Introduction
Hypoxia may result in an insufficient supply of
blood and nutrients to the heart, resulting in cardiomyocytes
undergoing apoptosis, which may potentially injure the cardiac
tissue (1). Myocardial infarction,
a normal ischemic heart disease, is one of the primary causes of
disability and the leading cause of mortality worldwide (2). At present, the most effective therapy
for treating patients following a myocardial infarction is
reperfusion, a process of rapidly restoring the blood flow through
the occluded coronary artery (3–5).
Reperfusion will result in myocardial ischemia/reperfusion (MI/R)
injury, which will result in additional death of cardiomyocytes and
will enlarge the size of the infarction (4,6,7). A
deeper understanding of the molecular mechanism underlying MI/R
injury may improve treatments and reduce MI/R-associated
damage.
There is increasing evidence illustrating the roles
of micro-RNAs (miRNAs) in the modulation of numerous biological
processes in diseases, including MI/R injury (8–10).
miRNAs, which are members of the non-coding small RNA family, are
involved in gene silencing by binding to the 3′-untranslated region
(3′-UTR) of its target genes (11,12).
Several differentially expressed miRNAs, including miR-21 (13), miR-24 (14) and miR-29 (15), have been demonstrated to be
involved in I/R-induced injury. miR-16 was identified as an
anti-apoptotic factor in glioblastoma multiforme (11).
In the present study, the mRNA expression levels of
miR-16 were significantly increased in H9c2(2-1) cardiac myoblast
cells following simulation of H/R, suggesting an important role of
miR-16 in H/R-injury response. In addition, the potential target
genes of miR-16 were examined. It was demonstrated that miR-16 may
reduce the damage of cardiac myoblast cells caused by H/R,
partially by downregulating the expression of cytokine-induced
apoptosis inhibitor 1 (CIAPIN1). To the best of our knowledge, the
present study is the first to study the specific role of miR-16 in
H/R injury.
Materials and methods
Cell culture and construction of an
H/R model
The H9c2(2-1) cardiac myoblasts cell line
(henceforth referred to as H9c2) was purchased from the American
Type Culture Collection (Manassas, VA, USA) and maintained in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
FBS, 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C with
95% atmosphere and 5% CO2. To construct the H/R model,
H9c2 cells were transferred into DMEM without FBS before being
subjected to hypoxia. The cells were incubated at 37°C for 6 h in
an anaerobic chamber containing 95% N2 and 5%
CO2. Subsequently, the cells were moved to a normoxic
incubator (5% CO2 and 95% atmosphere) with DMEM and
incubated for 24 h for re-oxygenation. The same treatment was
performed in the sham group without the hypoxic stimulus.
Transfection of miR-16
mimics/inhibitor
To investigate the role of miR-16, the miR-16
mimics, miR-16 inhibitor and their corresponding negative controls
(NC) were all synthesized by GenePharma Co., Ltd. (Shanghai,
China). The sequences of miR-16 mimics and the negative control
(NC) were: 5′-UAGCAGCACGUAAAUAUUGGUG-3′ and
5′-UACACCGAUCGAGUCAGGUTT-3′, respectively. The sequences of the
miR-16 inhibitor and its NC were 5′-CACCAAUAUUUACGUGCUGCUA-3′ and
5′-UCGAGACACGUACGCAGAATT-3′, respectively. A pcDNA3.1-CIAPIN1
plasmid was constructed by GenePharma Co., Ltd. to overexpress
CIAPIN1, and an empty pcDNA3.1 vector (GenePharma Co., Ltd.) was
used as the NC. H9c2 cells were seeded into 24-well plates and
grown to ~60% confluence. The miR-16 mimics, miR-16 inhibitor,
pcDNA3.1-CIAPIN1 and their corresponding NCs were transfected into
the cells using Lipofectamine® 3000 reagent (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham MA, USA) according to the
manufacturer's protocol. The final concentration of miR-16 mimics
and its NC was 50 nM, and the final concentration for miR-16
inhibitor and NC was 100 nM. The expression level of miR-16 was
detected by reverse transcription-quantitative (RT-q)PCR 24 h after
transfection.
Dual-luciferase reporter gene
assay
The potential targets of miR-16 were predicted using
the TargetScan website (http://www.targetscan.org/vert_72/). CIAPIN1 was
predicted as a target of miR-16. In order to confirm that miR-16
directly bound to CIAPIN1, a dual-luciferase reporter gene assay
was performed. The 3′-UTR of CIAPIN1 containing the miR-16-binding
sites (CIAPIN1-WT) and 3′-UTR of CIAPIN1 containing the mutant
miR-16-binding sites (CIAPIN1-Mut) were synthesized. The CIAPIN1-WT
and CIAPIN1-Mut were cloned into pmiR-RB-REPORT™ plasmid (Guangzhou
RiboBio Co., Ltd.). Each recombinant vector, pmiR-CIAPIN1-WT or
pmiR-CIAPIN1-Mut, along with miR-16 mimics or miR-16 mimics NC,
were co-transfected into the H9c2 cells using Lipofectamine™ 3000
reagent, according to the manufacturer's protocol. After 48 h of
transfection, the total protein was extracted, and the luciferase
activity was detected using a Dual-Luciferase Reporter assay kit
(Promega Corporation, Madison, WI, USA), according to the
manufacturer's protocol. The activity of Renilla luciferase was
used for normalization.
Cell Counting Kit-8 (CCK-8) assay
A CCK-8 assay (Beyotime Institute of Biotechnology,
Haimen, China) was used to examine the proliferative ability of
H9c2 cells after 24 h transfection. Single cell suspensions were
prepared and seeded into 96-well plates with 1×103
cells/well. The plates were incubated at 37°C with 5%
CO2. After a 24 h incubation, 10 µl CCK-8 reagent was
added to each well and incubated at 37°C for 1.5 h. Subsequently,
the absorbance was detected at 450 nm using an ELx800 microplate
reader (BioTek Instruments, Inc., Winooski, VT, USA).
RT-qPCR
Cells were harvested 24 h after transfection and the
total RNA was extracted using TRIzol® (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. For mRNA expression analysis, the isolated RNA was
reverse-transcribed to cDNA using a PrimeScript™ RT Reagent kit
(Takara Bio, Inc.). The RT procedure was: 37°C for 15 min and then
85°C for 15 sec. qPCR was performed using SYBR Premix Ex Taq II
(Takara Bio, Inc.) on a 7500 Real Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). To determine the level
of miR-16, Mir-X™ miRNA First Strand Synthesis kit (Takara Bio,
Inc.) was used to reverse-transcribe the RNA to cDNA according to
the supplier's instruction. qPCR was then performed using SYBR
PrimeScript™ miRNA RT-PCR kit (Takara Bio, Inc.). The PCR steps
including 95°C for 5 min, followed by 40 cycles of 5 sec at 95°C,
34 sec at 60°C, and then 72°C for 30 min. The expression of GAPDH
and U6 small nuclear RNA were used as internal references for mRNA
and miRNA accordingly. The relative expression of CIAPIN1 and
miR-16 was calculated using the 2−ΔΔCq method (16). All the primers used for qPCR are
presented in Table I. The U6 small
nuclear RNA primers and uni-miR qPCR primer were included with the
SYBR PrimeScript™ miRNA RT-PCR kit.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| A, mRNA primer
sequences |
|---|
| Primer | Sequence |
|---|
| CIAPIN1 F |
5′-GCTTGTGGCAGTGTTCTGTG-3′ |
| CIAPIN1 R |
5′-CACAGAACACTGCCACAAGC-3′ |
| GAPDH F |
5′-GCCAGCCTCGTCTCATAGAC-3′ |
| GAPDH R |
5′-AGTGATGGCATGGACTGTGG-3′ |
|
| B, miRNA primer
sequences |
|
| Primer |
Sequence |
|
| miR-16 F |
5′-GCGTAGCAGCACGTAAAT-3′ |
| miR-16 R | Uni-miR qPCR
Primer |
Western blotting
Total cellular proteins were extracted from H9c2
cells using the RIPA lysate with protease inhibitors (CWBIO,
Beijing, China. http://www.cwbiotech.com/) after 48 h transfection.
Proteins (20 µg each well) were then separated by SDS-PAGE with a
10% gel. Subsequently, the proteins from the gel were transferred
to a PVDF membrane (EMD Millipore, Billerica, MA, USA). Then the
membranes were blocked by 5% skimmed milk for 1 h at room
temperature. Then the membranes were incubated with primary
antibodies for 12 h, and then the secondary antibody for 1 h at
4°C. The signals were visualized using enhanced chemiluminescent
reagent, and the expression of β-actin was used for normalization.
The following primary antibodies were used, Bcl-2 (Abcam,
Cambridge, UK; cat. no. ab196495; 1:1,000), Bax (Abcam; cat. no.
ab53154; 1:1,000), cleaved caspase-3 (Abcam; cat. no. ab49822;
1:1,000), CIAPIN1 (Invitrogen; Thermo Fisher Scientific, Inc.; cat.
no. PA5-59903; 1:1,000), NF-κB (Abcam; cat. no. ab16502; 1:1,000),
p-NF-κB (CST Biological Reagents Co., Ltd., Shanghai, China; cat.
no. 3039, 1:1,000), IκBα (Abcam; cat. no. ab7217; 1:1,000), p-IκBα
(CST, cat. no. 2859; 1:1,000) and β-actin (CST Biological Reagents
Co., Ltd., cat. no. 4970; 1:1,000). The anti-rabbit immunoglobulin
G, horseradish peroxidase-linked antibody of CST Biological
Reagents Co., Ltd. (cat. no. 7074; 1:2,000) was used as the
secondary antibody. Quantity One 4.6.2 software (Bio-Rad
Laboratories, Inc.) was used for densitometry analysis.
Cell apoptosis analysis
Flow cytometry was used to evaluate the apoptosis of
H9c2 cells. Cells were harvested by centrifugation (300 × g, 5 min,
4°C) and washed using pre-cooled PBS. Cells were resuspended in
binding buffer(Sigma-Aldrich; Merck KGaA) at a final concentration
of 1.5×106 cells/ml. Double staining was performed with
an Annexin V-fluorescein isothiocyanate (FITC)/propidium staining
(PI) kit, according to the manufacturer's protocol (Sigma-Aldrich;
Merck KGaA; Darmstadt, Germany). Staining was measured using flow
cytometry (BD Biosciences) and analyzed with FlowJo software 7.2
(FlowJo LLC).
Statistical analysis
All the results are presented as the mean ± standard
deviation. Comparisons between two groups were analyzed using a
Student's t-test. Comparisons between multiple groups were analyzed
using a one-way ANOVA, followed by a post hoc Tukey's or Dunnett's
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
miR-16 expression is upregulated in
H9c2 cells following hypoxia
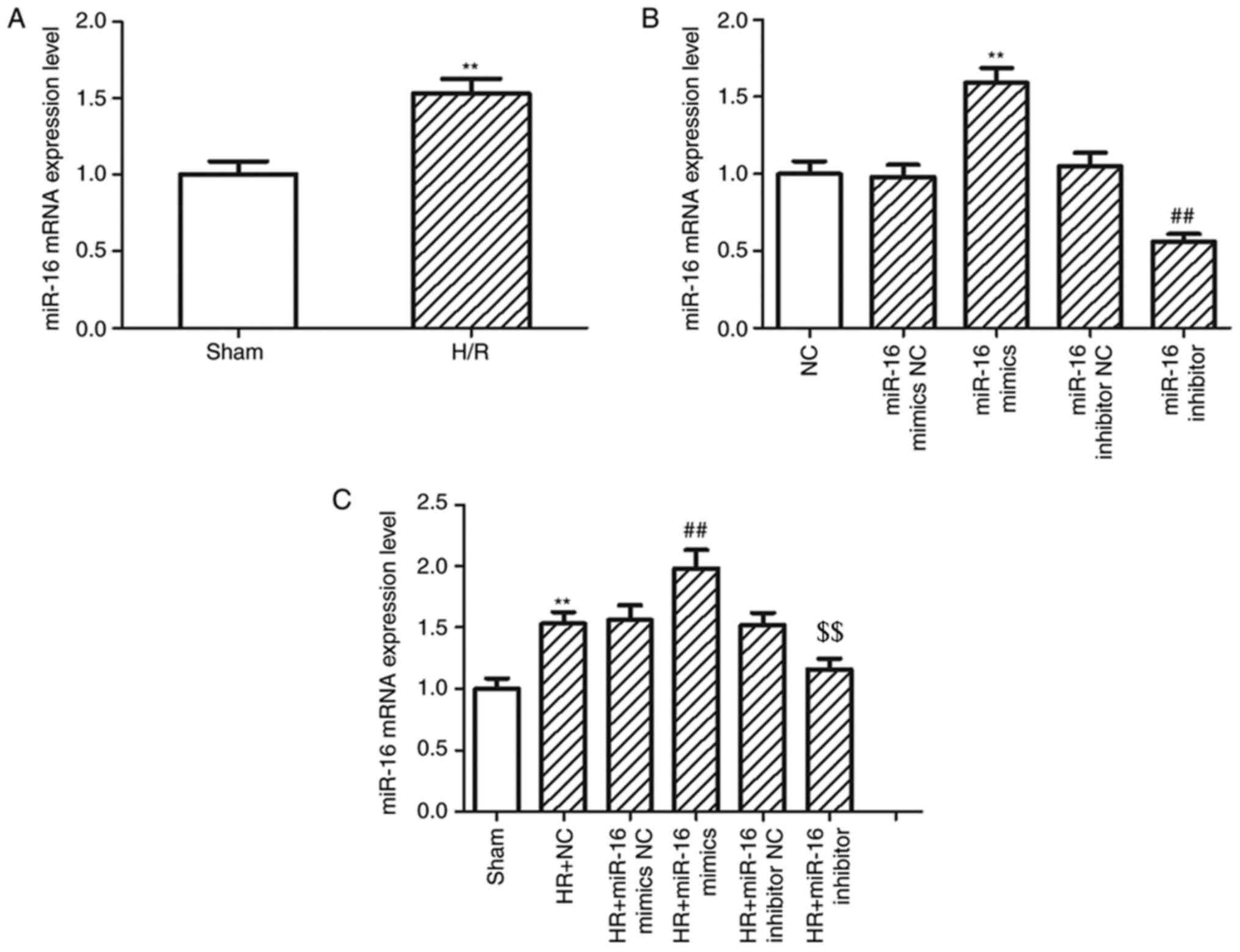
The mRNA expression levels of miR-16 in H9c2 cells
increased 1.53-fold following hypoxic treatment compared with the
H9c2 cells in the sham group (P=0.0019; Fig. 1A). To determine the possible
biological functions of miR-16, miR-16 mimics and miR-16 inhibitor
were used to upregulate or downregulate the expression of miR-16 in
H9c2 cells, respectively. The mRNA expression levels of miR-16 were
significantly increased in H9c2 cells transfected with miR-16
mimics (1.59±0.10 vs. 0.98±0.08; P=0.000026) and significantly
decreased in H9c2 cells transfected with miR-16 inhibitor
(0.56±0.05 vs. 1.05±0.09; P=0.000299; Fig. 1B) compared with the respective NC.
When H/R was simulated in the H9c2 cells, the expression of miR-16
was also increased in cells transfected with miR-16 mimics
(1.98±0.15 vs. 1.53±0.01; P=0.002205) and decreased by miR-16
inhibitors (1.16±0.09 vs. 1.52±0.01; P=0.006857 Fig. 1C) compared with the respective
NC.
miR-16 regulates the proliferation and
apoptosis of H9c2 cells following hypoxia
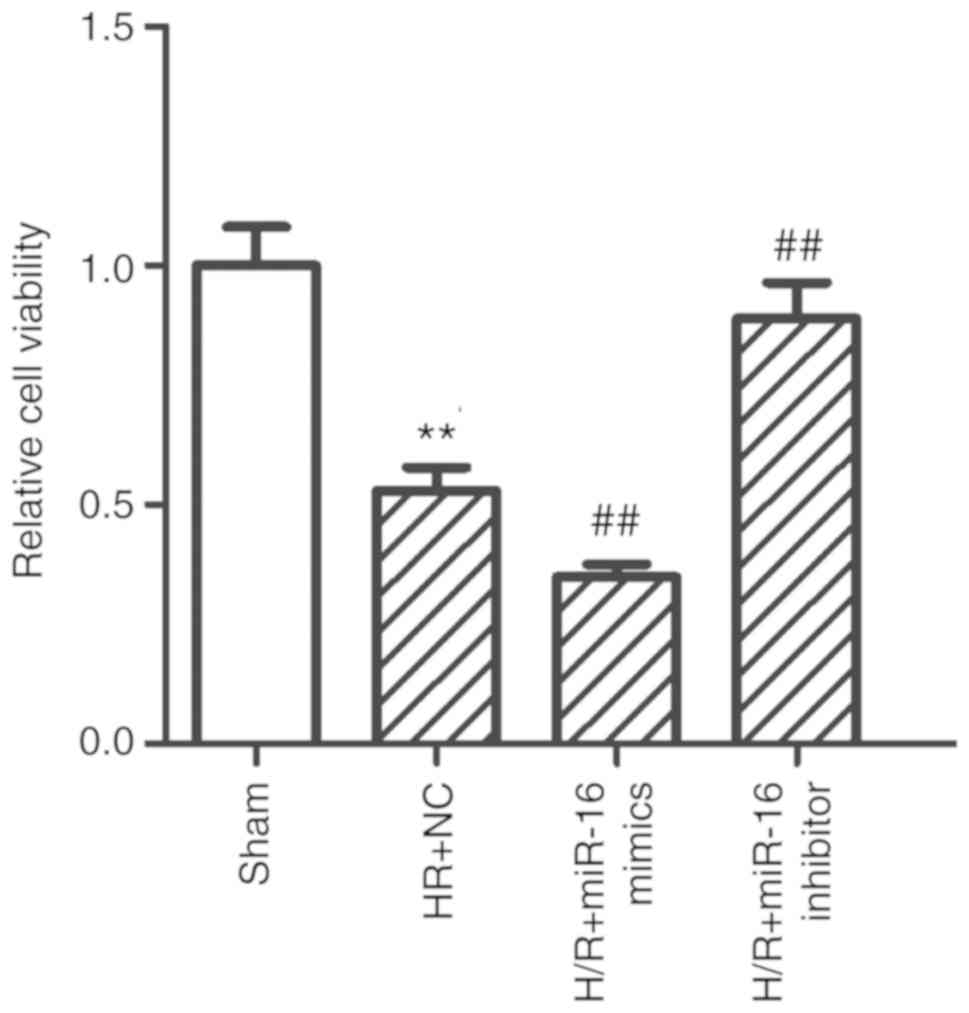
A CCK-8 assay and flow cytometry were performed to
examine the role exerted by miR-16 on the proliferation and
apoptosis of H9c2 cells following hypoxia. The viability of H9c2
cells was significantly reduced in the H/R+NC group compared with
the sham group (0.53±0.05 vs. 1.00±0.08; P=0.000003; Fig. 2). Upregulation of miR-16 using
miR-16 mimics (H/R+miR-16 mimics group) further decreased
proliferation of H9c2 cells compared with the H/R+NC group
(0.31±0.03 vs. 0.53±0.05; P=0.0097; Fig. 2). Downregulation of miR-16 using
the miR-16 inhibitor significantly increased the proliferation of
H9c2 cells compared with the H/R+NC group (0.89±0.08 vs. 0.53±0.05;
P=0.000385; Fig. 2).
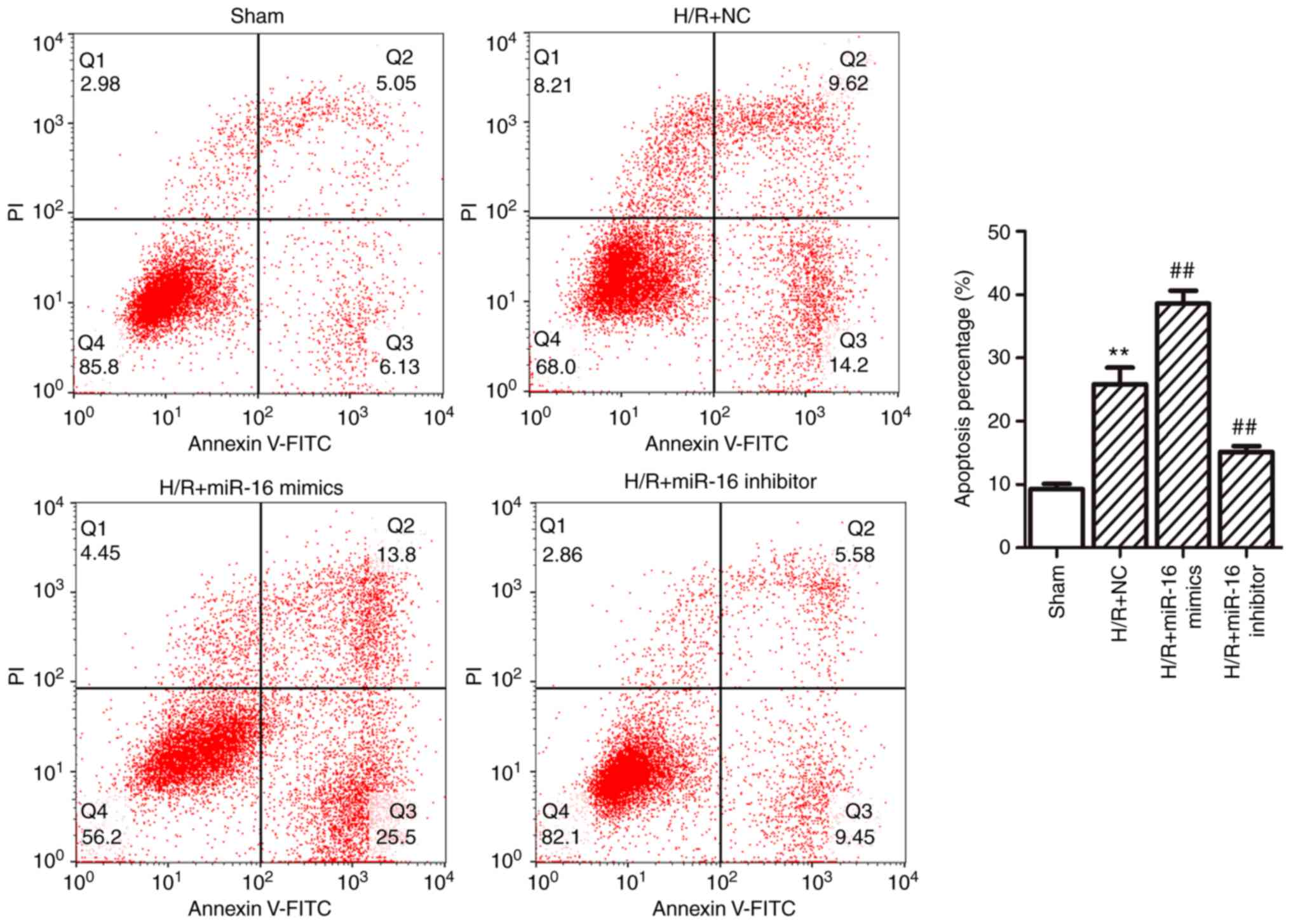
Furthermore, flow cytometric analysis demonstrated
that that the apoptotic percentage was significantly increased in
the H/R+NC group compared with the sham group (25.86±2.62% vs.
9.29±0.82%; P=0.000014; Fig. 3).
Compared with the H/R+NC group, the apoptotic percentage was
increased in the H/R+miR-16 mimics group (38.62±2.04% vs.
25.86±2.62%; P=0.000099; Fig. 3),
whereas in the H/R+miR-16 inhibitor group the apoptotic rate was
significantly decreased (15.14±0.92% vs. 25.86±2.62%; P=0.000343;
Fig. 3) compared with the H/R+NC
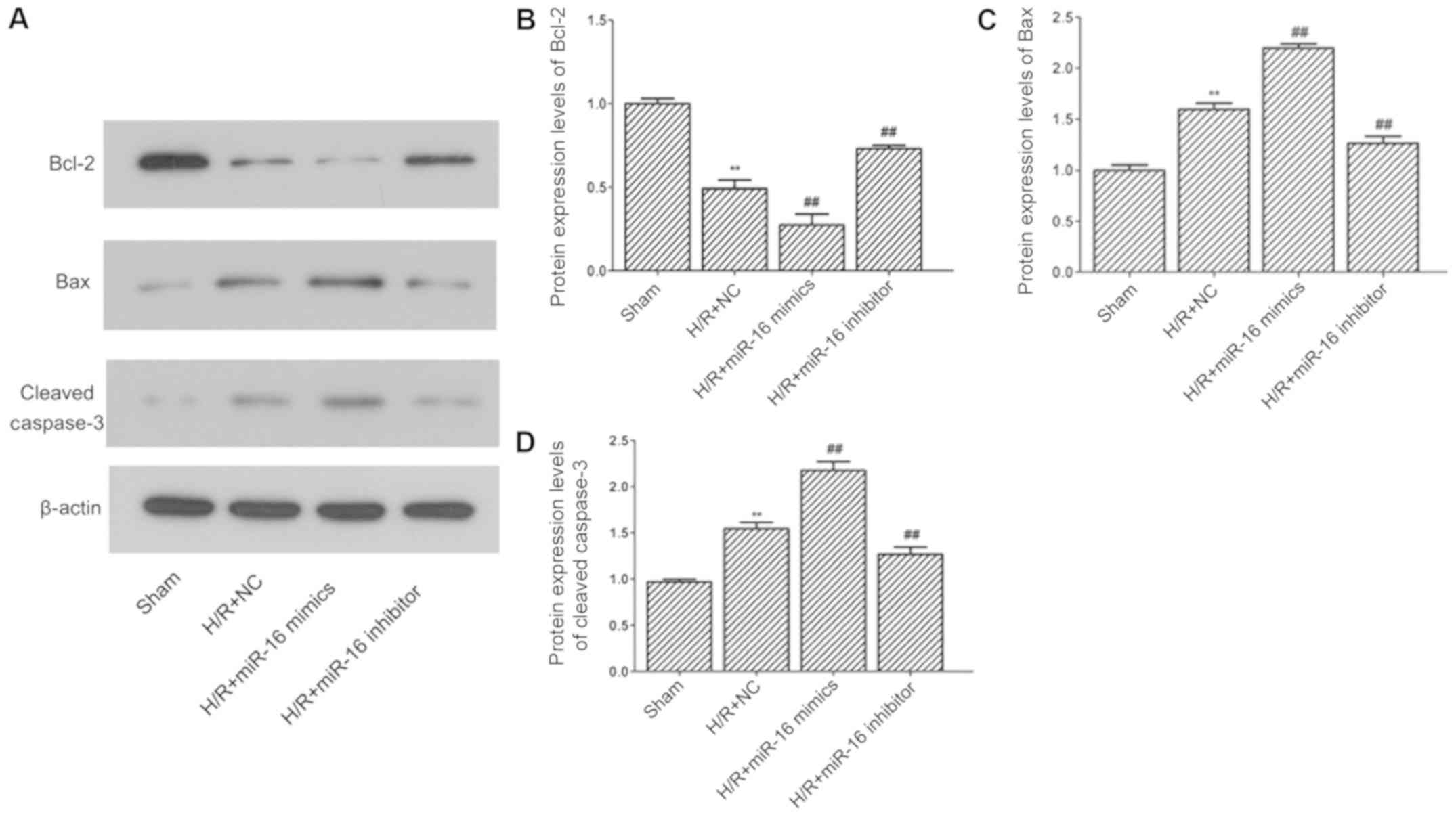
group. The expression levels of apoptosis-associated proteins were
determined by western blotting (Fig.
4A). The results demonstrated that the expression of Bcl-2
(0.49±0.05 vs. 1.00±0.03; P=0.000004; Fig. 4B) was decreased; whereas the levels
of Bax (1.60±0.06 vs. 1.00±0.05; P=0.000005; Fig. 4C) and cleaved caspase-3 (1.54±0.07
vs. 0.96±0.03; P=0.00001; Fig. 4D)
were increased in the H/R+NC group compared with the sham group.
Compared with the H/R+NC group, the expression of Bcl-2 (0.27±0.07
vs. 0.49±0.05; P=0.00193; Fig. 4B)
was decreased, whereas the levels of Bax (2.20±0.04 vs. 1.60±0.06;
P=0.000005; Fig. 4C) and cleaved
caspase-3 (2.17±0.09 vs. 1.54±0.07; P=0.000012; Fig. 4D) were increased in the H/R+miR-16
mimics group. In the H/R+miR-16 inhibitor group, the expression of
Bcl-2 (0.73±0.02 vs. 0.49±0.05; P=0.000983; Fig. 4B) was increased, whilst the levels
of Bax (1.26±0.07 vs. 1.60±0.06; P=0.000394; Fig. 4C) and cleaved caspase-3 (1.27±0.08
vs. 1.54±0.07; P=0.001939; Fig.
4D) were declined compared with the H/R+NC group.
miR-16 negatively regulates the
expression of CIAPIN1
To further examine how miR-16 affected cell
proliferation and apoptosis in H/R injury, the target was predicted
using TargetScan. CIAPIN1 was one of the targets of miR-16, and the
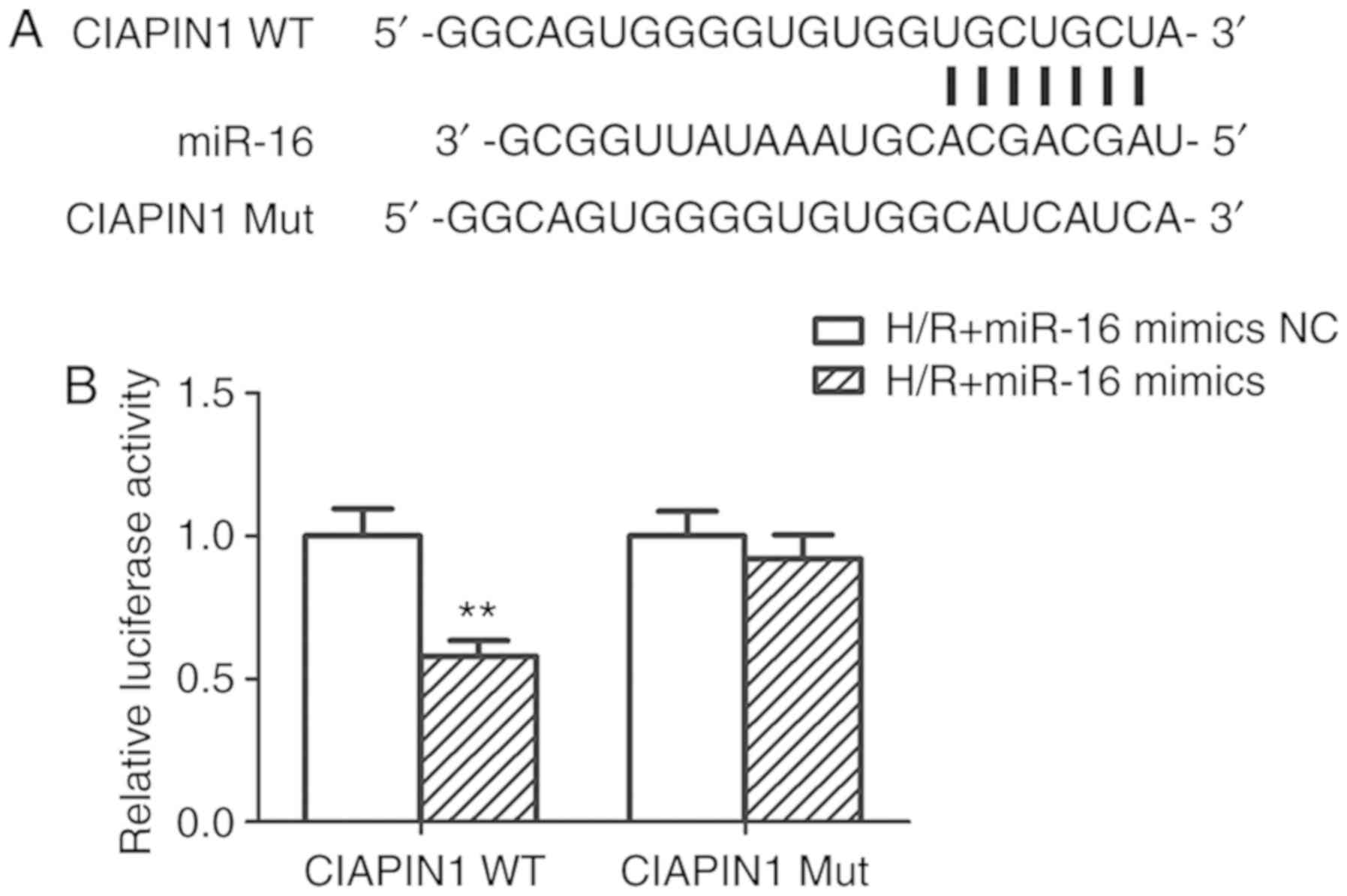
predicted binding sites are shown in Fig. 5A. The results of the luciferase
reporter assay revealed that the relative luciferase activity of
CIAPIN1-WT was dramatically decreased in the H/R+miR-16 mimics
group compared with the H/R+miR-16 mimics NC group (P<0.01;
Fig. 5B). However, upregulation of
miR-16 did not significantly influence the luciferase activity of
CIAPIN1-Mut (P>0.05; Fig. 5B).
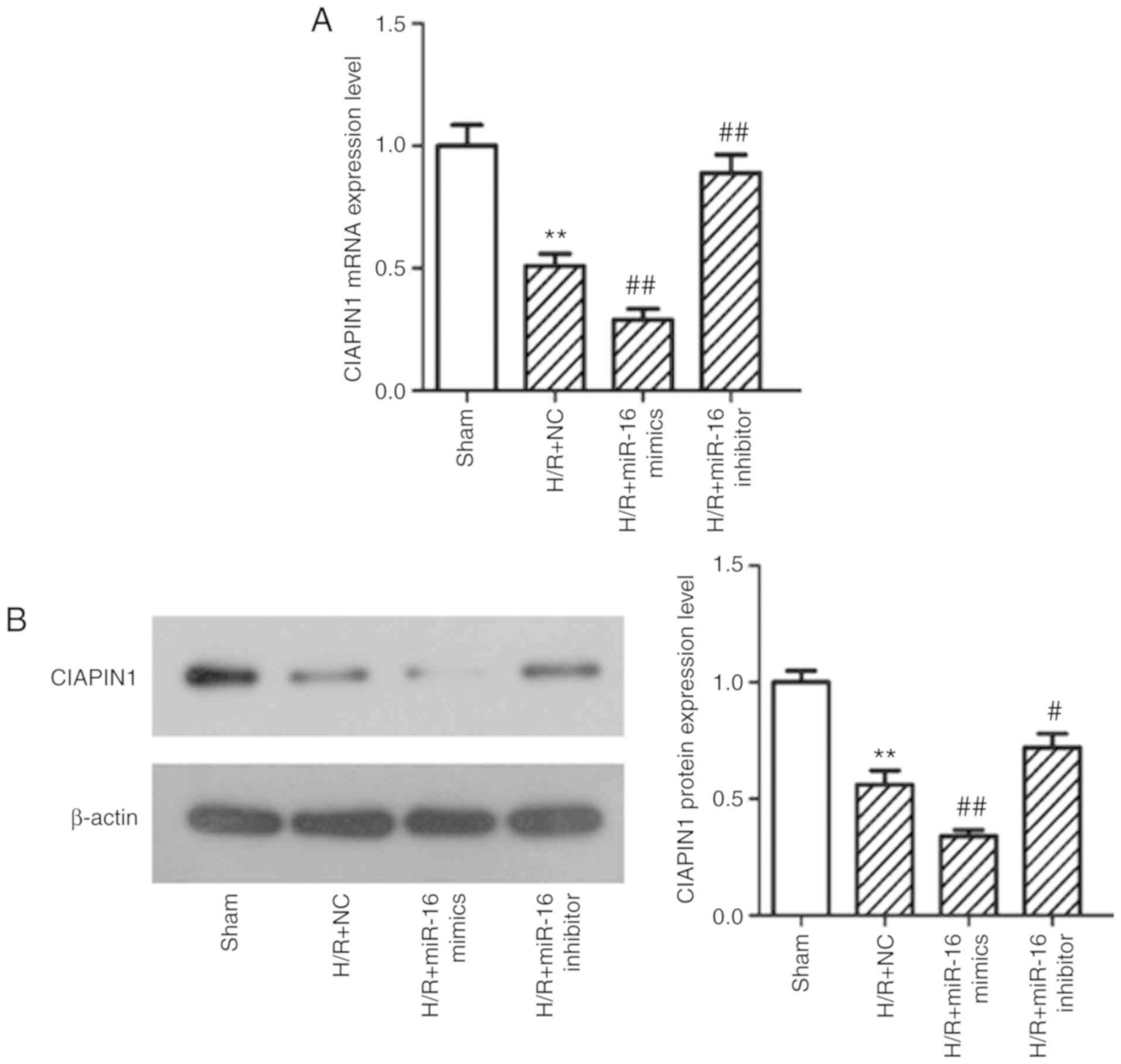
Therefore, the effect of miR-16 on CIAPIN1 expression was
determined by qPCR and western blotting. The mRNA and protein
expression levels of CIAPIN1 were decreased significantly in the
H/R+NC group compared with the sham group (both P<0.01; Fig. 6). Upregulation of miR-16 using
miR-16 mimics further decreased CIAPIN1 expression at both the mRNA
(Fig. 6A) and protein expression
levels (Fig. 6) in comparison with
H/R+NC group (both P<0.01). CIAPIN1 expression was enhanced in
the H/R+miR-16 inhibitor group compared with the H/R+NC group
(P=0.000858).
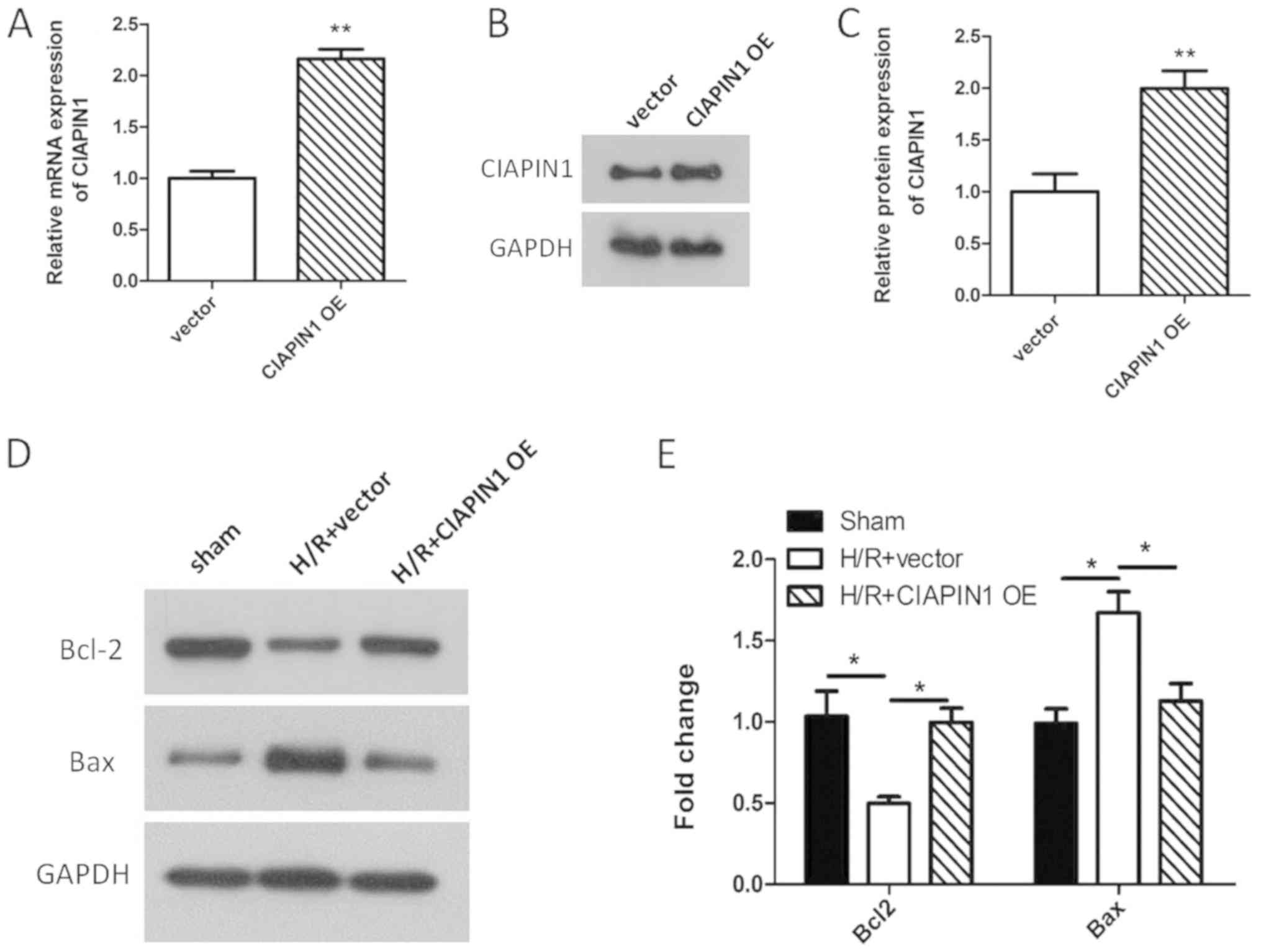
Overexpression of CIAPIN1 reverses the
changes of expression of apoptosis-associated proteins caused by
H/R
CIAPIN1 was overexpressed in H9c2 cells to determine
its effect on apoptosis. As shown in Fig. 7A-C, the mRNA and protein expression
levels of CIAPIN1 were significantly increased in H9c2 cells in the
CIAPIN1-overexpression group compared with the control (P<0.01).
The effect of CIAPIN1 overexpression on the expression of Bcl-2 and
Bax in H9c2 cells following H/R was determined by western blotting.
The results showed that the expression levels of Bcl-2 were
significantly increased, whereas the expression of Bax was
significantly decreased in H/R-damaged H9c2 cells transfected with
CIAPIN1-OE compared with the H/R+ CIAPIN1-OE group (Fig. 7D and E; P=0.035; P=0.032). These
data suggested that overexpression of CIAPIN1 reversed the changes
in expression of apoptosis-associated proteins caused by H/R,
suggesting that overexpression of CIAPIN1 reduced apoptosis induced
by H/R injury.
The NF-κB pathway may be involved in
miR-16-mediated protective effects following myocardial
ischemia
Western blotting was performed to determine the
influence of miR-16 expression on the NF-κB signaling pathway. The
results demonstrated that the expression levels of NF-κB and
inhibitor of NF-κB alpha (IκBα) remained relatively unchanged
despite the change in miR-16 expression. However, the expression
levels of p-NF-κB and p-IκBα were significantly upregulated in H9c2
cells following H/R (Fig. 8;
1.82±0.11 vs. 1.00±0.08, P=0.000152; and 1.77±0.07 vs. 1.00±0.00,
P=0.000024, respectively). Upregulation of miR-16 using miR-16
mimics further increased the levels of p-NF-κB and p-IκBα compared
with the H/R+NC group (Fig. 8;
3.10±0.14 vs. 1.82±0.11, P=0.000006; and 2.19±0.10 vs. 1.77±0.07,
P=0.0017, respectively). In contrast, the expression levels of
p-NF-κB and p-IκBα were decreased in the cells transfected with the
miR-16 inhibitor group compared with the H/R+NC group (Fig. 8; 1.26±0.13 vs. 1.82±0.11, P=0.002;
and 1.45±0.12 vs. 1.77±0.06, P=0.009).
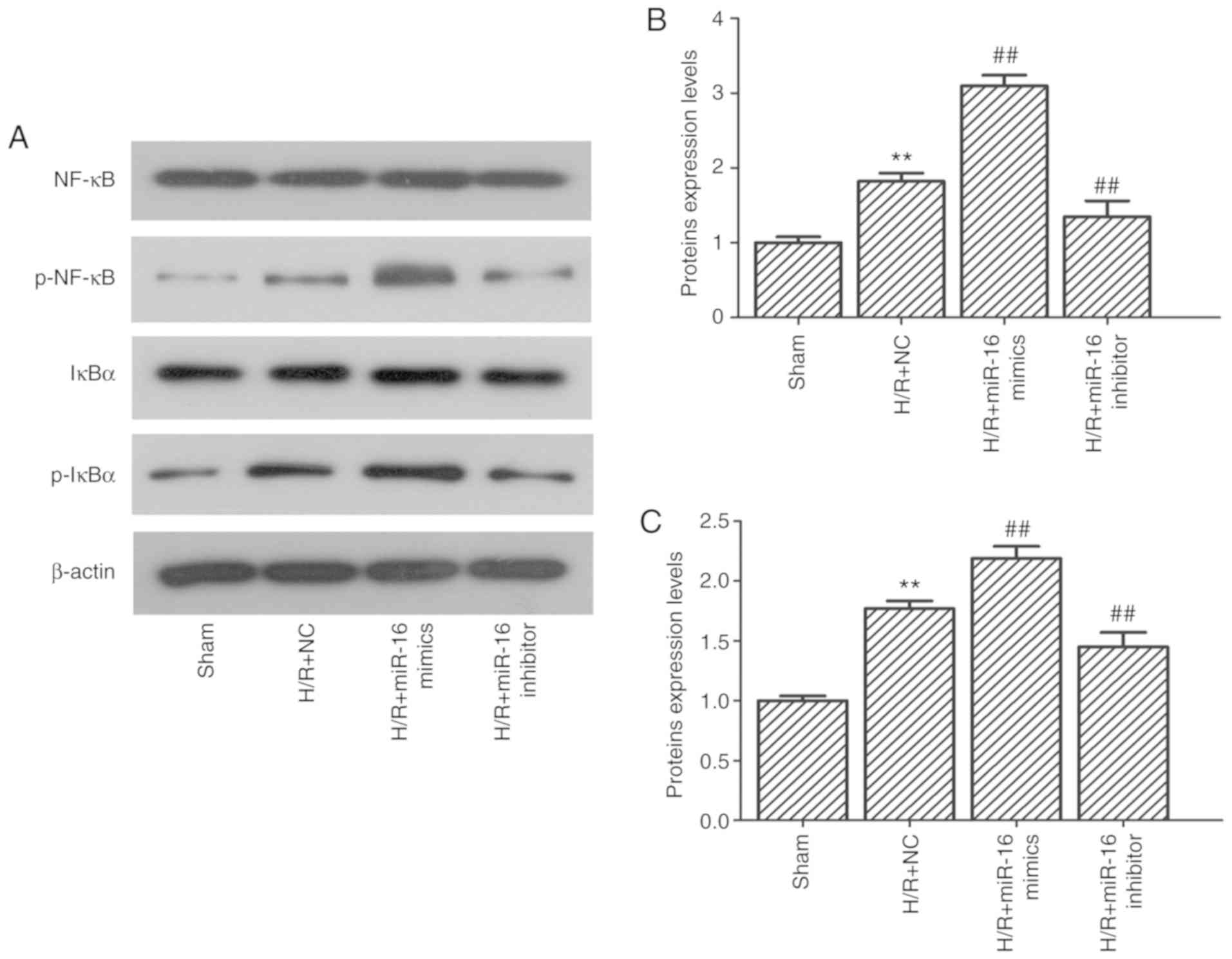 | Figure 8.Effect of miR-16 on the expression of
NF-κB, p-NF-κB, IκBα and p-IκBα. (A) Protein expression levels of
NF-κB, p-NF-κB, IκBα and p-IκBα were determined by western blot.
(B) Relative protein expression levels of p-NF-κB in cells
following H/R and/or transfection of the miR-mimics or inhibitors.
**P<0.01 vs. sham; ##P<0.01 vs. H/R+NC. (C)
Relative protein expression levels of p-IκBα in cells following H/R
and/or transfection of the miR-mimics or inhibitors. **P<0.01
vs. sham; ##P<0.01 vs. H/R+NC. miR, microRNA; H/R,
hypoxia/reoxygenation; p-, phospho; NC, negative control; NF-κB,
nuclear factor-κB; IκBα, NF-κB inhibitor α. |
Discussion
miRNAs have been demonstrated to serve important
roles in the regulation of MI/R injury (17). In the present study, miR-16
expression was enhanced in H9c2 cells following simulation of H/R.
Upregulation of miR-16 expression enhanced the inhibitory effects
on H9c2 cell proliferation caused by H/R. Downregulation of miR-16
expression reduced the suppressive effects of H/R on cell
proliferation as well as cell apoptosis. These observations
suggested that downregulation of miR-16 may mitigate H/R-induced
injury. Higher expression levels of miR-16 have been observed in
the urine of patients with acute kidney injury (AKI). The
researchers identified that miR-16 was transactivated by C/EBP-β,
which aggravated the I/R-induced AKI (18), consistent with the results of the
present study.
To date, the majority of the research surrounding
miR-16 has been focused on its role in cancer, and there are
conflicting reports on its function based on the type of cancer
studied (19–22), suggesting that it may target
distinct genes in different organs resulting in varied effects
(18). Among the numerous targets
of miR-16, a focus was placed on the CIAPIN1 gene in the recent
study. CIAPIN1 is a novel apoptosis inhibitor with no association
with apoptosis regulators in the Bcl-2 and caspase families
(23). Furthermore, it has been
identified as an important downstream effector of the Ras signaling
pathway (24). At present,
numerous studies have shown that CIAPIN1 exhibits different
functions based on the physiological and pathological conditions
(25–27). In large B-cell lymphoma and ovarian
cancer, CIAPIN1 exhibited anti-apoptotic effects and facilitated
proliferation (26). However, in
esophageal cancer and renal cell carcinoma, CIAPIN1 exhibited an
inhibitory effect on tumor cell viability (27). In hypoxia-induced cardiomyocytes,
CIAPIN1 has been illustrated to be a target of miR-182-5p, and the
expression of CIAPIN1 was dramatically decreased following hypoxia
(1). Furthermore, CIAPIN1
overexpression reduced the H/R-induced damage in H9c2 myocytes
(28). In the present study,
CIAPIN1 was predicted as one of the targets of miR-16. Using a
luciferase reporter assay, it was confirmed that miR-16 directly
regulated the expression of CIAPIN1 by binding to its 3′-UTR.
Expression of CIAPIN1 was downregulated in H9c2 cells following
H/R. Increasing expression of miR-16 using miR-16 mimics further
decreased both the mRNA and protein expression levels of CIAPIN1
following H/R compared with the H/R+NC group. In contrast,
downregulation of miR-16 using an inhibitor resulted in a
significant increase in the expression of CIAPIN1 compared with the
H/R+NC group. These observations illustrate that CIAPIN1 may be
negatively regulated by miR-16.
MI/R is associated with a serious inflammatory
response. NF-κB transcription factors take part in a number of
physiological processes, including inflammation, immunity and
tumorigenicity (29). In the
non-activated state, NF-κB exists as a heterotrimer composed of
p50, p65 and IκB subunits in the cytoplasm. Upon activation, IκBα
is phosphorylated, and subsequently degraded, allowing p65 to
translocate to the nucleus and bind to the DNA, leading to gene
transcription (29). Numerous
studies have suggested the involvement of the NF-κB signaling
pathway in the pathogenesis of MI/R (30–32).
In myocardial tissues of MI/R mice, the expression of IκBα was
significantly increased, and it was reported that downregulation of
miR-27a protected mice against MI/R injury, partially by modulating
the NF-κB signaling pathway (31).
In rats, myocardial NF-κB expression was significantly increased
following MI/R, highlighting its potential role in MI/R (30). In the present study, the relative
expression levels of p-NF-κB and p-IκBα were upregulated in H9c2
cells following H/R. Upregulation or downregulation of miR-16
significantly increased or decreased the expression levels of
p-NF-κB and p-IκBα levels. Therefore, it was hypothesized that
downregulation of miR-16 may reduce H/R injury, at least partially
through suppression of the NF-κB signaling pathway, which results
in decreased levels of inflammatory cytokines, and subsequently
reduced apoptosis. In ulcerative colitis, miR-16 was demonstrated
to participate in regulating the NF-κB signaling pathway by
modulating the expression of the adenosine A2a receptor (33). Upregulation of miR-16 increased
activation of the NF-κB signaling pathway in ulcerative colitis
(33), and this result was
consistent with the results of the present study. In the bladder
cancer cell line, T24, upregulation of miR-16 resulted in
deactivation of the NF-κB signaling pathway (34). In addition, miR-16 was reported to
decrease glioma malignancy by downregulating NF-κB (35). These seemingly opposite results may
result from differences in the tissues used and the diseases
studied. Reports regarding the association between CIAPIN1 and the
NF-κB signaling pathway are rare. In K562 chronic myeloid leukemia
cells, depletion of CIAPIN1 led to relatively lower levels of
p-IKKα/β and p-IκBα compared with CIAPIN1 scramble-transfected K562
cells (24). However, in the
present study, the expression levels of CIAPIN1 were relatively
higher in the miR-16 inhibitor group, whereas the expression levels
of p-IκBα and p-NF-κB were relatively lower in this group.
Additional studies are required to determine how miR-16 and CIAPIN1
interact with the NF-κB signaling pathway.
In conclusion, the present study highlights the
potential of targeting the miR-16/CIAPIN1 axis for treating
MI/R-induced injury. However, additional in vivo studies are
required to verify the present results. The association between
miR-16 and other targets, such as serotonin transporter (SERT)
(36) and B lymphoma mouse Moloney
leukemia virus insertion region (Bmi-1) (37), in MI/R injury may also be worth
studying to improve our understanding of the molecular mechanisms
underlying the role of miR-16 in MI/R-induced injury.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HJZ and YNZ conceived the study. HJZ and ZYT
conducted the experiments. HJZ wrote the manuscript. YNZ helped
interpret the results and revised the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhang Y, Fang J and Ma H: Inhibition of
miR-182-5p protects cardiomyocytes from hypoxia-induced apoptosis
by targeting CIAPIN1. Biochem Cell Biol. 96:646–654. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Forouzanfar MH, Moran AE, Flaxman AD, Roth
G, Mensah GA, Ezzati M, Naghavi M and Murray CJ: Assessing the
global burden of ischemic heart disease, part 2: Analytic methods
and estimates of the global epidemiology of ischemic heart disease
in 2010. Glob Heart. 7:331–342. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hillis LD and Lange RA: Myocardial
infarction and the open-artery hypothesis. N. Engl J Med.
355:2475–2477. 2006. View Article : Google Scholar
|
|
4
|
Liao YH, Xia N, Zhou SF, Tang TT, Yan XX,
Lv BJ, Nie SF, Wang J, Iwakura Y, Xiao H, et al: Interleukin-17A
contributes to myocardial ischemia/reperfusion injury by regulating
cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll
Cardiol. 59:420–429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xie J, Hu X, Yi C, Hu G, Zhou X and Jiang
H: MicroRNA-451 protects against cardiomyocyte anoxia/reoxygenation
injury by inhibiting high mobility group box 1 expression. Mol Med
Rep. 13:5335–5341. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zweier JL and Talukder MA: The role of
oxidants and free radicals in reperfusion injury. Cardiovasc Res.
70:181–190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ambrosio G and Tritto I: Myocardial
reperfusion injury. J Thromb Thrombolysis. 4:43–45. 2007.
|
|
8
|
Chen Z, Su X, Shen Y, Jin Y, Luo T, Kim
IM, Weintraub NL and Tang Y: miR322 mediates cardioprotection
against ischemia/reperfusion injury via FBXW7/notch pathway. J Mol
Cell Cardiol. 133:67–74. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng J, Li J, Kou B, Yi Q and Shi T:
MicroRNA-30e protects the heart against ischemia and reperfusion
injury through autophagy and the Notch1/Hes1/Akt signaling pathway.
Int J Mol Med. 41:3221–3230. 2018.PubMed/NCBI
|
|
10
|
Liu RR, Li J, Gong JY, Kuang F, Liu JY,
Zhang YS, Ma QL, Song CJ, Truax AD, Gao F, et al: MicroRNA-141
regulates the expression level of ICAM-1 on endothelium to decrease
myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ
Physiol. 309:H1303–H1313. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tian R, Wang J, Yan H, Wu J, Xu Q, Zhan X,
Gui Z, Ding M and He J: Differential expression of miR16 in
glioblastoma and glioblastoma stem cells: Their correlation with
proliferation, differentiation, metastasis and prognosis. Oncogene.
36:5861–5873. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Calin GA, Dumitru CD, Shimizu M, Bichi R,
Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L,
et al: Frequent deletions and down-regulation of micro-RNA genes
miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl
Acad Sci USA. 99:15524–15529. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tu Y, Wan L, Fan Y, Wang K, Bu L, Huang T,
Cheng Z and Shen B: Ischemic Postconditioning-mediated miRNA-21
protects against cardiac ischemia/reperfusion injury via PTEN/Akt
pathway. PLoS One. 8:e758722013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qian L, Van Laake LW, Huang Y, Liu S,
Wendland MF and Srivastava D: miR-24 inhibits apoptosis and
represses Bim in mouse cardiomyocytes. J Exp Med. 208:549–560.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ye Y, Hu Z, Lin Y, Zhang C and Perez-Polo
JR: Downregulation of microRNA-29 by antisense inhibitors and a
PPAR-gamma agonist protects against myocardial
ischaemia-reperfusion injury. Cardiovasc Res. 87:535–544. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang X, Zhang C, Wang N, Li Y, Zhang D
and Li Q: MicroRNA-486 alleviates Hypoxia-induced damage in H9c2
cells by targeting NDRG2 to inactivate JNK/C-Jun and NF-κB
signaling pathways. Cell Physiol Biochem. 48:2483–2492. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen HH, Lan YF, Li HF, Cheng CF, Lai PF,
Li WH and Lin H: Urinary miR-16 transactivated by C/EBPβ reduces
kidney function after ischemia/reperfusion-induced injury. Sci Rep.
6:279452016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu F, Zhang X, Lei Y, Liu X, Liu Z, Tong T
and Wang W: Loss of repression of HuR translation by miR-16 may be
responsible for the elevation of HuR in human breast carcinoma. J
Cell Biochem. 111:727–734. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu Y, Xia Y, Niu H and Chen Y: miR-16
induced the suppression of cell apoptosis while promote
proliferation in esophageal squamous cell carcinoma. Cell Physiol
Biochem. 33:1340–1348. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kang W, Tong JH, Lung RW, Dong Y, Zhao J,
Liang Q, Zhang L, Pan Y, Yang W, Pang JC, et al: Targeting of YAP1
by microRNA-15a and microRNA-16-1 exerts tumor suppressor function
in gastric adenocarcinoma. Mol Cancer. 14:522015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang CH, Fang XB, Li WF, Shi QY, Wu LP,
Chen XY, Huang ZX, Wu P, Wang ZZ and Liao ZS: Influence of
recombinant lentiviral vector encoding miR-15a/16-1 in biological
features of human nasopharyngeal carcinoma CNE-2Z cells. Zhonghua
Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 48:405–411. 2013.(In
Chinese). PubMed/NCBI
|
|
23
|
Shibayama H, Takai E, Matsumura I, Kouno
M, Morii E, Kitamura Y, Takeda J and Kanakura Y: Identification of
a cytokine-induced antiapoptotic molecule anamorsin essential for
definitive hematopoiesis. J Exp Med. 199:581–592. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang J, Li Q, Wang C, Xiong Q, Lin Y, Sun
Q, Jin H, Yang F, Ren X and Pang T: Knock-down of CIAPIN1
sensitizes K562 chronic myeloid leukemia cells to Imatinib by
regulation of cell cycle and apoptosis-associated members via NF-κB
and ERK5 signaling pathway. Biochem Pharmacol. 99:132–145. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang Z, Su G, Hu W, Bi XX, Zhang L and
Wan G: The study on expression of CIAPIN1 interfering
hepatocellular carcinoma cell proliferation and its mechanisms. Eur
Rev Med Pharmacol Sci. 21:3054–3060. 2017.PubMed/NCBI
|
|
26
|
Qu Y, Wang J, Ray PS, Guo H, Huang J,
Shin-Sim M, Bukoye BA, Liu B, Lee AV, Lin X, et al:
Thioredoxin-like 2 regulates human cancer cell growth and
metastasis via redox homeostasis and NF-κB signaling. J Clin
Invest. 121:212–225. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Santini D, Schiavon G, Vincenzi B, Gaeta
L, Pantano F, Russo A, Ortega C, Porta C, Galluzzo S, Armento G, et
al: Receptor activator of NF-κB (RANK) expression in primary tumors
associates with bone metastasis occurrence in breast cancer
patients. PLoS One. 6:e192342011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang Y: The effect of overexpressed
CIAPIN1on H9c2 myocytes H/R damageTianjin Medical University;
2016
|
|
29
|
Jin HR, Jin SZ, Cai XF, Li D, Wu X, Nan
JX, Lee JJ and Jin X: Cryptopleurine targets NF-κB pathway, leading
to inhibition of gene products associated with cell survival,
proliferation, invasion, and angiogenesis. PLoS One. 7:e403552012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li T, Yu J, Chen R, Wu J, Fei J, Bo Q, Xue
L and Li D: Mycophenolate mofetil attenuates myocardial
ischemia-reperfusion injury via regulation of the TLR4/NF-κB
signaling pathway. Pharmazie. 69:850–855. 2014.PubMed/NCBI
|
|
31
|
Liu JY, Shang J, Mu XD and Gao ZY:
Protective effect of down-regulated microRNA-27a mediating high
thoracic epidural block on myocardial ischemia-reperfusion injury
in mice through regulating ABCA1 and NF-κB signaling pathway.
Biomed Pharmacother. 112:1086062019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu Z, Du Q, Yan Y, Wang J, Dou S, Liu C
and Duan J: The protective effect of Luteolin on myocardial
ischemia/reperfusion (I/R) injury through TLR4/NF-κB/NLRP3
inflammasome pathway. Biomed Pharmacother. 91:1042–1052. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tian T, Zhou Y, Feng X, Ye S, Wang H, Wu
W, Tan W, Yu C, Hu J Zheng R, et al: MicroRNA-16 is putatively
involved in the NF-κB pathway regulation in ulcerative colitis
through adenosine A2a receptor (A2aAR) mRNA targeting. Sci Rep.
6:308242016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu X, Li S, Li Y, Cheng B, Tan B and Wang
G: Puerarin inhibits proliferation and induces apoptosis by
up-regulation of miR-16 in bladder cancer cell line T24. Oncol Res.
26:1227–1234. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang TQ, Luo XJ, Wu TF, Ding DD, Zhao ZH,
Chen GL, Xie XS, Li B, Wei YX, Guo LC, et al: miR-16 inhibits
glioma cell growth and invasion through the suppression of BCL2 and
NF-κB1/MMP-9 signaling pathway. Cancer Sci. 105:265–271. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Baudry A, Mouillet-Richard S, Schneider B,
Launay JM and Kellermann O: miR-16 targets the serotonin
transporter: A new Facet for adaptive responses to antidepressants.
Science. 329:1537–1541. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bhattacharya R, Nicoloso M, Arvizo R, Wang
E, Cortez A, Rossi S, Calin GA and Mukherjee P: miR-15a and miR-16
control Bmi-1 expression in ovarian cancer. Cancer Res.
69:9090–9095. 2009. View Article : Google Scholar : PubMed/NCBI
|