Introduction
The kidneys are the major osmoregulatory organs in
the body, which are not only involved in metabolic waste processing
and hormone secretion, but also maintain the water-electrolyte and
the acid-base balance. Research indicates that neighbor and distant
organs to the kidneys, including the heart, lung, liver, intestine
and brain, are severely damaged when renal function is impaired
(1).
Acute kidney injury (AKI) is a type of rapid renal
dysfunction caused by multiple etiological factors and pathological
syndromes, and is characterized by a high mortality (1). Kidneys, as hypertransfusion organs,
are extremely sensitive to ischemia, and ischemia-reperfusion (I/R)
injury (IRI) is common in kidneys (2). IRI is one of the major causes of AKI.
Renal IRI is a common pathological and physiological phenomenon.
Renal IRI frequently affects patients with hypertension or those
who have undergone renal surgery (3). Renal IRI is also a significant factor
that leads to delayed functional recovery after kidney
transplantation, poor outcomes and even acute renal failure
(4). Therefore, strategies to
effectively reduce renal IRI in clinical practice are urgently
required.
It is known that acute renal failure is
significantly associated with renal IRI, but the pathological
mechanisms of renal IRI are complex (5). Most of the recent studies on IRI
primarily focus on aspects including apoptosis and necrosis of
renal tubular epithelial cells, free radical-induced injury,
calcium overload, the obstacles of energy metabolism, adhesion
molecules and cell factors (6,7).
Once IRI has occurred, the prognosis of the patients is
significantly affected. However, there is currently a lack of
effective drugs. In clinical practice, affected patients are mainly
dependent on renal replacement therapies, including hemodialysis
and peritoneal dialysis, while passively waiting for the damaged
renal function to recover by itself (8). Therefore, the discovery of specific
therapeutic targets for treating renal IRI has great potential for
clinical application.
The mechanisms of renal IRI are complex, and mainly
involve the generation of numerous types of reactive oxygen
species, inflammatory damage and disturbance of the
microcirculation. The key link is disordered cell energy
metabolism. Mitochondria, which produce energy for the cell through
oxidative phosphorylation, are an important type of organelle. The
regulatory mechanisms of mitochondrial function are closely
associated with IRI (9).
Autophagy plays an important role in renal IRI, and
may even trigger apoptosis and necrosis. Autophagic cell death is a
type of non-apoptotic and programmed cell death induced by
oxidative stress and injury. Moderate autophagy is necessary for
cells to complete their own metabolism and organelle renewal. It is
important for maintaining the stability of the body's environment,
and for regulating cell damage and aging. Non-physiological and
excessive autophagy leads to disproportionate cell death, causing
pathological changes in tissues and organs. After reperfusion of
renal cells, autophagy is significantly increased. After drug
intervention or RNA interference is used to inhibit the expression
of autophagy-associated genes, serum creatinine (sCr) and urea
nitrogen levels are significantly decreased, and the pathological
changes in the kidneys are markedly reduced (10). Kimura et al (11) established a model of IRI in
autophagy related 5 (Atg5)-knockout mice and was the first to
demonstrate the protective effect of autophagy on renal IRI using
genetic evidence in vivo. Research has indicated that an
abnormal increase in autophagy in renal cells after reperfusion may
be one of the important triggering factors for structural and
functional damage of the kidney (12). Therefore, inhibition of autophagy
is expected to prevent renal IRI and even apoptosis.
Prostaglandin E2 (PGE2) is an important endogenous
regulatory factor in the body. When renal I/R occurs, PGE2 exerts
an important role in regulating the inflammatory reaction and
microcirculation. Cyclooxygenase-2 (COX-2) is an important
rate-limiting enzyme regulating the production of PGE2; it has been
proven that COX-2 has an important role in renal IRI (13).
PGE2 has four G protein-coupled receptors, namely
prostaglandin E2 receptor (EP)1-4. EP4 is one of the important G
protein-coupled receptors, and is a downstream molecule of COX-2.
After activation, EP4 may bind with intracellular G proteins and
upregulate the intracellular cyclic (c)AMP concentration to
influence the downstream signaling of extracellular
signal-regulated kinase 1/2. When IRI occurs, the gene expression
of EP4 increases, while that of EP3 decreases, while the expression
of EP1 and EP2 is not significantly affected, suggesting that EP4
and EP3 signals may be directly involved in the pathological
processes of IRI. Furthermore, EP4 agonists may significantly
reduce IRI. Since COX-2 is located upstream of EP4, COX-2
inhibitors suppress the opening of the mitochondrial permeability
transition pore during reperfusion by inhibiting the activity of
glycogen synthase kinase-3β, an important intracellular signaling
molecule. Studies on in vitro tumor cells have found that
EP4 protein can inhibit cell necrosis and apoptosis. Xiao et
al (14) suggested that EP4
signaling is involved in the process of myocardial I/R injury, and
the use of EP4 agonists can alleviate myocardial damage. Liang
et al (15) found that
activation of EP4 signaling can attenuate brain I/R injury.
CAY10598 is a commonly used EP4 agonist. In the present study, we
used it as a tool. The protective effect of EP4 signaling on IRI is
achieved by regulating the mitochondrial function of cells, but
this remains to be confirmed.
Although renal transplantation is a frontier
discipline, which has developed rapidly in recent years, IRI is an
inevitable adverse event associated with it. Current research
therefore has focused on the mechanisms of IRI with the aim to
identify approaches to significantly inhibit or prevent it. In this
light, the present study investigated the protective effect of EP4
signaling on renal mitochondria and IRI, and investigated the
underlying mechanisms in order to provide strategies to reduce IRI
in renal transplantation.
Materials and methods
Experimental grouping
A total of 18 male Sprague Dawley rats (weight,
250–300 g and age, 9 weeks) were used in the present study. The
rats were provided by the Animal Center of Xi'an Jiaotong
University. Rats were fed and housed in an animal room at
approximately 25°C, 3 per cage, with a natural circadian rhythm,
and a week of free eating. The rats were provided ad libitum
access to standard rat chow and water prior to and after the
experiment. The animals were randomly divided into three groups,
namely the sham group, the I/R group and the I/R+CAY10598 group,
with 6 rats in each group. In the Sham group, the abdominal cavity
was opened, but the pedicle of the blood vessels was not blocked.
In the I/R group, after 60 min of bilateral renal ischemia, the
vascular clamps were opened, resulting in reperfusion, which was
sustained for 24 h. In the I/R+CAY10598 group, the rats were
intraperitoneally injected with 1 mg/kg EP4 agonist CAYl0598 at 1.5
h prior to IRI, which was performed as in the I/R group. All
protocols were approved by the Institutional Animal Care and Use
Committee of the Xi'an Jiaotong University (Xi'an, China) (Permit
no. XJTULAC2016-558).
Rat model of renal IRI
The animals were fasted overnight prior to the
experiments with free access to water. They were anesthetized by an
intraperitoneal injection of 300 mg/kg 10% chloral hydrate. A
medial incision was performed in the back, and the renal as well as
the bilateral renal arteries were separated. Non-invasive
dissection of the artery clip was quickly performed for the
bilateral renal arteries at 60 min after opening the artery clip,
and blood flow perfusion was restored, followed by suturing of the
incision.
Primary culture and cell viability
determination of rat renal tubular epithelial cells
After establishing the rat model of renal IRI,
anaesthetic was intraperitoneally injected (10% chloral hydrate at
300 mg/kg). The rats were then sacrificed by cervical dislocation
and placed in 75% alcohol for 30 sec. After the rats were fixed on
the operating table, the abdominal cavity was opened with surgical
scissors and a surgical fistula, and the bilateral renal were taken
out and placed in pre-cooled phosphate-buffered saline (PBS). The
renal cortex was obtained and cut into small pieces, and the
resulting mixture was filtered through an 80-mesh filter. The
supernatant was filtered through a 100-mesh filter to leave the
tissue block. The cells were mixed with PBS, centrifuged for 5 min
at 1,200 rpm (300 × g), the supernatant was discarded, and
collagenase IV (C5138; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) (1 mg/ml) was added to the cells, followed by incubation
at 37°C for 15 min. Following centrifugation (with the
aforementioned conditions), the supernatant was discarded, and the
cells were suspended in serum-containing Dulbecco's modified
Eagle's F12 medium (12634-010; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The cells from each group were then inoculated
in a 96-well plate at 5.0×104 cells in 100 µl/well.
After 24 h of culture, the medium was discarded and 10 µl of Cell
Counting Kit-8 (CCK-8) (cat. no. 96992, USA, Sigma-Aldrich; Merck
KGaA) solution was added to each well. After further incubation at
37°C for 4 h, the light absorbance was measured at a wavelength of
450 nm using a microplate reader, and the relative viability of the
cells was calculated.
Determination of sCr, β2-microglobulin
(β2-MG) and neutrophil gelatinase-associated lipocalin (NGAL)
sCr (E-EL-S1181c), as well as serum concentrations
of β2-MG (E-EL-R1085c) and NGAL (E-EL-R0662c), were determined
using ELISA kits (Elabscience, Wuhan, China) according to the
manufacturer's instructions.
Hematoxylin and eosin (H&E)
staining
For histopathological analysis, the renal tissues
were paraffin-embedded and sectioned (5-mm in thickness), followed
by stepwise staining. In brief, the sections underwent dewaxing,
gradient alcohol hydration, washing, hematoxylin staining, washing,
eosin staining, gradient alcohol dehydration, transparency and
mounting/sealing with neutral resin. Finally, the pathological
changes in the kidneys were observed under an optical
microscope.
ATP determination
Renal tissue (100 mg) was accurately weighed and 1
ml pre-cooled ATP lysis solution was added, followed by
homogenization using an electric homogenizer. The tissue homogenate
was transferred to a 1.5-ml centrifuge tube, and centrifuged at 4°C
and 12,000 × g for 10 min. The supernatant remained. A total of 100
µl of the sample was added to each well of a microtiter plate, and
100 µl ATP assay solution was added. Following mixing, the BioTek
Synergy 4 Multiplate reader (BioTek Instruments, Inc., Winooski,
VT, USA) was immediately employed to determine the relative light
units. According to the standard curve, the ATP content of each
sample was interpolated.
JC-1 assay
The mitochondrial membrane potential of the renal
tubular epithelial cells was assessed using the Flow Cytometry
Mitochondrial Membrane Potential Detection kit (cat. no. 551302; BD
Biosciences, Franklin Lakes, NJ, USA). For each sample,
1×106 cells were transferred to a flow cytometry tube
and 400 µl binding buffer was added. The diluted conditioned
mitochondria were added to the tested mitochondria at a volume
ratio of 9:1. Following agitation, the cells were incubated at 37°C
for 15 min, re-suspended and analyzed using a flow cytometer (BD
Biosciences). The fluorescence intensity in A.U. was determined for
each sample.
Detection of mitochondrial (mt)DNA
fragments
Total DNA was isolated from the cytologic fractions
prepared from the renal tissues using a DNA extraction kit (Qiagen,
Hilden, Germany). The amount of mtDNA was determined by detecting
NADH dehydrogenase subunit 1 (NDl) with β-actin as the housekeeping
gene. The C1 mean difference between the reference gene and the
test sample was calculated. Finally, the ratio of the ND1 gene and
the nuclear β-actin gene copy was obtained.
Determination of glutathione (GSH),
malondialdehyde (MDA), superoxide dismutase (SOD) and COX levels by
ELISA
The content of GSH (cat. no. HS3275; Luzheng,
Shanghai, China), MDA (cat. no. HPBIO; Hepeng, Shanghai, China),
SOD (cat. no. BMS222; Biosource; Thermo Fisher Scientific, Inc.)
and COX (cat. no. HS2185; Luzheng) in renal tissues was determined
by double antibody sandwich ELISA. The procedures were performed in
strict accordance with the manufacturer's protocols. The
measurement of each sample and standard product was performed in 3
replicates. The absorbance was determined at a wavelength of 450
nm. The sample concentration was determined based on the standard
curve.
Microtubule-associated protein 1-light
chain 3B (LC3B) immunofluorescence analysis
The cells were washed with PBS, fixed with 4%
paraformaldehyde for 10 min at room temperature, washed and then
incubated with 0.1% Triton X-100 for 5 min. After blocking for 1 h
at room temperature, the cells were incubated with anti-LC3B
(dilution 1:100; cat. no. 700712; Invitrogen; Thermo Fisher
Scientific, Inc.) in the dark for 2 h at room temperature, followed
by incubation with the fluorescein isothiocyanate-conjugated
antibody (cat. no. 715-096-151, donkey anti-mouse; dilution 1:100;
Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) in
the dark for 30 min at room temperature. The cells were washed and
counterstained with DAPI for 1 min. Subsequently, the cells were
mounted with buffered glycerol and observed under a fluorescence
microscope (magnification ×200).
Western blot analysis
Lysates were prepared from renal tissues using
ice-cold lysis buffer containing a cocktail of protease inhibitors
(Sigma-Aldrich; Merck KGaA). Crude protein lysates (50 µg) were
separated by 12% SDS-PAGE, followed by transfer to a nitrocellulose
membrane (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and
blocking for 2 h with 5% non-fat powdered milk dissolved in
Tris-buffered saline with 0.05% Tween-20 at room temperature with
agitation. The membranes were then incubated overnight at 4°C with
antibodies to the following proteins: Cytochrome c (1:100,
cat. no. ab133504), caspase-3 (1:100, cat. no. ab197202), caspase-9
(1:100, cat. no. ab219590), LC3BII (1:200, cat. no. ab192890), Atg5
(1:100, cat. no. ab108327), lysosomal-associated membrane protein
(LAMP)1 (1:200, cat. no. ab108597), P62 (1:100, cat. no. ab109012),
protein kinase A (PKA) (1:100, cat. no. ab32390), phosphorylated
cAMP response element binding protein (p-CREB) (1:200, cat. no.
ab32096), aquaporin 5 (AQP5) (1:100, cat. no. 1b92320) (all from
Abcam, Cambridge, MA, USA) and anti-β-actin (dilution 1:5,000;
Sigma-Aldrich; Merck KGaA). Anti-mouse horseradish
peroxidase-conjugated secondary antibodies (cat. no. A8592,
Sigma-Aldrich; Merck KGaA) were used at 1:3,000 dilution.
Immunoreactive bands were visualized using a diaminobenzidine kit
(Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing,
China).
Statistical analysis
All experiments were repeated 3 times. Values are
expressed as the mean ± standard error of the mean. One-way
analysis of variance (ANOVA) was used for comparison between
groups, and least significant difference (LSD) or
Student-Newman-Keuls (SNK) tests were used for comparison between
groups. Analysis of variance was performed to assess the
differences among multiple groups using GraphPad Prism 6.0 software
(GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a significant difference between groups.
Results
Impaired renal function and altered
EP4 in renal IRI
Markers of renal function in the different groups
are provided in Fig. 1. The levels
of sCr, as well as serum β2-MG and NGAL, were significantly
increased in the IRI rats compared with those on day 0
(pre-operative; P<0.05). Within the same group, markers of renal
function gradually decreased with time (Fig. 1A-C). Histopathology confirmed that
in the I/R group, numerous renal tubular epithelial cells were
necrotic, and interstitial congestion and edema were observed
(Fig. 1D). The cell viability
assay confirmed that compared with the Sham group, the viability of
renal tubular epithelial cells was significantly decreased after
I/R (Fig. 1E). When the expression
of EP1-4 proteins was examined, it was identified that EP3 and EP4
exhibited minor changes after I/R. The expression of EP4 protein
was significantly increased on day 7 after surgery (Fig. 1F).
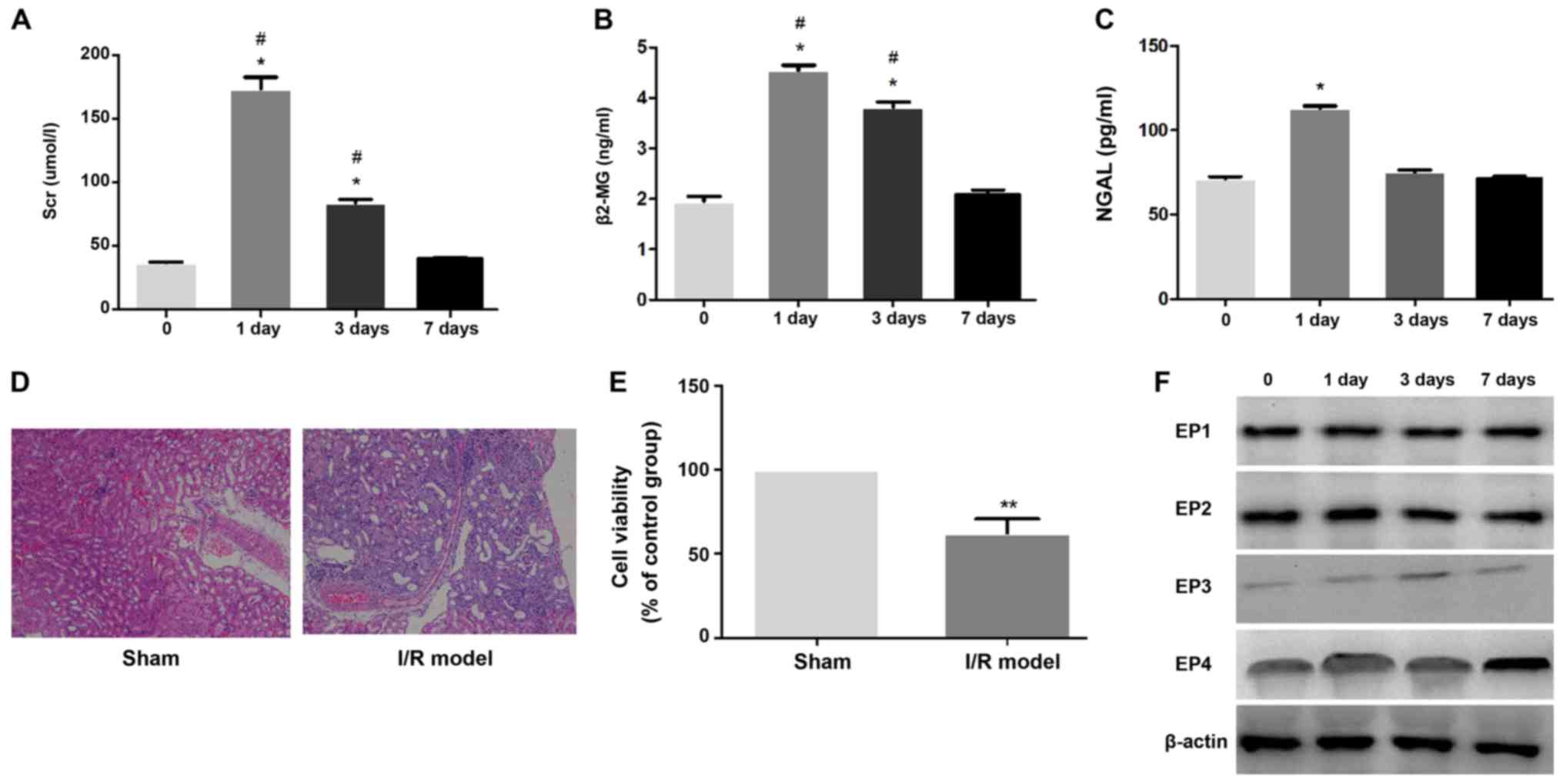 | Figure 1.(A-C) Kidney function parameters
(sCR, β2-MG and NGAL) in the serum of all groups in renal I/R
injury. Values are expressed as the means ± SD for group.
*P<0.05 indicates a significant difference when compared to the
0 day group; #P<0.05 indicates a significant
difference when compared to the day 7 group. (D) H&E staining
showed an increased tubular necrosis in renal I/R injury compared
with the Sham group. Original magnification, ×100. (E) Compared
with the Sham group, renal tubular epithelial cell viability was
significantly decreased after I/R; **P<0.01. (F) In the early
stage of renal I/R, the expression of EP4 exhibited only a minor
change, but with the prolongation of time, the expression of EP4
protein was significantly increased on day 7 after surgery. EP4,
prostaglandin E2 receptor 4; I/R, ischemia-reperfusion; sCr, serum
creatinine; NGAL, neutrophil gelatinase-associated lipocalin;
β2-MG, β2-microglobulin. |
EP4 agonist CAY10598 inhibits
mitochondrial membrane potential, cytochrome c release and
apoptosis associated with renal IRI
Ischemia-reperfusion injury can cause excessive
production of reactive oxygen species, calcium overload,
mitochondrial damage and cell death. Excessive reactive oxygen
species, as a stimulation of apoptosis, triggers the mitochondrial
apoptotic pathway, leading to the release of apoptotic factors such
as cytochrome c. Once released into the cytosol, cytochrome
c activates the downstream caspase family (16). Compared with that in the Sham
group, the mitochondrial membrane potential in the I/R group was
significantly decreased (P<0.001). However, administration of
CAY10598 prior to the induction of IRI inhibited this significant
decrease in the mitochondrial membrane potential (P<0.01;
Fig. 2A and B). The levels of
cytochrome c, caspase-3 and caspase-9 were significantly
increased in the I/R group compared with those in the Sham group
(P<0.01). The levels of cytochrome c, caspase-3 and
caspase-9 were significantly decreased in the I/R+CAY10598 group
compared with these levels in the I/R group (Fig. 2C-H). Thus, treatment with CAY10598
reversed the I/R-mediated increase in cytochrome c,
caspase-3 and caspase-9.
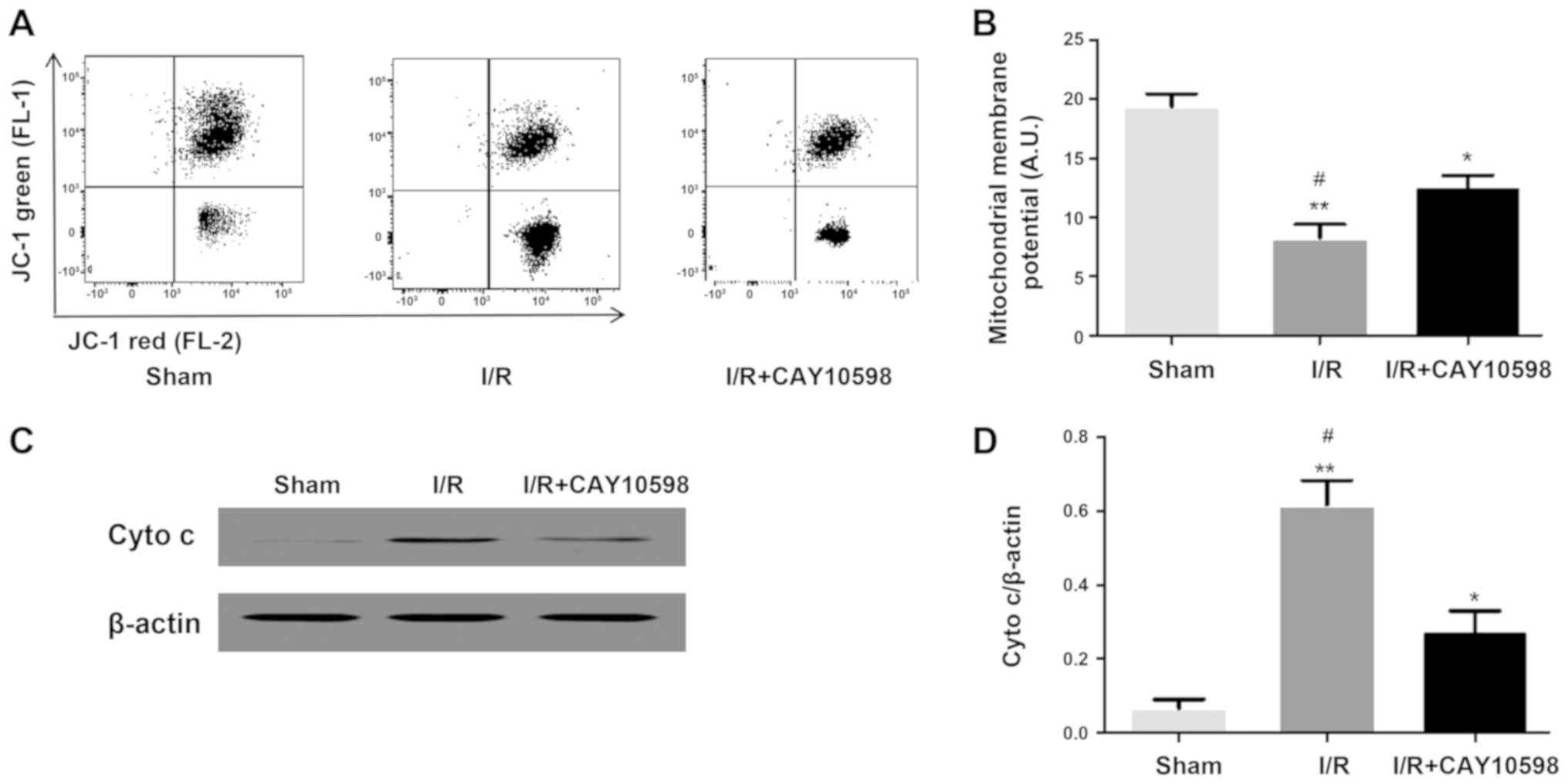 | Figure 2.EP4 agonist CAY10598 reverses
mitochondrial membrane potential downregulation, cyto c
release and increased apoptosis increase following renal I/R
injury. (A and B) FACS confirmed that compared with the Sham group,
mitochondrial membrane potential in the I/R injury group was
significantly decreased which was significantly rescued by
pre-treatment with CAY10598. *P<0.05, **P<0.01 vs. the Sham
group; #P<0.05 vs. the I/R+CAY10598 group. Western
blot analysis showed that I/R induced an increase in cyto c
(C and D); caspase-3 (E and F); caspase-9 (G and H) expression,
while CAY10598 is able to suppress the above changes. *P<0.05,
**P<0.01, ***P<0.001 vs. the Sham group;
#P<0.05, ##P<0.01 vs. the I/R+CAY10598
group. EP4, prostaglandin E2 receptor 4; cyto c, cytochrome
c; I/R, ischemia-reperfusion. |
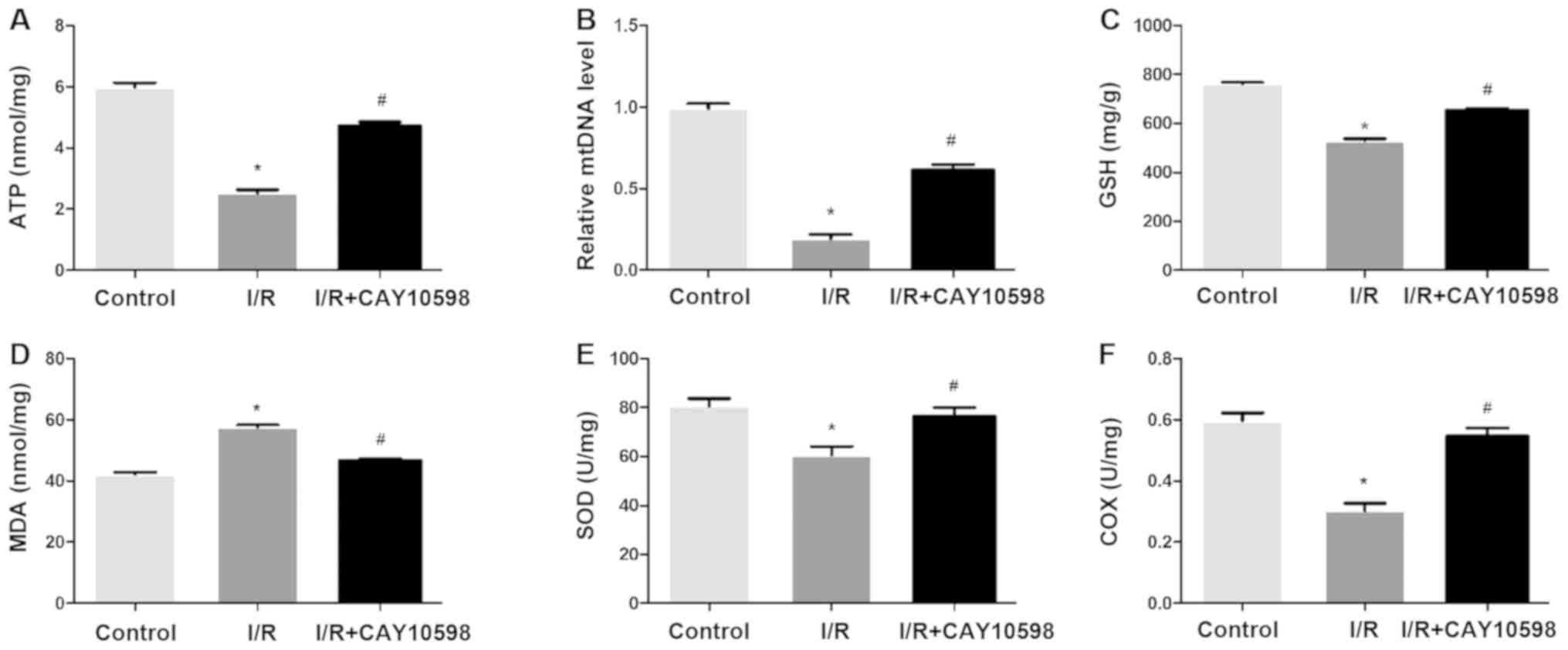
EP4 agonist CAY10598 has a protective
effect on mitochondrial dysfunction after renal IRI
As depicted in Fig.
3, I/R promoted a significant decrease in renal levels of ATP,
mtDNA, GSH, SOD and COX and a significant increase in MDA. A
significant increase was noted in the renal levels of ATP, mtDNA,
GSH, SOD and COX in the I/R+CAY10598 group compared with the levels
in the I/R group. The opposite trend was observed for the content
of MDA. Thus, CAY10598 rescued the I/R-promoted decrease in ATP,
mtDNA, GSH, SOD and COX and increase in MDA suggesting the
protective effect of the EP4 agonist on IRI.
EP4 agonist CAY10598 inhibits
mitochondrial autophagy after renal IRI
As depicted in Fig.
4A, after autophagy, microtubule-associated protein 1 light
chain 3B II (LC3BII) is transferred from the nucleus to the cell
membrane. At the protein level, the expression of LC3BII was
significantly increased in the I/R group. Pre-treatment with
CAY10598 reduced the I/R-mediated increase in LC3BII in renal
tissues of the I/R+CAY10598 group compared with the model I/R group
(Fig. 4B and C). The expression
levels of autophagy-associated factors, Atg5 and LAMP1 were
significantly increased following I/R but this I/R-promoted
increased was significantly abrogated in the IRI rats pre-treated
with CAY10598 compared with that in the model I/R group (Fig. 4D-G). The opposite trends were
observed for the levels of P62 (Fig.
4H and I).
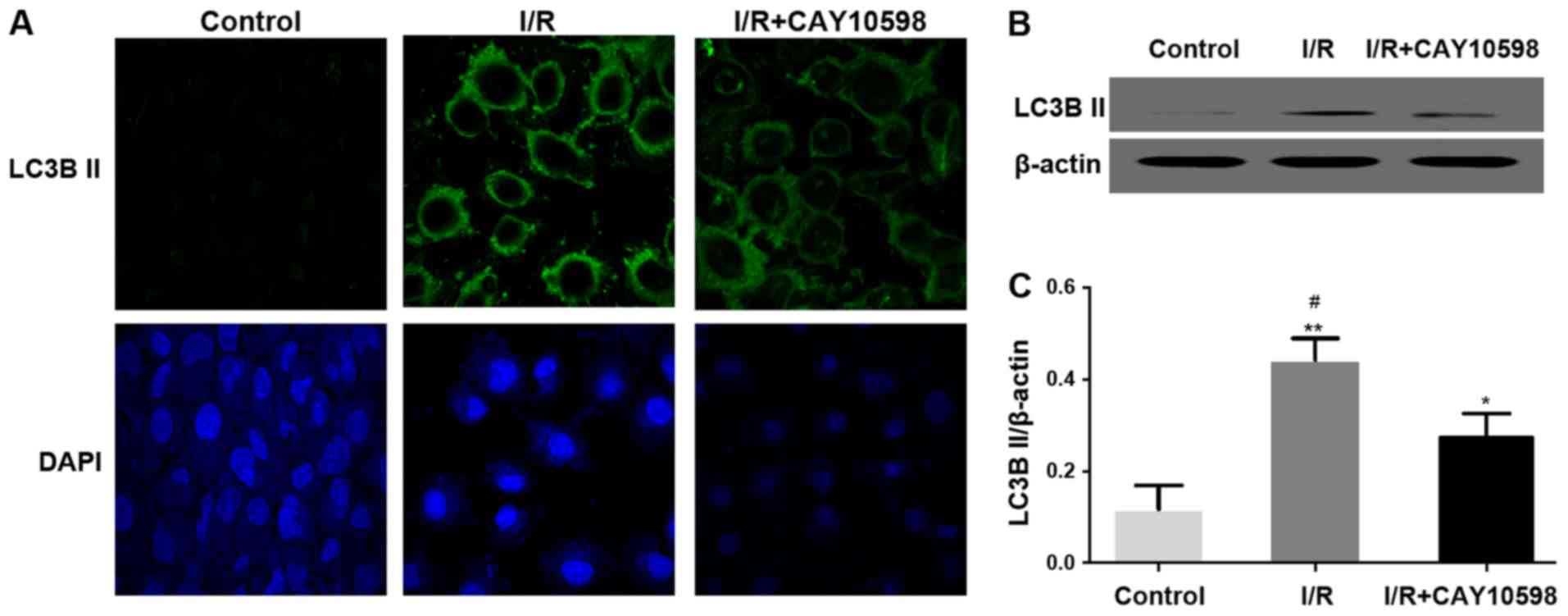 | Figure 4.EP4 agonist CAY10598 inhibits
mitochondrial autophagy after renal I/R injury. (A) Representative
image of immunofluorescence staining confirmed that LC3B II was
transferred from the nucleus to the cell membrane in the I/R injury
group. Magnification, ×400. Expression levels of LC3BII (B and C),
Atg5 (D and E), LAMP1 (F and G) and P62 (H and I) in the renal
tissue of all groups of rats. *P<0.05, **P<0.01,
***P<0.001 vs. the Sham group; #P<0.05,
##P<0.01 vs. the I/R+CAY10598 group. EP4,
prostaglandin E2 receptor 4; I/R, ischemia-reperfusion; LC3BII,
microtubule-associated proteins light chain 3B; Atg5, autophagy
related 5; LAMP1, lysosome associated membrane protein-1. |
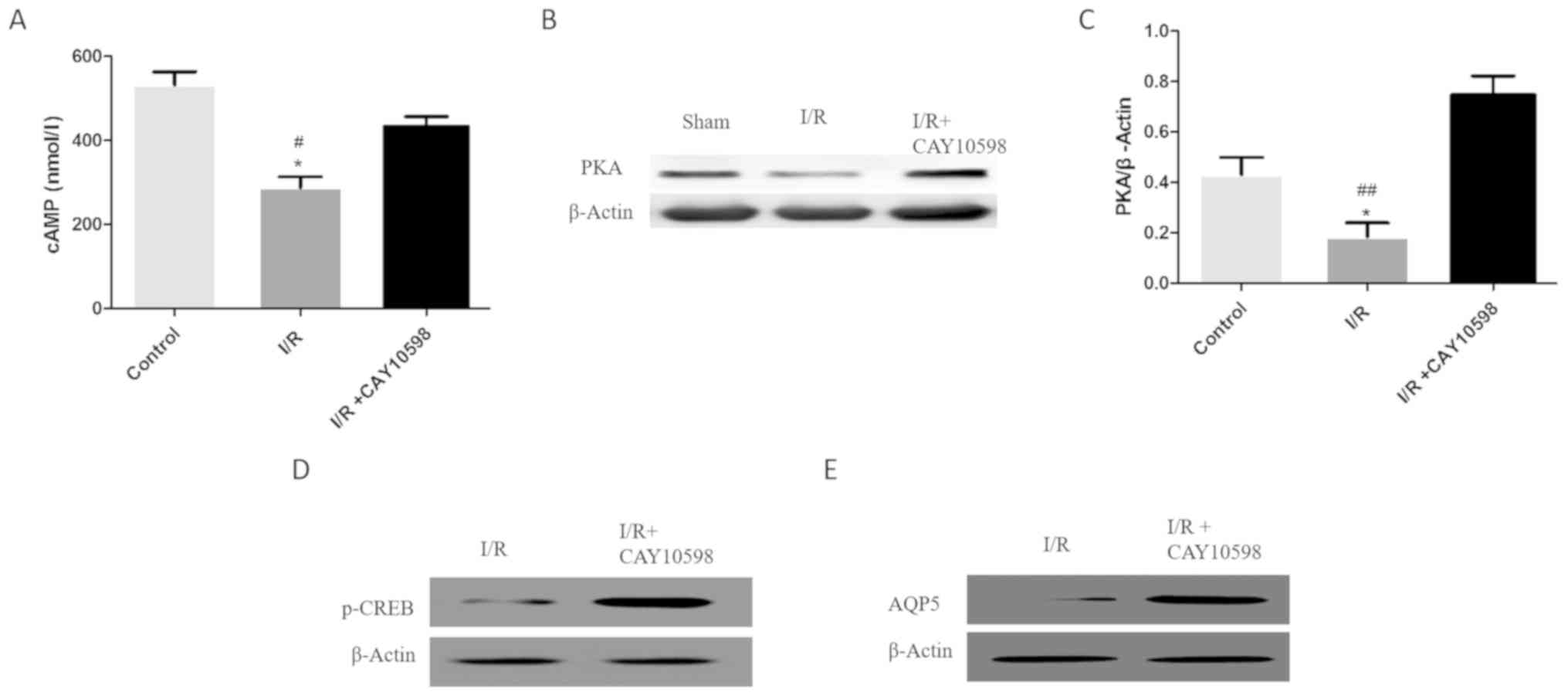
EP4 protects mitochondria from
autophagy by activating the cAMP/PKA signaling pathway
I/R promoted the decrease in the levels of cAMP,
PKA, p-CREB and AQP5. Administration of CAY10598 prior to I/R
resulted in a significant reversal of the I/R-mediated decrease in
these levels. Thus an increase in the levels of cyclic adenosine
monophosphate (cAMP), protein kinase A (PKA), phosphorylated cAMP
response element-binding protein (p-CREB) and aquaporin 5 (AQP5)
was observed in the I/R+CAY10598 group compared with the levels in
the I/R rats (P<0.01; Fig.
5).
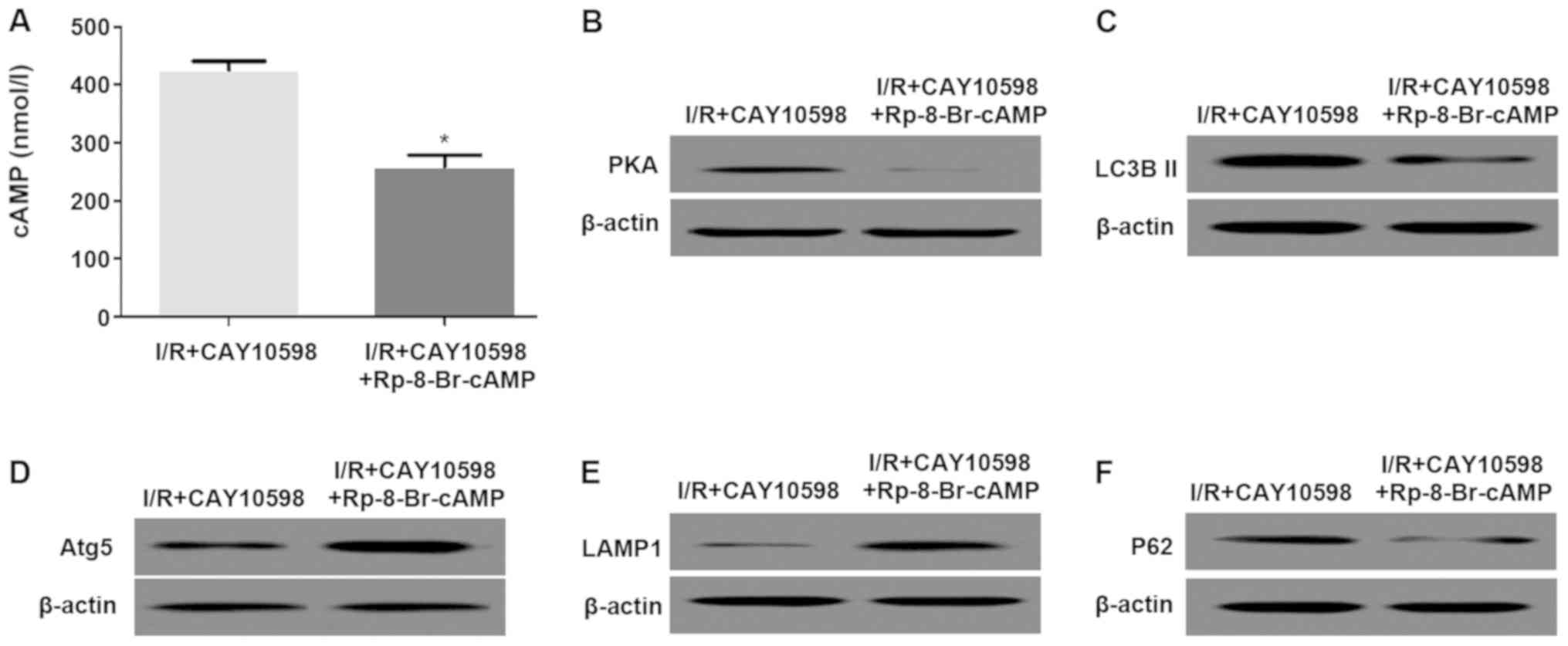
Blockage of the cAMP/PKA signaling
pathway reduces the inhibitory effect of CAY10598 on mitochondrial
autophagy after I/R
Pre-treatment with PAK inhibitor Rp-8-Br-cAMP and
CAY10598 significantly decreased cAMP and PKA levels compared with
those in the group treated with CAY10598 alone (Fig. 6A and B). Furthermore, the
expression levels of autophagy-associated factors LC3BII and P62
were decreased compared with those in the group treated with
CAY10598 alone. The expression of Atg5 and LAMP1 were opposite to
that of the above proteins (Fig.
6C-F).
Discussion
Ischemia-reperfusion (I/R) injury (IRI) is a common
clinicopathological phenomenon, and in severe cases, it may affect
the prognosis of patients. As a common reason for acute kidney
injury, renal IRI is also one of the major factors influencing a
kidney transplant patient's functional recovery in the early stage
and long-term survival (17).
Thus, the mechanism of renal IRI requires elucidation in order to
develop strategies to improve or reverse IRI. The mechanism of IRI
is complex and there are currently no effective measures for its
prevention. It has been suggested that IRI is closely linked to
cell necrosis, apoptosis and autophagy caused by energy metabolism
disorders (18).
Kidney damage mainly manifests as impaired renal
function and pathological changes in renal tissue morphology. Renal
function is mainly assessed by the sCr level, and dysfunction of
glomerular filtration may cause an elevated sCr. Levels of
β2-microglobulin (β2-MG) and neutrophil gelatinase-associated
lipocalin (NGAL) are also common parameters used in the evaluation
of renal function, with higher levels of β2-MG and NGAL indicating
more severe damage in regards to renal function (19). The present study indicated that
serum levels of Cr, β2-MG and NGAL in the I/R group were
significantly increased compared with the pre-operative levels,
indicating that renal dysfunction was present in this group and the
IRI model was successfully established. sCr, β2-MG and NGAL levels
gradually declined with time, indicating that renal function was
gradually restored. Histopathological changes may reflect the
severity of tissue damage (14),
and in the present study, morphological changes in the kidney
tissue were mainly assessed by histopathological H&E staining.
The results of the present study suggest that compared with those
at baseline, levels of sCr, β2-MG and NGAL were increased
significantly after renal IR, and the increase on day 1 was most
obvious. This result validated the successful establishment of the
model. On this basis, the morphological changes in renal tissues of
rats after renal I/R were observed. The results indicated that the
rats in the sham-operated group had a clear renal tissue structure,
and the renal tubular epithelial cells were intact and
well-arranged. In the model group, inflammatory cell infiltration
was observed and renal tubular epithelial cells were significantly
damaged, while the glomeruli were not obviously affected,
indicating renal tissue damage and inflammatory cell infiltration
after IRI.
The role of prostaglandin E2 receptor 4 (EP4), which
is an important downstream regulatory molecule of prostaglandin E2
signaling in vivo, in renal IRI remains to be fully
elucidated (20,21). It has been indicated that EP4
agonists are able to reduce myocardial IRI, but the exact mechanism
remains to be determined. It has also been suggested that EP4
agonists modulate the inflammatory response and reduce liver
damage. In the present study, it was revealed that the expression
of EP4 was significantly decreased after IRI. Furthermore, the
expression of EP4 protein was significantly increased on the day 7
after surgery.
Autophagy is a process in which cells degrade their
own damaged organelles by lysosomal enzymes under the action of
autophagy-associated genes, and it participates in a variety of
pathological and physiological activities within the cell (22,23).
In 2010, Jiang et al (24)
constructed a mouse model of renal IRI. It was confirmed that
autophagy has a protective role in renal IRI. In 2011, Kimura et
al (11) reported that after
IRI in mice lacking the autophagy-associated gene Atg5, the amount
of renal tubular epithelial cell apoptosis was significantly higher
than that in normal mice, indicating that autophagy maintains the
homeostasis of renal tubular epithelial cells and protect against
renal IRI. The results of the present study suggested that the EP4
agonist CAY10598 inhibited mitochondrial membrane potential
decreases, as well as cytochrome c release and apoptosis
following renal IRI.
ATP is the most direct source of energy in the body.
The mitochondrial ATP content is an important indicator of
mitochondrial function (25). The
present study indicated that the expression of EP4 was inhibited
during renal IRI, and furthermore, collapse of the mitochondrial
membrane potential and O2 utilization disorders may have
significantly reduced ATP synthesis. After pre-treatment with EP4
agonist, the mitochondrial ATP content was significantly increased,
suggesting that EP4 is beneficial for maintaining mitochondrial ATP
(26). A normal number, structure
and function of mitochondria are required for the maintenance of
the biological repair of the mitochondria. mtDNA regulates the
expression of mitochondrial structural and functional proteins, and
thus, it plays a crucial role in maintaining the integrity of
mitochondrial morphology and function (27–29).
The amount of mtDNA reflects the mitochondrial bioremediation
function. The results of the present study indicated that the renal
mitochondria were damaged by renal IRI and that the copy number of
mtDNA was decreased. This result may illustrate that IRI of the
kidney may lead to impairment of mitochondrial bio-regeneration. In
the group pre-treated with EP4 agonists, the mtDNA copy number
gradually recovered. This indicated that EP4 has a protective role
to sustain the mitochondrial bio-repair function in renal tubular
epithelial cells damaged by renal IRI.
Mitochondrial autophagy is a mechanism of
mitochondrial degradation under various stress conditions, which
has been demonstrated to occur in a variety of mitochondrial
dysfunction-associated diseases (30). Renal IRI leads to mitochondrial
damage in renal tubular epithelial cells (31). Activation of caspase-associated
signaling leading to cell death has been confirmed in the above
studies. In the present study, mitochondrial autophagy in renal IRI
was further explored. Detection of autophagy-associated factors
revealed excessive mitochondrial autophagy in renal IRI. The
results also indicated that mitochondrial autophagic death was
attributed to excessive mitochondrial damage. IRI may damage the
mitochondria directly or indirectly, thus inducing excessive
mitochondrial autophagic clearance. However, the activation of
mitochondrial autophagy as a secondary response to mitochondrial
damage induced by renal IRI cannot be excluded. In turn, the loss
of mitochondrial function may further exacerbate mitochondrial
autophagy, which may induce further mitochondrial loss. This
self-amplifying circle continues until the cells die. This is
probably why inhibition of mitochondrial autophagy is protective in
the initial stage, whereas accumulation of damaged mitochondria
later in life promotes cell death (32,33).
Finally, the present study further explored the
signaling mechanism by which EP4 regulates mitochondrial autophagy.
The detection of cAMP levels and PKA signaling downstream of EP4
revealed that IRI reduces the cAMP concentration and the expression
of proteins associated with the PKA signaling pathway, while EP4
agonist CAY10598 inhibited this phenomenon (34). The study found that the cAMP-PKA
signaling pathway did not only mediate the trafficking and gating
of AQPs, but also mediated its redistribution, and found that this
signaling pathway regulates the expression pathway of AQP5 which
may be related to the CREB transcription initiation site upstream
of the AQP5 open reading window; point-related, suggesting that the
cAMP/PKA-CREB transcriptional response may be involved in the
expression of AQP5 through this site. Stimulated by certain
factors, CREB is phosphorylated, cAMP is activated, and the amount
of cAMP in the cells is increased, and then PKA is activated. PKA
further catalyzes the phosphorylation of serine on CREB, which
makes AQP5 highly expressed, and finally increases the permeability
of the cell membrane to water. In the present study, an increase in
AQP5 expression was also detected after CAY10598 pre-treatment. In
addition, the results of the present study also indicated that EP4
protected the mitochondria from autophagy by activating the
cAMP/PKA signaling pathway, while blockage of the cAMP/PKA
signaling pathway reversed the inhibitory effect of CAY10598 on
mitochondrial autophagy after IRI. These results confirmed that EP4
protects the kidneys against IRI, and this effect may be achieved
by activating the cAMP/PKA signaling pathway and eventually
inhibiting excessive mitophagy.
In conclusion, the present study demonstrated the
protective effect of EP4 signaling on mitochondria and renal IRI,
as well as the underlying mechanisms. It was revealed that renal
IRI reduced the renal mitochondrial mass, decreased the copy number
of mtDNA and inhibited ATP production. The loss of mitochondria was
attributed to the excessive mitochondrial autophagy induced by
renal IRI. The EP4 agonist inhibited excessive mitochondrial
autophagy, the loss of mitochondria and energy imbalances within
the cell. This further confirms that excessive mitochondrial
autophagy is instigated by renal I/R, which is one of the important
causes of renal dysfunction.
Acknowledgements
Not applicable.
Funding
The present study was supported by The Scientific
and Technological Project of Shaanxi Province (grant no.
2016SF-246); and The National Nature Science Foundation of China
(grant nos. 81670681 and 81870514); and The Major Clinical Research
Projects of the First Affiliated Hospital of Xi'an Jiao Tong
University (XJTU1AF-CRF-2015-005).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
CD, WX and PT designed the experiments; CD, FH, HX,
YW, YL, XD and JZ performed the experiments. MD and XX were
responsible for data processing and statistical analyses. YL and JZ
were responsible for writing the manuscript, and XD was responsible
for modification of the manuscript. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All protocols were approved by the Institutional
Animal Care and Use Committee of the Xi'an Jiaotong University
(Xi'an, China; permit no. XJTULAC2016-558).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nezu M, Souma T, Yu L, Suzuki T, Saigusa
D, Ito S, Suzuki N and Yamamoto M: Transcription factor Nrf2
hyperactivation in early-phase renal ischemia-reperfusion injury
prevents tubular damage progression. Kidney Int. 91:387–401. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Malek M and Nematbakhsh M: Renal
ischemia/reperfusion injury; from pathophysiology to treatment. J
Renal Inj Prev. 4:20–27. 2015.PubMed/NCBI
|
|
3
|
Zhu J, Chen X, Wang H and Yan Q: Catalpol
protects mice against renal ischemia/reperfusion injury via
suppressing PI3K/Akt-eNOS signaling and inflammation. Int J Clin
Exp Med. 8:2038–2044. 2015.PubMed/NCBI
|
|
4
|
Kim N, Woo DC, Joo SJ, Song Y, Lee JJ, Woo
CW, Kim ST, Hong S, Cho YM and Han DJ: Reduction in renal
ischemia-reperfusion injury in mice by a phosphoinositide 3-kinase
p110gamma-specific inhibitor. Transplantation. 99:2070–2076. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meng QH, Liu HB and Wang JB: Polydatin
ameliorates renal ischemia/reperfusion injury by decreasing
apoptosis and oxidative stress through activating sonic hedgehog
signaling pathway. Food Chem Toxicol. 96:215–225. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ozbilgin S, Ozkardesler S, Akan M, Boztas
N, Ozbilgin M, Ergur BU, Derici S, Guneli ME and Meseri R: Renal
ischemia/reperfusion injury in diabetic rats: The role of local
ischemic preconditioning. Biomed Res Int. 2016:85804752016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Akan M, Ozbilgin S, Boztas N, Celik A,
Ozkardesler S, Ergur BU, Guneli E, Sisman AR, Akokay P and Meseri
R: Effect of magnesium sulfate on renal ischemia-reperfusion injury
in streptozotocin-induced diabetic rats. Eur Rev Med Pharmacol Sci.
20:1642–1655. 2016.PubMed/NCBI
|
|
8
|
Wu SH, Chen XQ, Lü J and Wang MJ: BML-111
Attenuates renal ischemia/reperfusion injury via peroxisome
proliferator-activated receptor-α-regulated heme oxygenase-1.
Inflammation. 39:611–624. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu D, Jin F, Shu G, Xu X, Qi J, Kang X,
Yu H, Lu K, Jiang S, Han F, et al: Enhanced efficiency of
mitochondria-targeted peptide SS-31 for acute kidney injury by
pH-responsive and AKI-kidney targeted nanopolyplexes. Biomaterials.
211:57–67. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu JB, Li SJ, Kang XQ, Qi J, Wu JH, Wang
XJ, Xu XL, Ying XY, Jiang SP, You J and Du YZ: CD44-targeted
hyaluronic acid-curcumin prodrug protects renal tubular epithelial
cell survival from oxidative stress damage. Carbohydr Polym.
193:268–280. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kimura T, Takabatake Y, Takahashi A,
Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T,
Soga T, et al: Autophagy protects the proximal tubule from
degeneration and acute ischemic injury. J Am Soc Nephrol.
22:902–913. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hamzawy M, Gouda SAA, Rashed L, Morcos MA,
Shoukry H and Sharawy N: 22-oxacalcitriol prevents acute kidney
injury via inhibition of apoptosis and enhancement of autophagy.
Clin Exp Nephrol. 23:43–55. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pang L: Inhibition of COX-2/PGE2/EP4
signaling protects against post-hypoxic apoptosis in H9C2
cardiomyocytes. Faseb J. 30:2016.
|
|
14
|
Xiao CY, Yuhki K, Hara A, Fujino T,
Kuriyama S, Yamada T, Takayama K, Takahata O, Karibe H, Taniguchi
T, et al: Prostaglandin E2 protects the heart from
ischemia-reperfusion injury via its receptor subtype EP4.
Circulation. 109:2462–2468. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liang X, Lin L, Woodling NS, Wang Q,
Anacker C, Pan T, Merchant M and Andreasson K: Signaling via the
prostaglandin E2 receptor EP4 exerts neuronal and
vascular protection in a mouse model of cerebral ischemia. J Clin
Invest. 121:4362–4371. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C,
Karasuyama H, Su MS, Rakic P and Flavell RA: Reduced apoptosis and
cytochrome c-mediated caspase activation in mice lacking caspase 9.
Cell. 94:325–337. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wiktorowska-Owczarek A and Owczarek J: The
effect of hypoxia on PGE2-stimulated cAMP generation in HMEC-1.
Cell Mol Biol Lett. 20:213–221. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Y, Zhong D, Lei L, Jia Y, Zhou H and
Yang B: Propofol prevents renal ischemia-reperfusion injury via
inhibiting the oxidative stress pathways. Cell Physiol Biochem.
37:14–26. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou S, Sun Y, Zhuang Y, Zhao W and Chen
Y, Jiang B, Guo C, Zhang Z, Peng H and Chen Y: Effects of
kallistatin on oxidative stress and inflammation on renal
ischemia-reperfusion injury in mice. Curr Vasc Pharmacol.
13:265–273. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cursio R, Colosetti P and Gugenheim J:
Autophagy and liver ischemia-reperfusion injury. Biomed Res Int.
2015:4175902015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Decuypere JP, Ceulemans LJ, Agostinis P,
Monbaliu D, Naesens M, Pirenne J and Jochmans I: Autophagy and the
kidney: Implications for ischemia-reperfusion injury and therapy.
Am J Kidney Dis. 66:699–709. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li H, Liu X, Zhu Y, Liu Y and Wang Y:
Magnolol derivative 002C-3 protects brain against
ischemia-reperfusion injury via inhibiting apoptosis and autophagy.
Neurosci Lett. 588:178–183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qiao PF, Yao L, Zhang XC, Li GD and Wu DQ:
Heat shock pretreatment improves stem cell repair following
ischemia- reperfusion injury via autophagy. World J Gastroenterol.
21:12822–12834. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang M, Liu K, Luo J and Dong Z:
Autophagy is a renoprotective mechanism during in vitro hypoxia and
in vivo ischemia-reperfusion injury. Am J Pathol. 176:1181–1192.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tan KY, Li CY, Li YF, Fei J, Yang B, Fu YJ
and Li F: Real-time monitoring ATP in mitochondrion of living
cells: A specific fluorescent probe for ATP by dual recognition
sites. Anal Chem. 89:1749–1756. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lewis SC, Uchiyama LF and Nunnari J:
ER-mitochondria contacts couple mtDNA synthesis with mitochondrial
division in human cells. Science. 353:aaf55492016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lauritzen KH, Kleppa L, Aronsen JM, Eide
L, Carlsen H, Haugen ØP, Sjaastad I, Klungland A, Rasmussen LJ,
Attramadal H, et al: Impaired dynamics and function of mitochondria
caused by mtDNA toxicity leads to heart failure. Am J Physiol Heart
Circ Physiol. 309:H434–H449. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cheng N, Lo YS, Ansari MI, Ho KC, Jeng ST,
Lin NS and Dai H: Correlation between mtDNA complexity and mtDNA
replication mode in developing cotyledon mitochondria during mung
bean seed germination. New Phytol. 213:751–763. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hämäläinen RH: Mitochondria and mtDNA
integrity in stem cell function and differentiation. Curr Opin
Genet Dev. 38:83–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Baruffini E, Ferrari J, Dallabona C,
Donnini C and Lodi T: Polymorphisms in DNA polymerase γ affect the
mtDNA stability and the NRTI-induced mitochondrial toxicity in
Saccharomyces cerevisiae. Mitochondrion. 20:52–63. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sokolowska M, Chen LY, Liu Y,
Martinez-Anton A, Qi HY, Logun C, Alsaaty S, Park YH, Kastner DL,
Chae JJ and Shelhamer JH: Prostaglandin E2 inhibits NLRP3
inflammasome activation through EP4 receptor and intracellular cAMP
in human macrophages. J Immunol. 194:5472–5487. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chang HH, Young SH, Sinnett-Smith J, Chou
CE, Moro A, Hertzer KM, Hines OJ, Rozengurt E and Eibl G:
Prostaglandin E2 activates the mTORC1 pathway through an
EP4/cAMP/PKA- and EP1/Ca2+-mediated mechanism in the human
pancreatic carcinoma cell line PANC-1. Am J Physiol Cell Physiol.
309:C639–C649. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chang HH, Young S, Sinnett-Smith J, Chou
CEN, Hines OJ, Rozengurt E and Eibl G: Abstract 1028: Prostaglandin
E2 activates the mTORC1 pathway through an EP4/cAMP/PKA and
EP1/calcium-mediated mechanisms in human pancreatic carcinoma
cells. Am J Physiol Cell Physiol. 309:2015.
|
|
34
|
Xu S, Ge JP, Zhang ZY and Zhou WQ: AB063.
A prostaglandin E (PGE) receptor EP4 is involved in the cell growth
and invasion of prostate cancer via the cAMP-PKA/PI3K-AKT signaling
pathway. Transl Androl Urol. 6 (Suppl 3):2017. View Article : Google Scholar : PubMed/NCBI
|