared with the control (Fig. 1B).
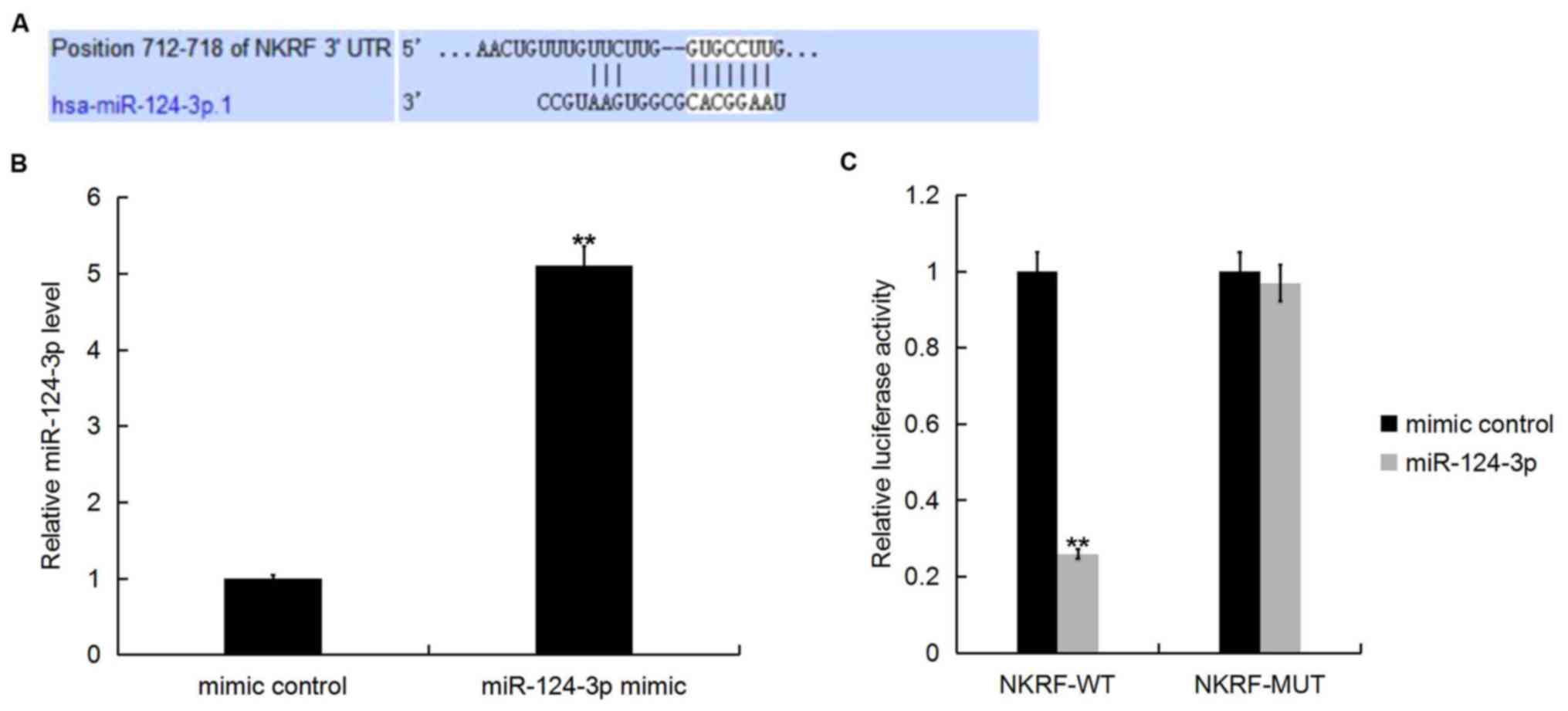
NKRF is a target of miR-124-3p
TargetScan was employed to predict potential targets
of miR-124-3p. Various target genes of miR-124-3p, including NKRF
(Fig. 2A). Previous studies
indicated that NKRF is a silencer protein that binds negative
regulatory elements (NRE) to repress the transcription of various
NF-κB-regulated genes (24).
Therefore, NKRF serves important roles in inflammation responses
and apoptosis; however, the involvement of this gene in AMI remains
unclear. Thus, NKRF was selected for further investigation in the
present study.
To determine whether miR-124-3p directly modulates
the expression of NKRF via interactions with potential binding
sites in the 3′-UTR, a luciferase reporter assay was performed.
Transfection of H9c2 cells with miR-124-3p mimic upregulated the
expression of miR-124-3p compared with the mimic control (Fig. 2B). Furthermore, co-transfection of
H9c2 cells with miR-124-3p mimic and NKRF-WT plasmid significantly
decreased the luciferase activity compared with co-transfection
with miR-124-3p mimic and NKRF-MUT plasmid (Fig. 2C). The results indicated that
miR-124-3p directly targeted NKRF mRNA.
miR-124-3p inhibition suppresses the
hypoxia-induced inhibition of the viability of H9c2 cells
To investigate the effects of miR-124-3p in AMI,
H9c2 cells were transfected with NC, miR-124-3p inhibitor,
control-siRNA, NKRF-siRNA or miR-124-3p inhibitor + NKRF-siRNA, and
the transfection efficiency was determined 48 h following
transfection. Transfection with miR-124-3p inhibitor significantly
downregulated the expression of miR-124-3p compared with the
control (Fig. 3A), whereas
NKRF-siRNA significantly downregulated the expression of NKRF mRNA
(Fig. 3B) and protein (Fig. 3C and D) in H9c2 cells compared with
control-siRNA. It was further demonstrated that miR-124-3p
inhibitor significantly increased the expression levels of NKRF at
the mRNA (Fig. 3E) and protein
(Fig. 3F and G) levels; these
effects were eliminated by transfection with NKRF-siRNA.
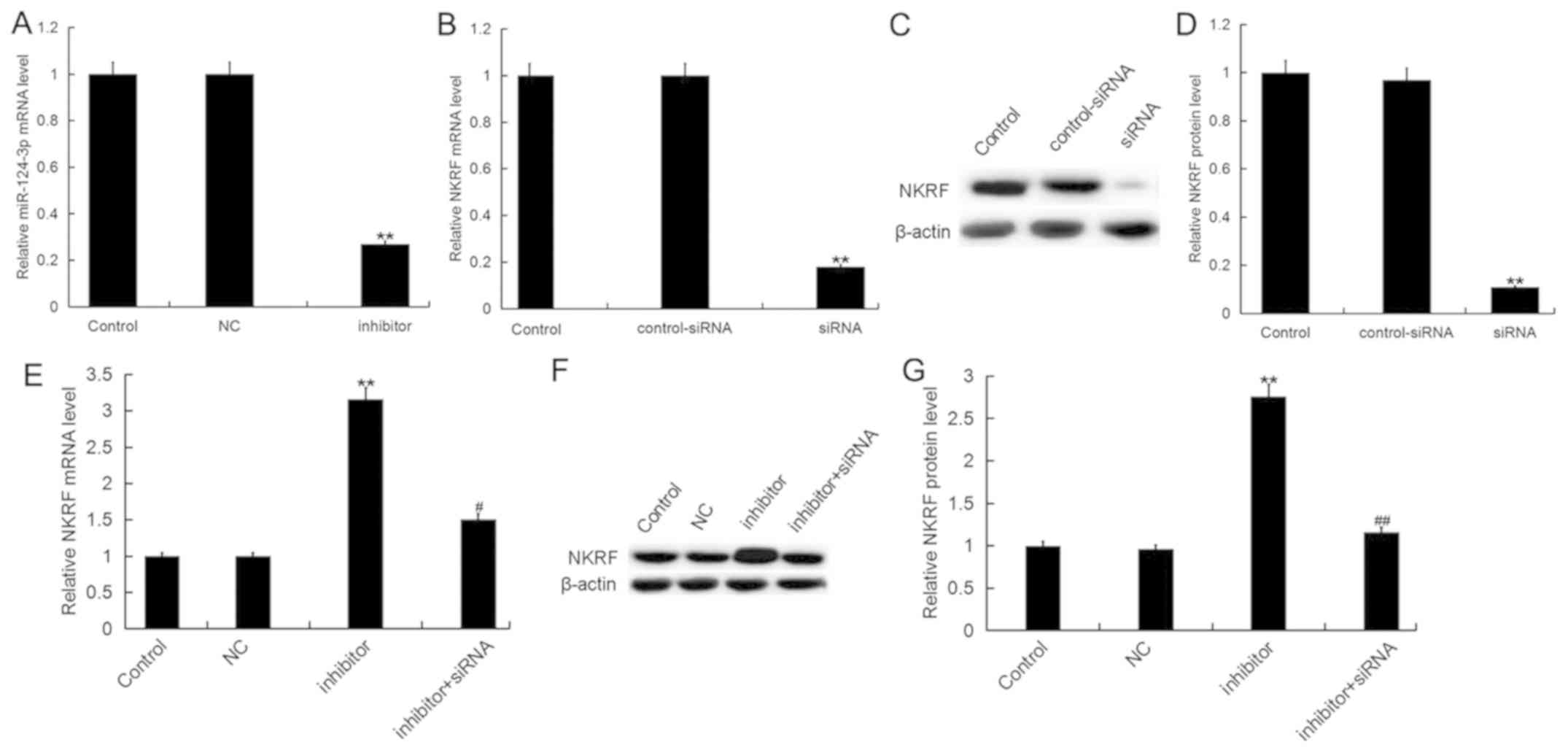 | Figure 3.Downregulation of miR-124-3p promotes
NKRF expression in H9c2 cells. (A) Expression of miR-124-3p in H9c2
cardiomyocytes transfected with inhibitor or NC. Expression of NKRF
(B) mRNA or protein, as measured by (C) western blotting and (D)
quantified, in H9c2 cells transfected with NKRF-siRNA or
control-siRNA. Expression of NKRF (E) mRNA or protein, as measured
by (F) western blotting and (G) quantified, in H9c2 cells
transfected with inhibitor in the presence and absence of
NKRF-siRNA. Data are presented as the mean ± standard deviation of
three independent experiments. **P<0.01 vs. control;
#P<0.05, ##P<0.01 vs. inhibitor.
miR-124-3p, microRNA-124-3p; inhibitor, miR-124-3p inhibitor; NC,
negative control; NKRF, nuclear factor κ-light-chain-enhancer of
activated B cells repressing factor; siRNA, small interfering
RNA. |
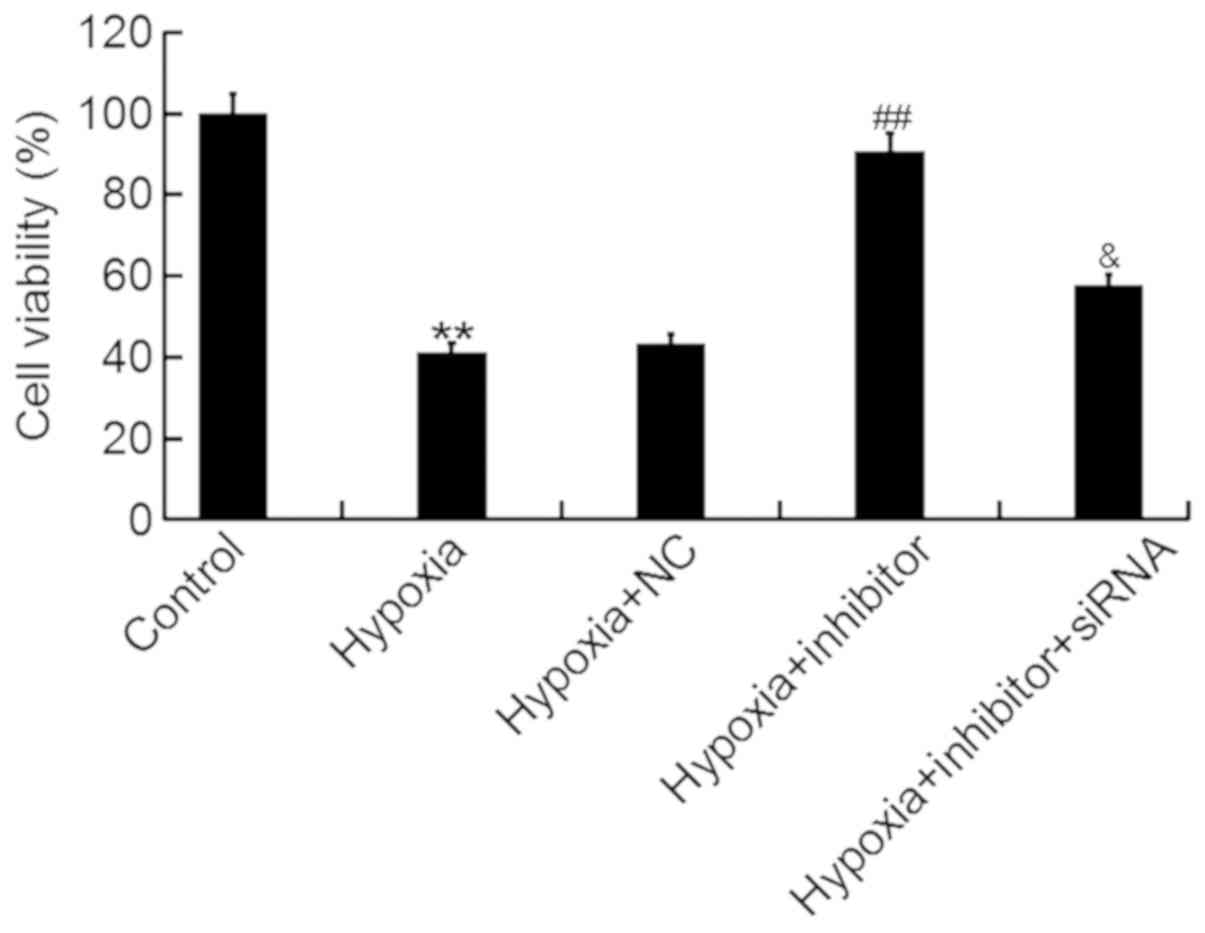
An MTT assay was performed to determine the effects
of miR-124-3p on the viability of H9c2 cells following hypoxia. It
was revealed that miR-124-3p inhibitor significantly attenuated the
hypoxia-induced decrease in the viability of H9c2 cells, whereas
co-transfection with inhibitor and NKRF-siRNA eliminated the
effects of the inhibitor (Fig.
4).
miR-124-3p inhibition suppresses the
hypoxia-induced apoptosis of H9c2 cells
The effects of miR-124-3p on the apoptosis of H9c2
cells were determined via flow cytometry. It was demonstrated that
hypoxia significantly promoted H9c2 cell apoptosis, whereas
transfection with miR-124-3p inhibitor attenuated the effects of
hypoxia (Fig. 5A and B).
Furthermore, co-transfection with NKRF-siRNA significantly
eliminated the effects of miR-124-3p inhibitor on the apoptosis of
H9c2 cells.
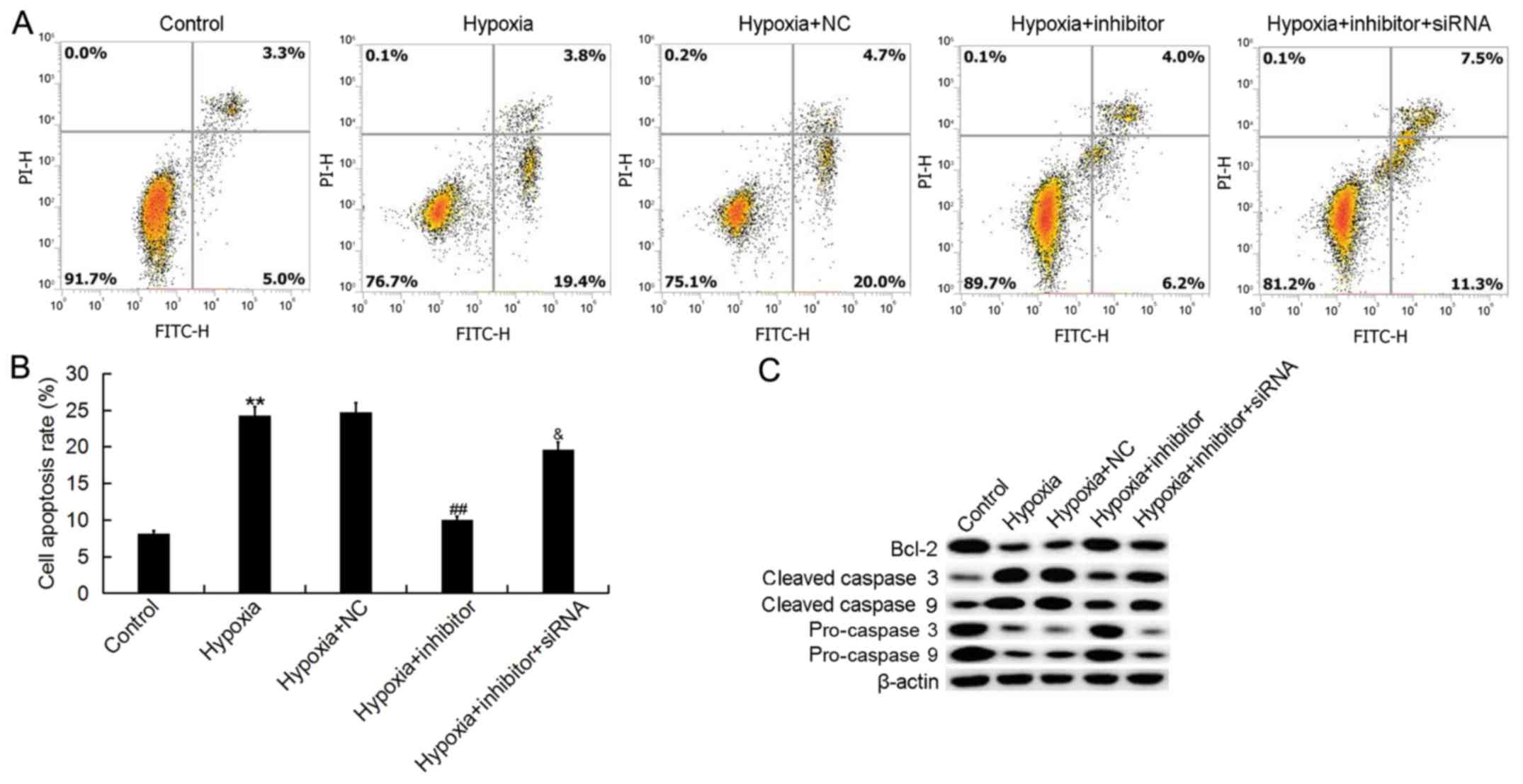 | Figure 5.Effects of microRNA-124-3p expression
on the apoptosis of H9c2 cells. (A) Flow cytometry was performed to
determine the apoptosis of H9c2 cardiomyocytes following exposure
to hypoxic or control conditions for 48 h, and transfection with
NC, inhibitor or inhibitor + NKRF-siRNA. (B) Quantification of the
apoptosis of H9c2 cells. (C) Expression of Bcl-2, and cleaved and
procaspases 3 and 9 following the aforementioned treatments. Data
are presented as the mean ± standard deviation of three independent
experiments. **P<0.01 vs. control; ##P<0.01 vs.
hypoxia; &P<0.05 vs. hypoxia + inhibitor. Bcl-2,
B-cell lymphoma 2; FITC, fluorescein isothiocyanate; Inhibitor,
miR-124-3p inhibitor; NC, negative control; NKRF, nuclear factor
κ-light-chain-enhancer of activated B cells repressing factor; PI,
propidium iodide; siRNA, small interfering RNA. |
To further investigate the antiapoptotic effects of
miR-124-3p inhibitor, the expression of apoptosis-associated
proteins was determined, including Bcl-2, cleaved caspase 3,
cleaved caspase 9, procaspase 3 and procaspase 9. It was observed
that the protein expression levels of Bcl-2, procaspase 3 and
procaspase 9 were markedly decreased in H9c2 cells following
hypoxia, and those of cleaved caspase 3 and cleaved caspase 9 were
significantly increased in H9c2 cells treated with hypoxia,
compared with the control; transfection with miR-124-3p inhibitor
reversed the hypoxia-induced effects (Fig. 5C). Furthermore, the effects of
miR-124-3p inhibitor on the expression of Bcl-2, cleaved caspase 3,
cleaved caspase 9, procaspase 3 and procaspase 9 in H9c2 cells were
eliminated by the silencing of NKRF.
miR-124-3p inhibition suppresses
hypoxia-induced inflammatory responses in H9c2 cells
As inflammation serves important roles in the
development of AMI, the effects of miR-124-3p on inflammatory
responses during AMI were investigated. As presented in Fig. 6, the protein and mRNA expression
levels of TNF-α, IL-1β and IL-6 were significantly increased
following hypoxia; however, transfection with miR-124-3p inhibitor
significantly inhibited the hypoxia-induced upregulation of
inflammatory cytokines, effects which were eliminated following
co-transfection with NKRF-siRNA. The results indicated that
miR-124-3p inhibitor suppressed hypoxia-induced inflammatory
responses in cardiomyocytes.
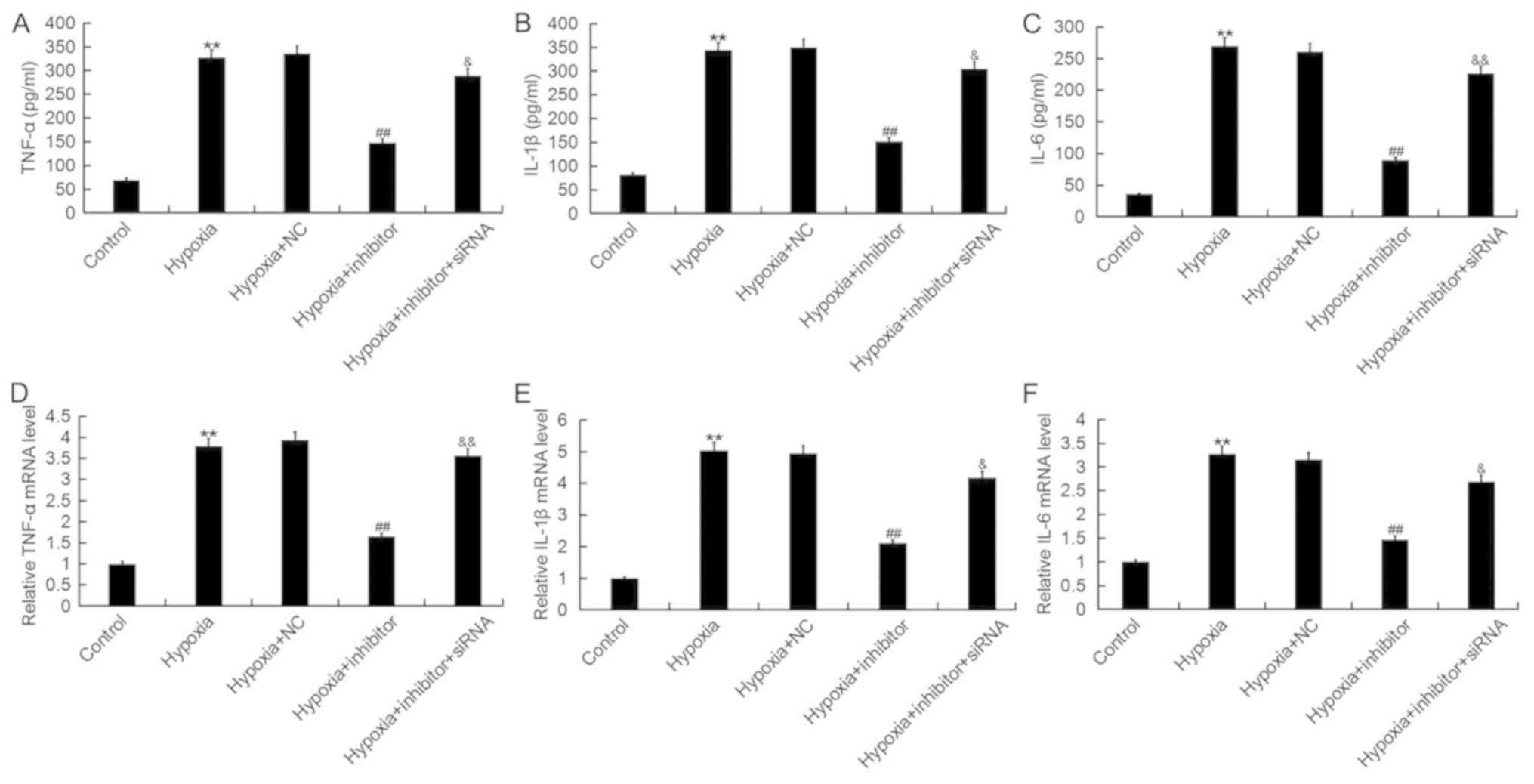 | Figure 6.Effects of microRNA-124-3p expression
on the levels of TNF-α, IL-1β and IL-6 in H9c2 cells. Expression of
(A) TNF-α, (B) IL-1β and (C) IL-6 protein in H9c2 cardiomyocytes
following exposure to hypoxic or control conditions for 48 h, and
transfection with NC, inhibitor or inhibitor + NKRF-siRNA, as
determined by ELISA. Expression of (D) TNF-α, (E) IL-1β and (F)
IL-6 mRNA in H9c2 cells following the aforementioned treatments, as
determined by reverse transcription-quantitative polymerase chain
reaction analysis. Data are presented as the mean ± standard
deviation of three independent experiments. **P<0.01 vs.
control; ##P<0.01 vs. hypoxia;
&P<0.05, &&P<0.01 vs.
hypoxia + inhibitor. IL, interleukin; Inhibitor, miR-124-3p
inhibitor; NC, negative control; NKRF, nuclear factor
κ-light-chain-enhancer of activated B cells repressing factor;
siRNA, short interfering RNA; TNF-α, tumor necrosis factor-α. |
miR-124-3p inhibitor suppresses the
hypoxia-induced activation of NF-κB signaling in H9c2 cells
To investigate the mechanisms underlying the effects
of miR-124-3p inhibitor on the cardiomyocyte cell line H9c2, the
activity of the NF-κB pathway was analyzed. As presented in
Fig. 7, hypoxia significantly
downregulated the expression of NKRF (Fig. 7A-C) and promoted the
phosphorylation of NF-κB p65 in H9c2 cells (Fig. 7D-F); transfection with miR-124-3p
inhibitor significantly attenuated the effects of hypoxia on NKRF
expression and p65 phosphorylation. Additionally, it was
demonstrated that NKRF-siRNA significantly eliminated the effects
of miR-124-3p inhibitor on NKRF and p-p65 levels in H9c2 cells. The
expression of p65 protein in H9c2 cells was not notably different
across the various groups.
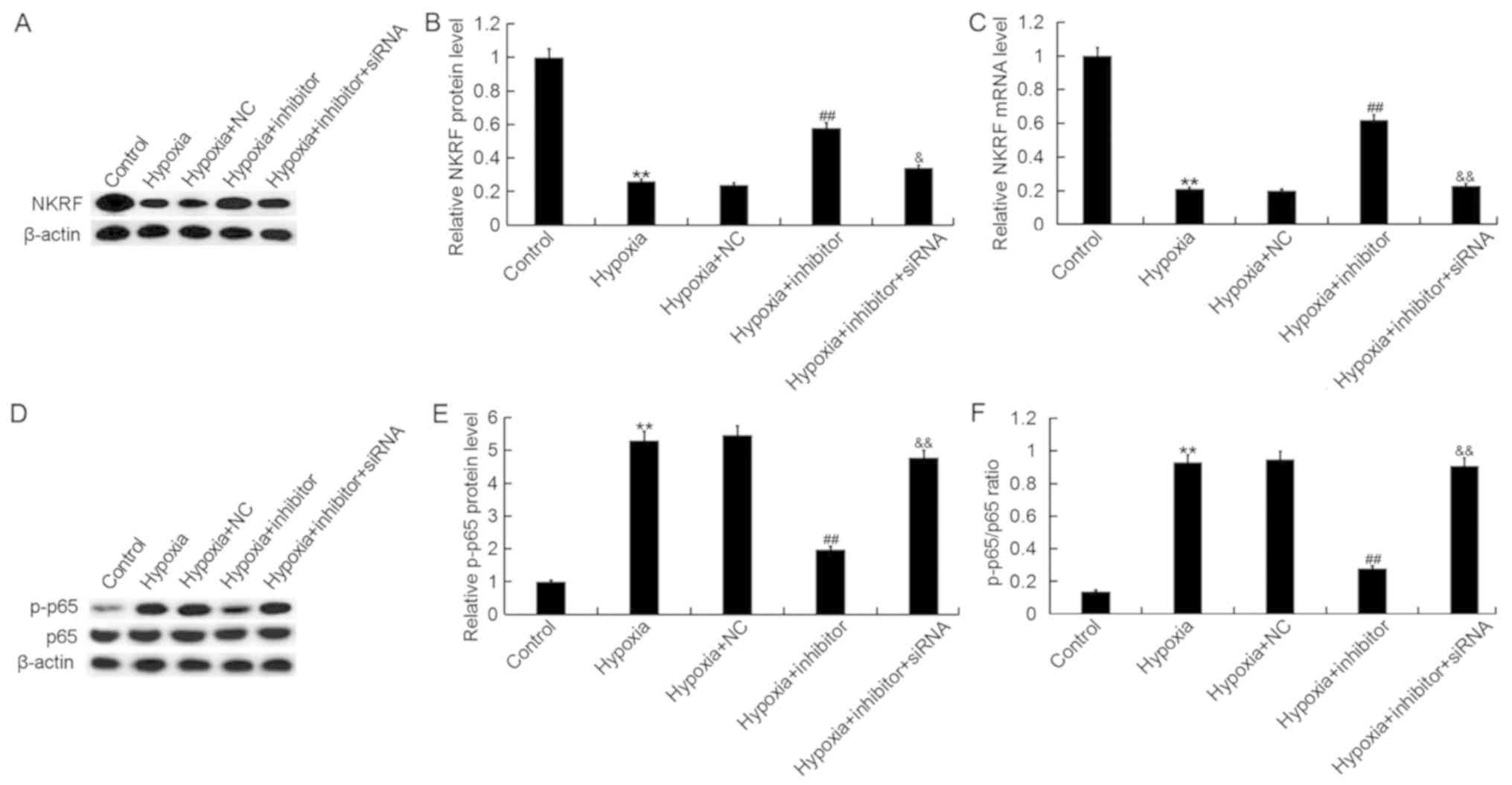 | Figure 7.Effects of miR-124-3p expression on
the NF-κB pathway in H9c2 cells. Expression of NKRF protein,
assessed by (A) western blotting and (B) quantification, and (C)
mRNA in H9c2 cardiomyocytes following exposure to hypoxic or
control conditions for 48 h, and transfection with NC, inhibitor or
inhibitor + NKRF-siRNA, as determined by western blot and reverse
transcription-quantitative polymerase chain reaction analyses,
respectively. (D) Expression of NF-κB p65 and p-p65 in H9c2 cells
following the aforementioned treatments, as determined via western
blotting. Expression of p-p65 normalized to (E) the internal
control and (F) p65 expression. Data are presented as the mean ±
standard deviation of three independent experiments. **P<0.01
vs. control; ##P<0.01 vs. hypoxia;
&P<0.05, &&P<0.01 vs.
hypoxia + inhibitor. Inhibitor, miR-124-3p inhibitor; NC, negative
control; NF-κB, nuclear factor κ-light-chain-enhancer of activated
B cells; NKRF, NF-κB repressing factor; p, phosphorylated; siRNA,
small interfering RNA. |
Discussion
In the present study, it was demonstrated that
expression of miR-124-3p was significantly increased during AMI.
Downregulation of miR-124-3p opposed the effects of hypoxia on the
viability, apoptosis and inflammatory responses of H9c2 cells by
targeting NKRF. In addition, the results suggested that miR-124-3p
downregulation inhibited the hypoxia-induced activation of the
NF-κB pathway. The findings of the present study indicated that
miR-124-3p inhibitor protected H9c2 cardiomyocytes against hypoxia,
and that miR-124-3p may be a promising therapeutic target in the
treatment of AMI.
AMI is an acute necrotic myocardial infarction
induced by persistent and severe ischemia, and is one of the most
common cardiovascular diseases globally (25). At present, there are widely
available treatments; however, the prevalence of cardiovascular
diseases, mortality and treatment costs continue to increase in
developed and developing countries (26). To effectively reduce the occurrence
and development of AMI, it is necessary to identify novel targets
and methods for the diagnosis and treatment of AMI.
miRNAs have received increasing attention in various
research fields, such as cancer and cardiovascular diseases
(27,28). The abnormal expression of miRNAs
has been closely associated with the pathophysiological processes
of AMI (20,21). A previous study reported the
upregulation of miR-124 in the blood of patients with AMI (22); however, the expression and role of
miR-124-3p in the development of AMI remain unclear. Therefore, the
present study was conducted.
The levels of miR-124-3p during AMI were determined;
it was observed that miR-124-3p levels were significantly increased
in the blood of patients with AMI and hypoxia-treated H9c2 cells,
indicating the potential role of miR-124-3p in AMI. It was
subsequently identified that NKRF was a direct target of
miR-124-3p.
Persistent inflammatory responses and the necrosis
of ischemic tissue are two prominent features that are mutually
reinforced during AMI-induced heart damage, eventually leading to
heart failure (29). Inflammation
is the principal pathological process that occurs during early MI;
inflammatory factors serve important roles in ventricular
remodeling and the progression of heart failure (30,31).
Apoptosis and necrosis are the two notable events during the
development of AMI (32,33). Therefore, determination of the
molecular mechanisms underlying the apoptosis and inflammatory
responses of cardiomyocytes is required to develop effective
treatment strategies for ischemic heart disease. Numerous studies
have reported that miR-124-3p was involved in the development of
various diseases by regulating the proliferation and apoptosis of
cells (34–37). miR-124-3p has also been reported to
contribute to the regulation of inflammatory responses (38). A recent study demonstrated that the
long non-coding RNA taurine upregulated gene 1 alleviated
hypoxia-induced injury (as determined by an increase in the
viability and a decrease in the apoptosis of cells) by targeting
miR-124 in H9c2 cells, and that miR-124 promoted hypoxia-induced
effects in H9c2 cells by regulating the expression of hydrogen
peroxide-inducible clone-5 (39).
The present study reported that hypoxia-induced reductions in the
viability and increases in the apoptosis of H9c2 cells were opposed
by miR-124-3p downregulation. Furthermore, the hypoxia-induced
upregulation of TNF-α, IL-1β and IL-6 was inhibited by miR-124-3p
downregulation. Conversely, silencing NKRF eliminated all the
effects of miR-124-3p inhibitor on H9c2 cells.
It was revealed that NKRF was a direct target of
miR-124-3p. NKRF is a silencer protein that binds NREs to suppress
the basal transcription of NF-κB-regulated genes (40). Therefore, the effects of miR-124-3p
on the NF-κB pathway were investigated in cardiomyocytes, and the
findings suggested that miR-124-3p inhibitor inhibited the
hypoxia-induced activation of the NF-κB pathway in H9c2 cells; this
inhibition was eliminated by NKRF silencing.
In conclusion, it was demonstrated that the
expression of miR-124-3p was abnormally high in AMI, and its
inhibition suppresses inflammatory responses and apoptosis in a
cell model of AMI in an NKRF-dependent manner. miR-124-3p may be a
novel therapeutic target in the treatment of AMI; however, as this
is a preliminary study into the role of miR-124-3p in AMI, further
investigation is required. For example, the expression of NKRF in
the blood of patients with AMI should be determined. Furthermore,
as a cellular model of AMI is notably different to the human
disease, in vivo and clinical studies are required to
demonstrate the role of miR-124-3p in AMI and supporting the
findings observed in vitro.
Acknowledgements
Not applicable.
Funding
The present study was supported by Heilongjiang
Province Science Fund for returning to the country (grant no.
LC2013C37) and Heilongjiang Provincial Education Office Project
(No. 12541437).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GH designed the current study, collected and
analyzed the data, performed statistical analysis, searched the
literature, and prepared the manuscript. LM, FD and XH contributed
to data collection and data interpretation. SL and HS contributed
to statistical analyses and interpreted the data.
Ethics approval and consent to
participate
The present study was approved by the Ethical
Committee of The Second Affiliated Hospital of Harbin Medical
University. All patients provided written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Roger VL, Go AS, Lloyd-Jones DM, Benjamin
EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al:
Executive summary: Heart disease and stroke statistics-2012 update:
A report from the American Heart Association. Circulation.
125:188–197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reed GW, Rossi JE and Cannon CP: Acute
myocardial infarction. Lancet. 389:197–210. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
White HD, Thygesen K, Alpert JS and Jaffe
AS: Clinical implications of the third universal definition of
myocardial infarction. Heart. 100:424–432. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schüssler-Lenz M, Beuneu C,
Menezes-Ferreira M, Jekerle V, Bartunek J, Chamuleau S, Celis P,
Doevendans P, O'Donovan M, Hill J, et al: Cell-based therapies for
cardiac repair: A meeting report on scientific observations crud
European regulatory viewpoints. Eur J Heart Fail. 18:133–141. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Frangogiannis NG: Regulation of the
inflammatory response in cardiac repair. Circ Res. 110:159–173.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Feng Y, Zhao J, Hou H, Zhang H, Jiao Y,
Wang J, Wang Y and Sun Y: WDR26 promotes mitophagy of
cardiomyocytes induced by hypoxia through Parkin translocation.
Acta Biochim Biophys Sin (Shanghai). 48:1075–1084. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Swynghedauw B: Molecular mechanisms of
myocardial remodeling. Physiol Rev. 79:215–262. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Minicucci MF, Azevedo PS, Polegato BF,
Paiva SA and Zornoff LA: Heart failure after myocardial infarction:
Clinical implications and treatment. Clin Cardiol. 34:410–414.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Whelan RS, Kaplinskiy V and Kitsis RN:
Cell death in the pathogenesis of heart disease: Mechanisms and
significance. Annu Rev Physiol. 72:19–44. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goretti E, Wagner DR and Devaux Y: miRNAs
as biomarkers of myocardial a step forward towards personalized
medicine? Trends Mol Med. 20:716–725. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fathil MF, Md Arshad MK, Gopinath SC,
Hashim U, Adzhri R, Ayub RM, Ruslinda AR, Nuzaihan MNM, Azman AH,
Zaki M and Tang TH: Diagnostics on acute myocardial infarction:
Cardiac troponin biomarkers. Biosens Bioelectron. 70:209–220. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim J, Yao F, Xiao Z, Sun Y and Ma L:
MicroRNAs and metastasis: Small RNAs play big roles. Cancer
Metastasis Rev. 37:5–15. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kozomara A and Griffiths-Jones S: miRBase:
Integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res 39 (Database Issue). D152–D157. 2011. View Article : Google Scholar
|
|
15
|
Chen CZ, Li L, Lodish HF and Bartel DP:
MicroRNAs modulate hematopoietic lineage differentiation. Science.
303:83–86. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fujii T, Shimada K, Nakai T and Ohbayashi
C: MicroRNAs in smoking-related carcinogenesis: Biomarkers,
functions, and therapy. J Clin Med. 7(pii): E982018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu X and Li H: Integrated microRNA-gene
analysis of coronary artery disease based on miRNA and gene
expression profiles. Mol Med Rep. 13:3063–3073. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harada M, Luo X, Murohara T, Yang B,
Dobrev D and Nattel S: MicroRNA regulation and cardiac calcium
signaling: Role in cardiac disease and therapeutic potential. Circ
Res. 114:689–705. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Matkovich SJ and Hu Y: Regulation of
cardiac microRNAs by cardiac microRNAs. Circ Res. 113:62–71. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen Z, Li C, Lin K, Zhang Q, Chen Y and
Rao L: MicroRNAs in acute myocardial infarction: Evident value as
novel biomarkers? Anatol J Cardiol. 19:140–147. 2018.PubMed/NCBI
|
|
21
|
Paiva S and Agbulut O: MiRroring the
multiple potentials of MicroRNAs in acute myocardial infarction.
Front Cardiovasc Med. 4:732017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo ML, Guo LL and Weng YQ: Implication of
peripheral blood miRNA-124 in predicting acute myocardial
infarction. Eur Rev Med Pharmacol Sci. 21:1054–1059.
2017.PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dreikhausen U, Hiebenthal-Millow K,
Bartels M, Resch K and Nourbakhsh M: NF-kappaB-repressing factor
inhibits elongation of human immunodeficiency virus type 1
transcription by DRB sensitivity-inducing factor. Mol Cell Biol.
25:7473–7483. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
White HD and Chew DP: Acute myocardial
infarction. Lancet. 372:570–584. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al:
Heart disease and stroke statistics-2014 update: A report from the
American Heart Association. Circulation. 129:e28–e292. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kulkarni S, Qi Y, O'HUigin C, Pereyra F,
Ramsuran V, McLaren P, Fellay J, Nelson G, Chen H, Liao W, et al:
Genetic interplay between HLA-C and MIR148A in HIV control and
Crohn disease. Proc Natl Acad Sci USA. 110:20705–20710. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Finn NA and Searles CD: Intracellular and
extracellular miRNAs in regulation of angiogenesis signaling. Curr
Angiogenes. 4:299–307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang YY, Li T, Liu YW, Wang Y, Hu XM, Gao
WQ, Wu P, Li X, Peng WJ, Gao W, et al: Ischemic postconditioning
before percutaneous coronary intervention for acute ST-segment
elevation myocardial infarction reduces contrast-induced
nephropathy and improves long-term prognosis. Arch Med Res.
47:483–488. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Marchant DJ, Boyd JH, Lin DC, Granville
DJ, Garmaroudi FS and McManus BM: Infammation in myocardial
diseases. Circ Res. 110:126–144. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
He Q, Zhou W, Xiong C, Tan G and Chen M:
Lycopene attenuates inflammation and apoptosis in post-myocardial
infarction remodeling by inhibiting the nuclear factor-κB signaling
pathway. Mol Med Rep. 11:374–378. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheng P, Zeng W, Li L, Huo D, Zeng L, Tan
J, Zhou J, Sun J, Liu G, Li Y, et al: PLGA-PNIPAM microspheres
loaded with the gastrointestinal nutrient NaB ameliorate cardiac
dysfunction by activating Sirt3 in acute myocardial infarction. Adv
Sci (Weinh). 3:16002542016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Santana ET, Feliciano RD, Serra AJ,
Brigidio E, Antonio EL, Tucci PJ, Nathanson L, Morris M and Silva
JA Jr: Comparative mRNA and MicroRNA profling during acute
myocardial infarction induced by coronary occlusion and ablation
radio-frequency currents. Front Physiol. 7:5652016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Geng L, Liu W and Chen Y: miR-124-3p
attenuates MPP+-induced neuronal injury by targeting
STAT3 in SH-SY5Y cells. Exp Biol Med (Maywood). 242:1757–1764.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yuan Q, Sun T, Ye F, Kong W and Jin H:
MicroRNA-124-3p affects proliferation, migration and apoptosis of
bladder cancer cells through targeting AURKA. Cancer Biomark.
19:93–101. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kang Q, Xiang Y, Li D, Liang J, Zhang X,
Zhou F, Qiao M, Nie Y, He Y, Cheng J, et al: miR-124-3p attenuates
hyperphosphorylation of Tau protein-induced apoptosis via
caveolin-1-PI3K/Akt/GSK3β pathway in N2a/APP695swe cells.
Oncotarget. 8:24314–24326. 2017.PubMed/NCBI
|
|
37
|
Dong RF, Zhang B, Tai LW, Liu HM, Shi FK
and Liu NN: The neuroprotective role of miR-124-3p in a
6-hydroxydopamine-induced cell model of parkinson's disease via the
regulation of ANAX5. J Cell Biochem. 119:269–277. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang S, Ge X, Yu J, Han Z, Yin Z, Li Y,
Chen F, Wang H, Zhang J and Lei P: Increased miR-124-3p in
microglial exosomes following traumatic brain injury inhibits
neuronal inflammation and contributes to neurite outgrowth via
their transfer into neurons. FASEB J. 32:512–528. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiang N, Xia J, Jiang B, Xu Y and Li Y:
TUG1 alleviates hypoxia injury by targeting miR-124 in H9c2 cells.
Biomed Pharmacother. 103:1669–1677. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nourbakhsh M and Hauser H: The
transcriptional silencer protein NRF: A repressor of NF-kappa B
enhancers. Immunobiology. 198:65–72. 1997. View Article : Google Scholar : PubMed/NCBI
|