Introduction
Leukaemia, a clonal proliferative neoplasm derived
from haematopoietic stem cells, is commonly caused by various
genetic mutations (1). There are a
variety of clinical therapies involving chemotherapy used to treat
leukaemia (2). After prolonged
exposure to chemotherapeutic drugs, some patients may develop
multidrug resistance, which is defined by resistance to a wide
range of functionally unrelated chemotherapeutic agents. Drug
resistance hinders chemotherapeutic efficacy and leads to poor
prognosis in patients with leukaemia (3). Therefore, there is an urgent
requirement for more effective and less toxic drugs to manage
chemotherapy-induced multidrug resistance.
The exact mechanism of multidrug resistance is not
clear, and previous studies have suggested that it may be partially
due to the overexpression of ATP-binding cassette (ABC)
transporters, alteration of metabolised enzymes, impaired apoptosis
and autophagy, and the existence of leukaemia stem cells (4). P-glycoprotein (P-gp) is one of the
most studied ABC transporters (5),
and functions as a transporter to move substrates across cellular
membranes using the energy released from ATP hydrolysis (6). The mdr1 (multidrug-resistance)
gene is located on chromosomal region 7q21 and encodes an amino
acid sequence that forms P-gp (170 kDa) after glycosylation
(7). P-gp is constitutively
expressed in various adult tissues, such as those of the intestine,
liver, kidney and brain, where it serves an important role in drug
excretion and protects cells from external threats (8). Overexpression of P-gp in cancer cells
often leads to multidrug resistance by pumping agents out of cells
(9).
Apoptosis is a caspase-3-dependent programmed cell
death process (10) that is
regulated by a variety of elements such as the Bcl-2 protein family
(11). The Bcl-2 family includes
both pro-apoptotic proteins, such as Bax and Bcl-2 homologous
antagonist killer (Bak), and anti-apoptotic proteins, such as Bcl-2
and Bcl-xL (12). Bax/Bak promotes
apoptosis by forming homogenous or heterogeneous dimers, and
enhancing the permeabilization of the outer mitochondrial membrane.
Bcl-2/Bcl-xL can combine with Bax/Bak to suppress the formation of
dimers, resulting in apoptosis inhibition (13,14).
A number of studies have reported that the ratio of anti-apoptotic
to pro-apoptotic protein levels, rather than a single Bcl-2 family
protein, determines apoptosis susceptibility (15). Once cancer cells become
unsusceptible to chemotherapy-induced apoptosis, they may acquire
drug resistance (16).
Autophagy is an evolutionarily conserved process of
eukaryotic cells. It enables cells to sequester cellular components
by forming a double-membrane vacuole, called an autophagosome, and
subsequently to degrade those components after the fusion of
autophagosomes with lysosomes (17). The degradation products, including
amino acids, fatty acids, nucleotides and ATP, are reused by cells
to maintain the cell structure and metabolism (18). In addition to participating in
physiological processes, autophagy also serves a vital role in
various pathological conditions, such as neurodegenerative
disorders, autoimmunity disease, inflammation and cancer (19–22).
Evidence suggests that autophagy emerges in a context-dependent
role in cancer. On the one hand, autophagy inhibits tumour
initiation by cleaning up oncogenic protein substrates, toxic
misfolded proteins and dysfunctional organelles (23). On the other hand, once a tumour has
been established, autophagy can facilitate tumour survival in an
environment of nutrient depletion or hypoxia by catabolising
unnecessary proteins into amino acids and generating the energy
needed for tumour cell survival, which is associated with the drug
resistance of cancer cells (24).
However, it has been reported that hyperactive autophagy can induce
apoptosis or degrade cytoplasmic contents, thereby resulting in the
death of tumour cells (25,26).
In our previous study, it was determined that
multidrug-resistant leukaemia cells (K562/ADM cells) demonstrated a
higher level of autophagy than drug-sensitive leukaemia cells (K562
cells), both at the basic metabolic state and when under nutrient
deprivation stress, implying that autophagy is associated with the
drug resistance of leukaemia cells (27). To clarify the mechanism of drug
resistance in leukaemia cells, hydroxychloroquine (HCQ), a classic
autophagy inhibitor, was used to reverse the drug resistance of
K562/ADM cells, increase their apoptosis level and inhibit their
P-gp expression. This observation indicated the complicated
relationship between autophagy and multidrug resistance, and
suggested a novel target for leukaemia intervention.
Materials and methods
Chemicals and antibodies
HCQ was purchased from the Tokyo Chemical Industry
Co., Ltd. Adriamycin (ADM) was obtained from the Kangbaotai
Biochemical Industry Company. Newborn bovine serum was obtained
from the Rongye Biotech Company (http://royabio.800400.net). RPMI-1640 medium was
acquired from Gibco (Thermo Fisher Scientific, Inc.), MTT from
Sigma-Aldrich (Merck KGaA); and TRIzol® was from
Invitrogen (Thermo Fisher Scientific, Inc.). Specific primers for
mdr1 and β-actin were synthesised by the Takara Bio, Inc.
Antibodies against P62 (cat. no. 88588) and light chain (LC)3 (cat.
no. 4108) were obtained from Cell Signaling Technology, Inc.;
anti-β-actin antibody (cat. no. A1813) was from BioVision, Inc.;
antibodies against Bax (cat. no. TA346891), Bcl-2 (cat. no.
TA803003) and cleaved caspase-3 (cat. no. TA336455) were purchased
from Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.; anti-P-gp
antibody (cat. no. BM4508), horseradish peroxidase (HRP)-linked
anti-rabbit (cat. no. BA1054) and anti-mouse (cat. no. BA1050) IgG
antibodies were from the Wuhan Boster Biological Technology,
Ltd.
Cell lines and culture
The human ADM-resistant leukaemia cell line
(K562/ADM cells) and its parental subline (K562 cells) were both
provided by the Medical Experimental Center of Lanzhou University.
K562/ADM and K562 cells were maintained in RPMI-1640 medium,
supplemented with 10% inactivated newborn bovine serum and 2 mmol/l
L-glutamine, at 37°C in a cell incubator with 5% CO2. Experiments
were performed when the cells reached the mid-log phase.
Cell viability assay (MTT assay)
K562/ADM and K562 cells were seeded at a density of
1×105 cells/ml in 96-well plates. The cells were treated
with 0, 2, 4, 8, 16, 20 and 40 µmol/l of HCQ for 24 h at 37°C. MTT
solution (10 µl; 5 g/l) was added to each well before the cells
were incubated at 37°C for a further 4 h. Then, 100 µl of 10%
acidulated sodium dodecyl sulfate (SDS) were added to each well,
which was incubated for 12 h at 37°C to dissolve the formazan
crystals. Cell proliferation was then detected by MTT colorimetric
assay. The optical density (OD) was measured at 570 nm using a
Powerwave X plate reader (BioTek Instruments, Inc.). Cell
proliferation inhibition rates were calculated using the following
formula: Cell proliferation inhibition rate =
[(ODcontrol-ODexperiment)/ODcontrol] × 100%. 4 and 16 µmol/l of HCQ
were selected as the maximum non-toxic concentration to treat with
K562 and K562/ADM cells in the following experiments. Cells were
seeded in 96-well plates at a density of 1×105 cells/ml
in triplicate and were cultured with ADM or/and HCQ at the
indicated concentrations at 37°C for 12, 24 or 48 h. K562 cells
were treated with 0, 0.09, 0.18, 0.375, 0.75, 1.5 µmol/l of ADM or
pretreated with 4 µmol/l of HCQ for 3 h before exposure to ADM.
K562/ADM cells were treated with 0, 1.5, 3, 6, 12, 24 µmol/l of ADM
or pretreated with 16 µmol/l of HCQ for 3 h before exposure to ADM,
and then MTT assay was executed as above. The half-maximal
inhibitory concentration (IC50) was calculated using cell
proliferation inhibition rate by SPSS 17.0 software (SPSS, Inc.).
All samples were prepared in triplicate.
Acridine orange (AO) and ethidium
bromide (EB) staining
AO and EB staining have been used to discriminate
normal cells from those undergoing apoptosis and death (28). K562/ADM and K562 cells were treated
with 35 and 0.75 µmol/l of ADM, respectively, for 24 h or
pretreated with HCQ (16 and 4 µmol/l, respectively) for 3 h prior
to exposure to ADM. Cells were harvested and washed twice with PBS.
After being suspended in 100 µl of PBS, the cells were stained with
5 µl of EB (200 mg/l) and 5 µl of AO (200 mg/l) for 30 min at 37°C
in the dark. Subsequently, the cells were rinsed with PBS three
times, and a drop of suspension was placed on a glass slide. Cells
were observed under a fluorescence microscope (Olympus Corporation)
with a blue filter in fields with ×400 magnification.
Detection by transmission electron
microscopy
K562/ADM and K562 cells were treated with 35 and
0.75 µmol/l of ADM, respectively, for 24 h or pretreated with HCQ
(16 and 4 µmol/l, respectively) for 3 h prior to exposure to ADM.
Cells were fixed with glutaraldehyde at 4°C overnight. The next
day, after rinsing with PBS three times for 5 min, cells were fixed
with 1% OsO4 at room temperature for 1.5 h. After dehydration,
cells were embedded in epoxy resin and then solidified for 12 h at
45°C. After being sliced into 70 nm-thick sections, the cells were
stained with 2% uranyl acetate for 30 min and 2% lead citrate for
15 min at 37°C. Finally, the ultrastructure of the cells was
observed under a JEM1230 transmission electron microscope in fields
with ×8,000 magnification (JEOL, Ltd.).
Flow cytometry
K562/ADM and K562 cells were treated with 35 and
0.75 µmol/l of ADM, respectively, for 24 h or pretreated with HCQ
(16 and 4 µmol/l, respectively) for 3 h prior to exposure to ADM.
For the determination of Bax and Bcl-2 levels, 1×106
cells were fixed with 200 µl of an acetone and glutaraldehyde
mixture (1:1) for 20 min at 4°C. After rinsing with PBS, each
sample was incubated with anti-Bax FITC (1:20, cat. no. DB1010, DB
Biotech) or anti-Bcl-2 FITC (1:20, cat. no. DB1011) for 15 min in
100 µl of PBS in the dark at room temperature, and then suspended
in 500 µl of PBS. For detection of P-gp expression, cells were
collected and washed with PBS, and then incubated with antibodies
against P-gp (1:20, cat. no. ab93590, Abcam) for 15 min in the
dark. After rinsing with PBS, cells were suspended in 500 µl of
PBS. To analyse caspase-3 activity, 1 µl of FITC-DEVD-FMK (1:100,
cat. no. QIA911KIT, Sigma-Aldrich; Merck KGaA) was added to cell
suspensions in 100 µl of washing buffer. After incubation for 30
min at 37°C, the cells were washed and suspended in 500 µl of
washing buffer. After treatment, these samples were analysed using
a flow cytometer (Beckman Coulter, Inc.) and analysed using Windows
Multiple Document Interface for Flow Cytometry software (version
2.8; The Scripps Research Institute, La Jolla, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
K562/ADM and K562 cells were treated with 35 and
0.75 µmol/l of ADM, respectively, for 24 h or pretreated with HCQ
(16 and 4 µmol/l, respectively) for 3 h prior to exposure to ADM. A
total of 1×106 cells were collected, and then total RNA
was extracted from cells with a TRIzol kit according to the
manufacturer's protocols. Both the concentrations and purity of the
extracted RNA samples were determined by spectrophotometry.
Subsequently, 500 ng of each RNA sample was converted into cDNA
using a Prime Script RT Master Mix (Takara Bio, Inc.). Temperature
protocol: 70°C for 30 min, 37°C for 15 min, 95°C for 5 min. qPCR
assays were performed on a Rotor-Gene 3000 quantitative PCR
amplifier (Corbett, Australia) with a SYBR Premix Taq II kit
(Takara) and primers. The following primers were used: mdr1
forward, 5′-CTCATAATTCCATTAGGACG-3′; mdr1 reverse,
5′-GCTCACGCTACAGGTCCTGT-3′; β-actin forward,
5′-TGCTCCTCCTGAGCGCAAGTA-3′; and β-actin reverse,
5′-CCACATCTGCTGGAAGGTGGA-3′. The conditions included an initial
denaturing step at 95°C for 10 sec, then 40 cycles of denaturing at
95°C for 5 sec and annealing at 60°C for 30 sec. The relative
expression of each mRNA was calculated by comparison to β-actin
mRNA using the 2−ΔΔCq method (29). All samples were prepared in
triplicate.
Western blotting
K562/ADM and K562 cells were treated with 35 and
0.75 µmol/l of ADM, respectively, for 24 h or pretreated with HCQ
(16 and 4 µmol/l, respectively) for 3 h prior to exposure to ADM.
Cells were lysed in RIPA lysis buffer with phenylmethanesulfonyl
fluoride (PMSF; PMSF/RIPA=1:100) for 30 min on ice and then
centrifuged at 16,000 × g and 4°C for 15 min. The protein
concentration of the supernatant was measured using a bicinchoninic
acid protein assay kit. Equal amounts of protein (40 µg) from cell
extracts were separated via 10% SDS-PAGE and transferred onto PVDF
membranes. After blocking with 5% defatted milk in PBST at 24°C for
1 h, the membranes were incubated with primary antibodies including
anti-P62 (1:500), anti-LC3 (1:2,000), anti-Bax (1:1,000),
anti-Bcl-2 (1:1,000), anti-cleaved Caspase-3 (1:1,000), anti-P-gp
(1:500) and anti-β-actin (1:1,000) at 4°C overnight and then with
HRP-conjugated secondary antibodies (1:10,000). Finally, the
protein bands of the immunoblot were determined using a
chemiluminescent approach in a dark room. Protein bands were
visualized using enhanced chemiluminescence reagents (EMD
Millipore). Western blots were scanned using an Infrared Imaging
System (LI-COR Biosciences) and the bands were quantified using
ImageJ software (version 1.45S; National Institutes of Health).
Statistical analysis
Student's t-test or one-way ANOVA was conducted to
compare the differences in continuous data. Bonferroni correction
was employed to determine the significance of differences between
two groups. All statistical analyses were conducted by using SPSS
17.0 software (SPSS, Inc.). All statistical analyses were
two-sided, and the results are all presented as the mean ± the
standard deviation of at least three independent experiments.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Overexpression of P-gp and increased
autophagic activity co-occur in K562/ADM cells
To investigate the differences in P-gp levels
between K562/ADM and K562 cells, P-gp protein expression levels
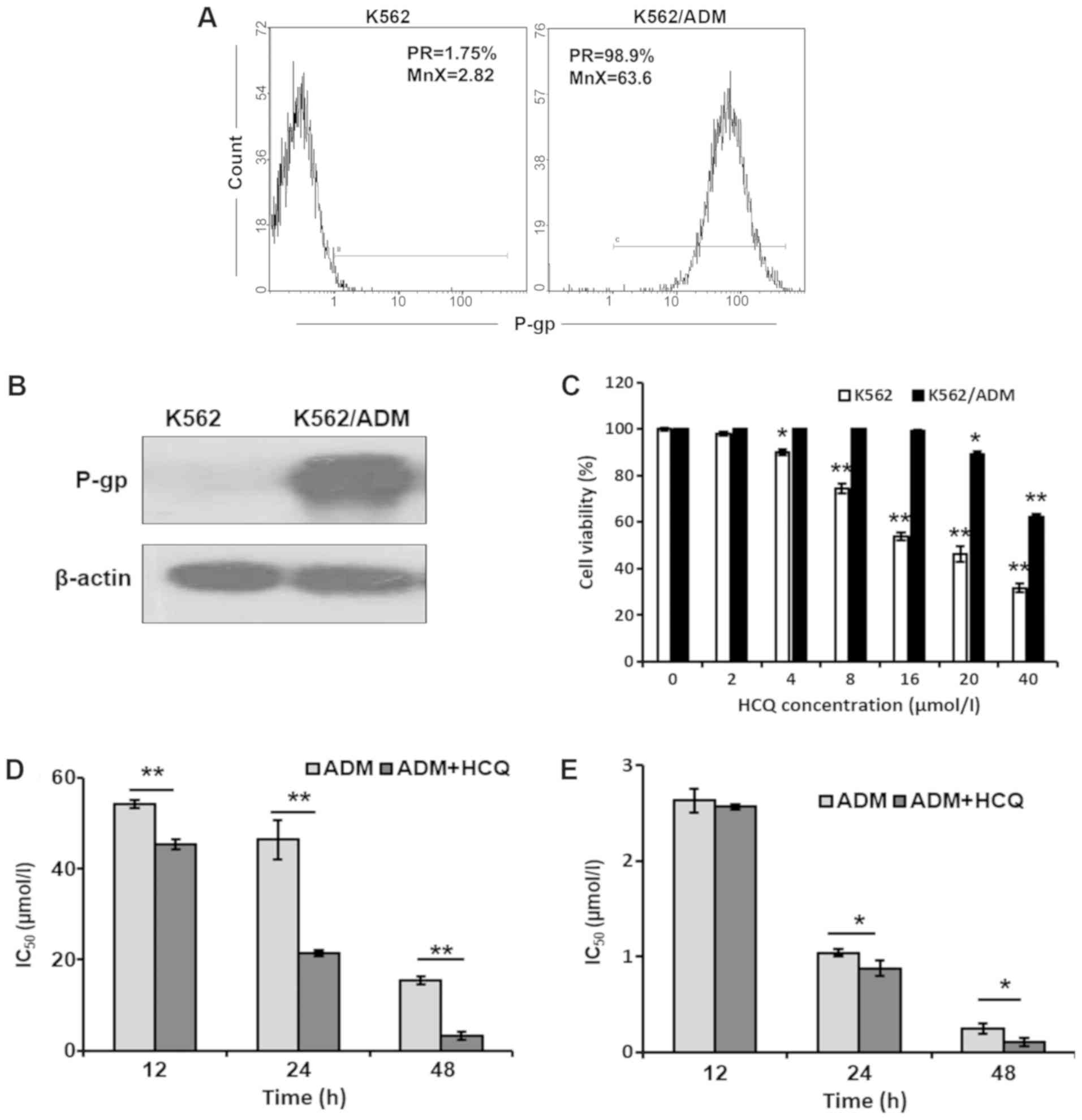
were detected via flow cytometric analysis. As shown in Fig. 1A, the positive rate and mean
fluorescence intensity of P-gp were both markedly elevated in
K562/ADM cells compared with K562 cells. Similar results were
observed with a western blotting assay (Fig. 1B), which indicated that K562/ADM
cells exhibited notably higher P-gp protein expression levels
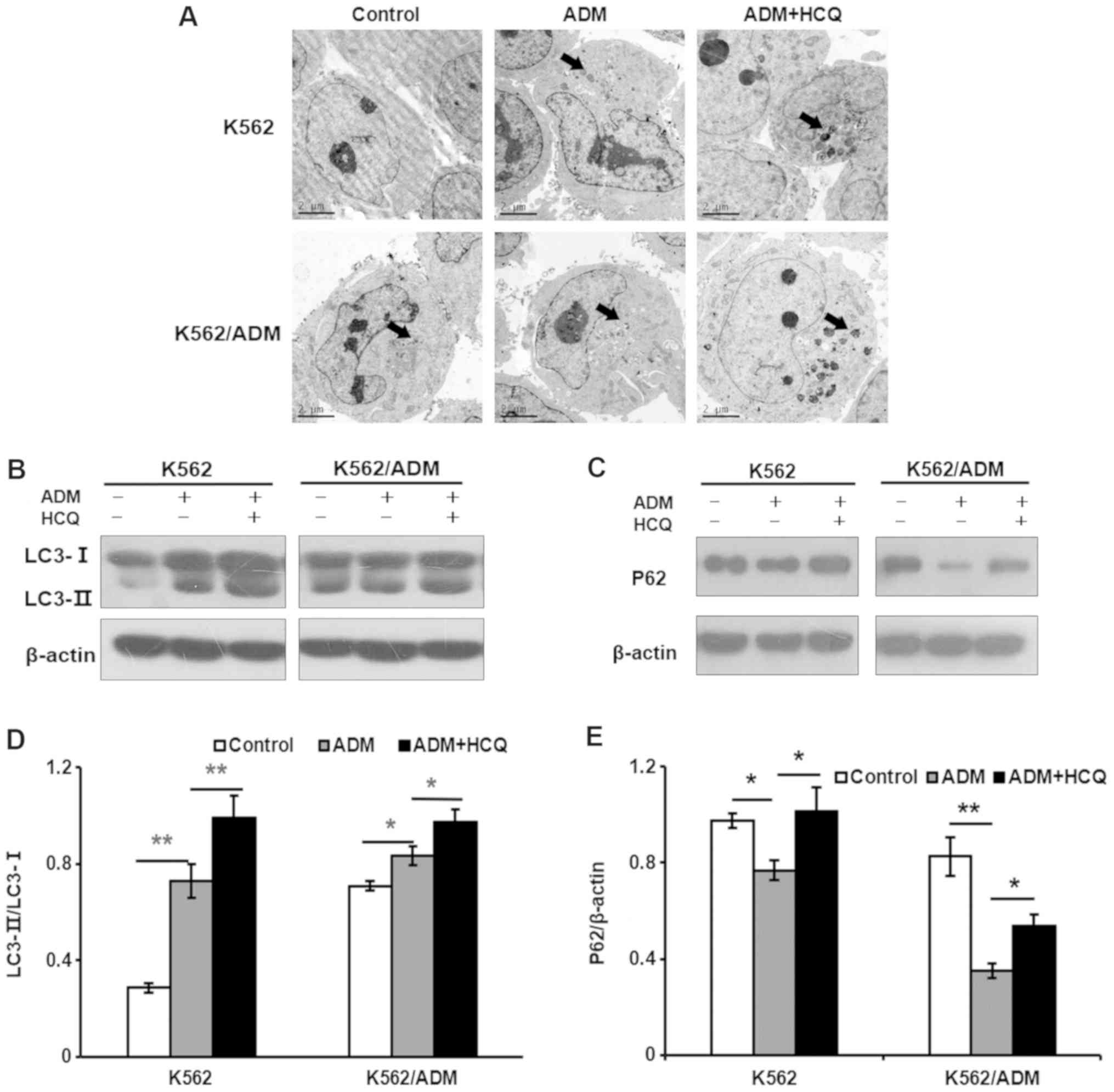
compared with K562 cells. Furthermore, the basic autophagic
activities of K562/ADM and K562 cells were measured. After
observation of autophagosomes in cells under a transmission
electron microscope, more cytosolic contents-packaged autophagic
vacuoles were identified in K562/ADM cells compared with K562 cells
(Fig. 2A). To further verify the
results described above, other autophagic indicators were examined,
such as LC3 and P62 (also known as sequestosome-1). As we know, the
ratio of LC3-II to LC3-I will become larger when the quantity of
autophagosomes increases, and P62, a ubiquitination substrate, has
a contrary relationship with autophagic activity (27). As presented in Fig. 2B and C, K562/ADM cells demonstrated
a notably increased LC3-II/LC3-I ratio and decreased P62 levels
compared with K562 cells, indicating that K562/ADM cells exhibited
a higher level of autophagic flux.
HCQ enhances the cytotoxic effects of
ADM in K562/ADM cells
Different concentrations of HCQ were used to
investigate its cytotoxic effects on K562/ADM and K562 cells using
an MTT assay. As presented in Fig.
1C, HCQ inhibited the viability of K562/ADM and K562 cells in a
dose-dependent manner. K562 and K562/ADM cells demonstrated almost
no cytotoxicity (cell viability >80%) following treatment with 4
and 16 µmol/l of HCQ, respectively, which were selected as the
maximum non-toxic concentrations in the following experiments.
Then, K562/ADM and K562 cells were treated with different
concentrations of ADM alone or following pre-treatment with HCQ (16
and 4 µmol/l, respectively) for 3 h prior to exposure to ADM for
12, 24 or 48 h. It was identified that the IC50 values for ADM in
K562/ADM cells following HCQ treatment for 12, 24 and 48 h were
45.66±5.08, 21.44±0.59 and 3.26±0.86 µmol/l, respectively, which
were 0.84-, 0.46- and 0.21-fold less than those values after only
ADM treatment (Fig. 1D). By
contrast, HCQ slightly decreased the IC50 values for ADM in K562
cells (Fig. 1E). These results
indicated that HCQ could effectively reverse the ADM resistance of
K562/ADM cells.
HCQ reduces ADM-induced autophagy in
K562 and K562/ADM cells
To investigate the effect of ADM on autophagy,
K562/ADM and K562 cells were treated with 35 and 0.75 µmol/l of
ADM, respectively, for 24 h or pretreated with HCQ (16 and 4
µmol/l, respectively) for 3 h prior to exposure to ADM. Then, they
were observed under a transmission electron microscope for
autophagic vacuoles. As presented in Fig. 2A, more autophagic vacuoles
(indicated by black arrows) were found in ADM-treated cells than in
the controls, and HCQ increased the number of autophagic vacuoles
in K562 and K562/ADM cells treated with ADM. To further validate
these results, the levels of the autophagy markers LC3 and P62 were
analysed using a western blotting assay. It was found that ADM
induced autophagy in K562 and K562/ADM cells, as indicated by the
increased LC3-II/LC3-I ratios (P=0.001 and 0.023, respectively;
Fig. 2B and D) and a decrease in
the P62 level (P=0.023 and 0.001, respectively; Fig. 2C and E). Furthermore, HCQ increased
the accumulation of LC3-II (P=0.009 and 0.012, respectively) and
P62 (P=0.012 and 0.022, respectively) in both K562 and K562/ADM
cells exposed to ADM, indicating that HCQ reduced autophagy
activity induced by ADM. Collectively, these findings indicated
that HCQ can inhibit the ADM-induced autophagy response in K562 and
K562/ADM cells by preventing the degradation of autophagic
vacuoles.
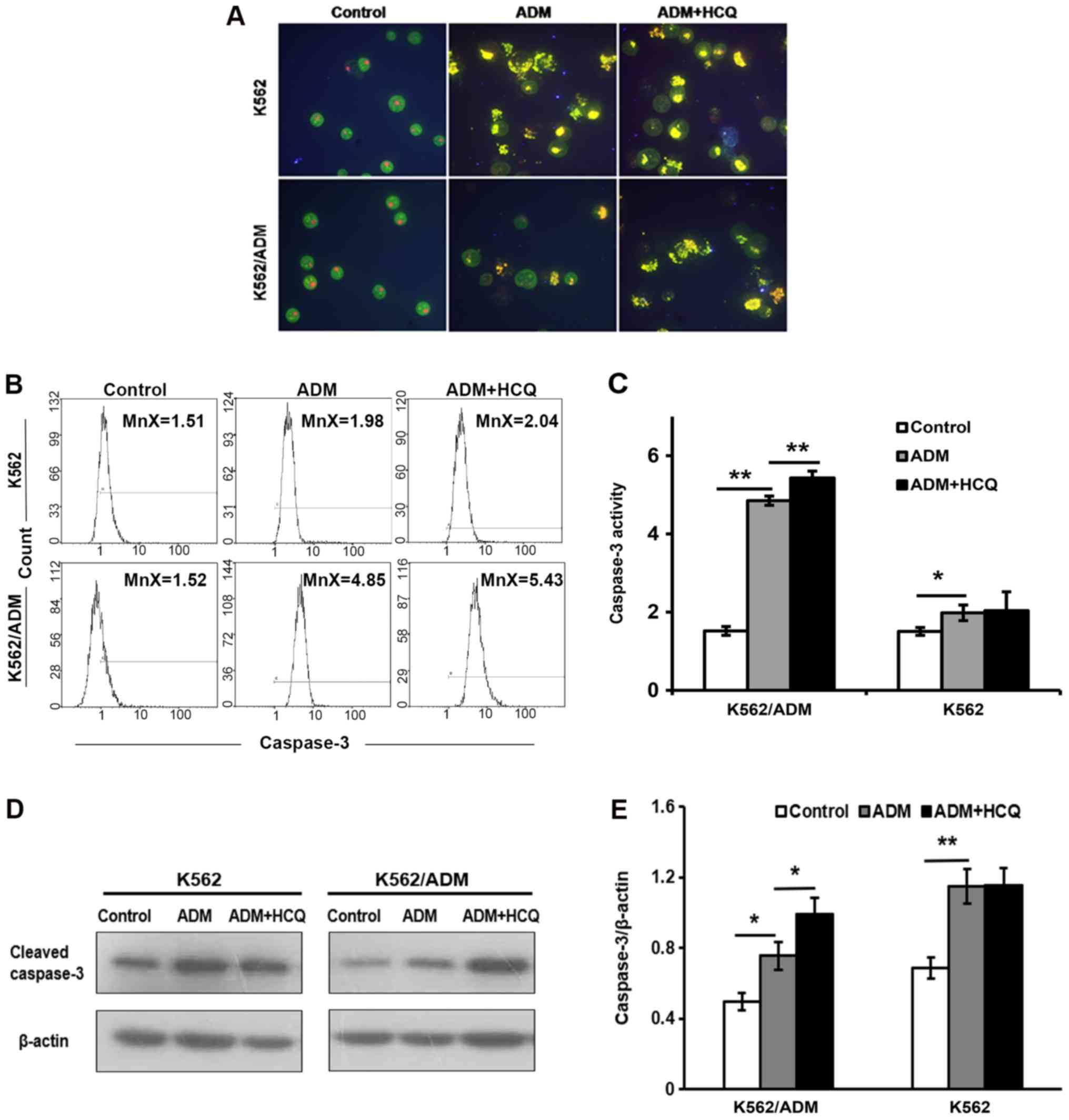
HCQ potentiates ADM-induced apoptosis
in K562/ADM cells
To explore the apoptosis response in two cell lines
following ADM exposure, K562/ADM and K562 cells were treated with
35 and 0.75 µmol/l of ADM separately for 24 h and stained with
AO/EB. Observed under a fluorescence microscope, the controls
exhibited green-stained integral nuclei, whereas yellow or orange
nuclei with condensed and fragmentary chromatin were found in cells
treated with ADM, suggesting that ADM induced apoptosis in the two
cell lines. Furthermore, when cells were pretreated with HCQ for 3
h prior to exposure to ADM, the proportion of apoptotic K562/ADM
cells was notably increased compared with those treated only with
ADM; however, the effects of HCQ pre-treatment on the apoptosis of
K562 cells were not pronounced (Fig.
3A). Morphological changes indicated that HCQ upregulated
ADM-induced apoptosis in K562/ADM.
To confirm these apparent changes in apoptosis,
caspase-3 activity was evaluated using flow cytometry, and the
expression of the cleaved forms of caspase-3 was detected via
western blotting. As presented in Fig.
3B and C, ADM led to an increased mean fluorescence intensity
for activated caspase-3 in K562 (P=0.032) and K562/ADM (P=0.001)
cells. K562/ADM cells pre-treated with HCQ prior to exposure to ADM
exhibited a significantly greater mean fluorescence intensity for
activated caspase-3 compared with cells exposed to only ADM
(P=0.007), whereas K562 cells were not significantly affected by
HCQ pre-treatment. Similar results were also obtained from the
western blot assay (Fig. 3D and
E).
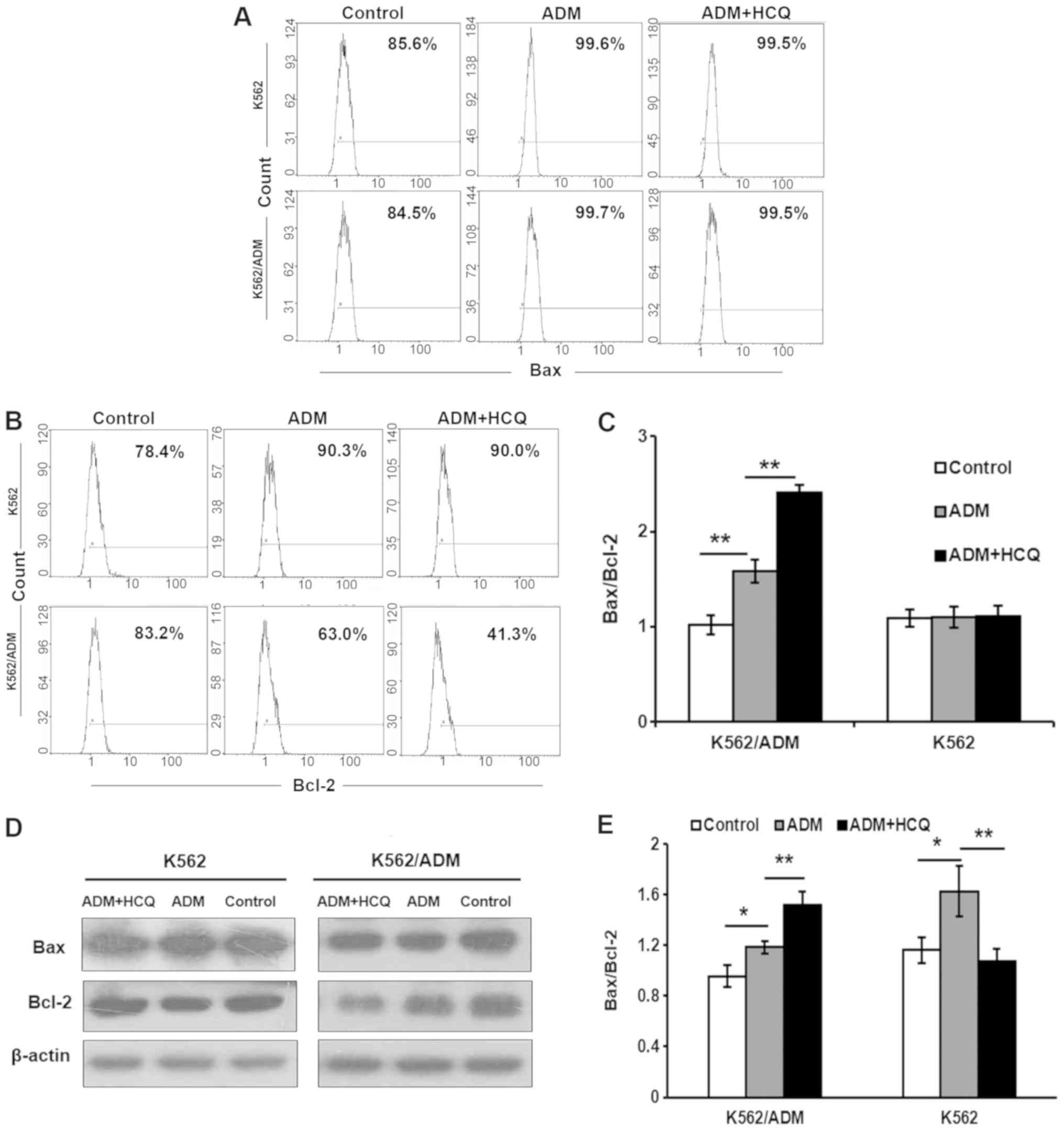
These results were verified by the determination of
the protein expression levels of Bax/Bcl-2. Flow cytometry
demonstrated that the ratio of Bax/Bcl-2 was upregulated in
K562/ADM cells by ADM (P=0.001), which could be further enhanced
following 3 h of HCQ pre-treatment (P=0.001; Fig. 4A-C). However, there were no notable
effects on K562 cells. The results of the western blot assay of
K562/ADM cells were also consistent with these observations, but
the situations of K562 cells were incompatible between the flow
cytometry and western blotting results (Fig. 4D and E). The change of K562 cells
may be induced by other causes in addition to apoptosis.
Collectively, these findings indicate that HCQ could sensitise
K562/ADM cells to caspase-dependent apoptosis induced by ADM,
thereby reversing multidrug resistance.
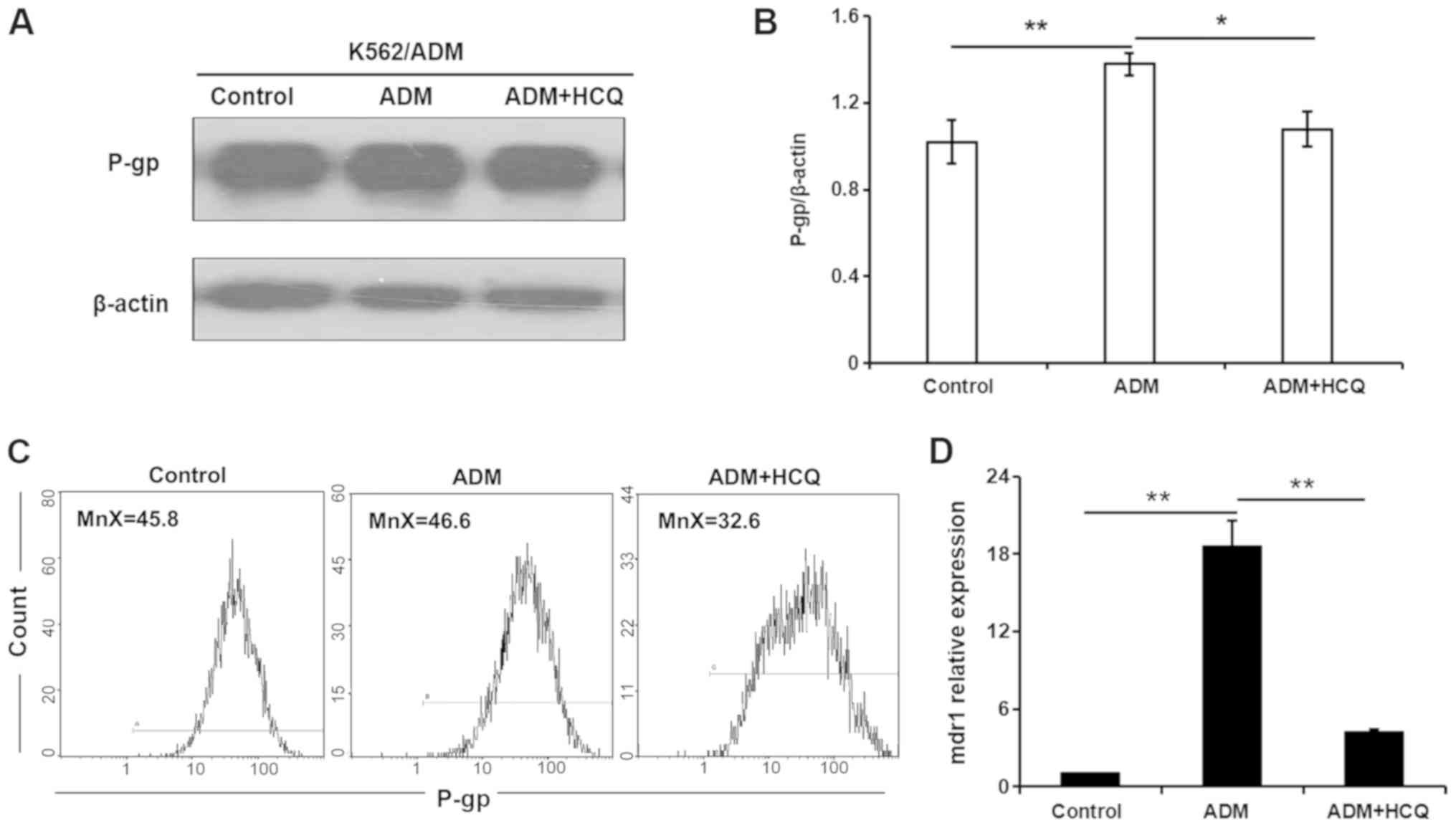
HCQ diminishes the expression of P-gp
in K562/ADM cells exposed to ADM
To further clarify the role of autophagic activity
in multidrug resistance, the present study modulated autophagy and
observed alterations in the expression of the drug
resistance-associated protein P-gp during the autophagy process.
ADM (35 µmol/l) was used to induce the autophagic response, and 16
µmol/l HCQ was used to block autophagy in K562/ADM cells. As
demonstrated by a western blot assay, P-gp expression in K562/ADM
cells was increased in the presence of ADM (P=0.004), and 3 h HCQ
treatment decreased ADM-induced P-gp expression to a level similar
to that of control treatment (P=0.011; Fig. 5A and B). A flow cytometric assay
validated this observation, as a slight increase in the mean
fluorescence intensity for P-gp was found in K562/ADM cells
following ADM incubation, which fell with HCQ treatment (Fig. 5C). In addition, the mRNA expression
profile of the mdr1 gene in K562/ADM cells following the
aforementioned treatments, as determined via RT-qPCR analysis, was
consistent with P-gp expression (Fig.
5D). Collectively, these findings indicated that the inhibition
of autophagy diminished the expression of P-gp, which may be
involved in autophagic regulation in K562/ADM cells.
Discussion
Leukaemia is a highly malignant cancer of the blood
system characterised by rapid onset, poor prognosis and high cost
of treatment (30). It is known
that leukaemia is a malignancy with one of the highest mortality
rates (31). Although there are a
variety of chemotherapy drugs for use against leukaemia,
chemotherapy-induced multidrug resistance is still a challenging
issue during drug treatment (31).
Therefore, more effective clinical therapies are needed to conquer
the drug resistance induced by chemotherapy.
K562/ADM cells are a multidrug-resistant leukaemia
cell line with high expression of P-gp, and are acquired by
exposing K562 cells to step-wise increasing concentrations of ADM
(32). They are characterised by
their resistance not only to ADM but also to other anticancer drugs
with varying structures and functions (32). The results of the present study
demonstrated that K562/ADM cells exhibited a high level of P-gp
expression, while K562 cells expressed minimal levels of P-gp,
indicating that K562/ADM cells are a multidrug-resistant cell line
and that K562 is a sensitive cell strain. In addition, it was
identified that basic autophagy activity in K562/ADM cells was
distinctly higher than it was in K562 cells, which suggests that
autophagy may be involved in multidrug resistance. This result is
consistent with our previous study (27).
It is well known that autophagy serves a dual role
in tumorigenesis. It has been suggested that, in cancer cells,
certain chemotherapeutics are able to induce autophagic cell death
(ACD), a cell death pathway distinct from apoptosis. Puissant et
al (26) reported that
resveratrol treatment could lead to ACD in chronic myelogenous
leukaemia (CML) cells, and that autophagy inhibition decreased the
sensitivity of CML cells to resveratrol. Li et al (33) suggested that 3-methyladenine, an
autophagy inhibitor, could promote Raji cell proliferation by
restraining the ACD induced by arsenic trioxide. However, in the
majority of established tumours, autophagy protects against the
survival of cancer cells and confers resistance to chemotherapy.
Han et al (34) reported
that daunorubicin initiated protective autophagy in acute
myelocytic leukaemia (AML) via the mitogen-activated protein kinase
(MAPK) kinase/ERK pathway, which resulted in the daunorubicin
resistance of AML cells. This pro-survival role of autophagy has
also been supported by other studies showing that autophagy
inhibition elevated sensitivity to chemotherapy in multiple
myeloma, breast cancer, colorectal cancer and prostate cancer cells
(35–38).
Chemotherapy drugs can induce autophagy in leukaemia
cells. Li et al (39)
indicated that macroautophagy activity in Raji cells was
significantly enhanced by arsenic trioxide. Consistent with this
result, the present study demonstrated that autophagy in K562/ADM
and K562 cells was increased with ADM treatment. It was also found
that HCQ could inhibit the ADM-induced autophagy response in K562
and K562/ADM cells by restraining the degradation of autophagic
vacuoles. HCQ belongs to the 4-amino quinoline family, which is
used for antimalarial medications (40). HCQ was employed to inhibit
autophagy due to its well-known safety profile. Moreover, when
cells were treated with HCQ at nontoxic concentrations, the IC50
values of ADM in K562/ADM cells at different time points were
significantly reduced, indicating that HCQ could effectively
reverse the ADM resistance of K562/ADM cells, and that increased
autophagy is one of the mechanisms of multidrug resistance in
K562/ADM cells.
In addition to autophagy, it has been suggested that
apoptosis is involved in drug resistance (41). Over the years, evidence has
accumulated demonstrating that apoptosis and autophagy may share
the same signalling pathways, such as p38-MAPK and JNK (42), and there is an interconnected
relationship between apoptosis and autophagy (43). For example, a previous study
described that autophagy inhibited apoptosis by decreasing
Bcl-2-associated death promoter and Bcl-2-like protein 11
expression in hepatocellular carcinoma cells (44). Another study suggested that
autophagy was involved in the early stage of apoptosis, leading to
the death of acute lymphocytic leukaemia cells (45). The data from the present study
demonstrated that the proportion of apoptotic cells induced by ADM
was prominently upregulated following pre-treatment with HCQ in
K562/ADM cells, which was also confirmed by an increase in
caspase-3 activity and the enhanced ratio of Bax/Bcl-2; two classic
hallmark proteins for apoptosis. Therefore, autophagy inhibition by
HCQ may potentiate ADM-induced apoptosis and contribute to
overcoming ADM resistance in K562/ADM cells.
P-gp functions as an ATP-dependent drug ejector pump
and decreases the intracellular concentrations of its substrates
with different chemical structures (46). Consequently, the enhanced
expression of P-gp may lead to resistance to a wide range of drugs
(47). It has been suggested that
P-gp may be involved in regulating molecular metabolism,
proliferation and differentiation in cells (48), indicating that P-gp might
potentially possess multiple physiological functions. A previous
study demonstrated the synergistic roles of P-gp, autophagy and
NF-κB pathways in the epirubicin resistance of triple-negative
breast cancer (49). However,
another study suggested that the role of P-gp in the development of
multidrug resistance in breast cancer may be independent of
autophagy (50). The present study
preliminarily demonstrated an association between autophagy and
P-gp expression. The results demonstrated that ADM treatment
resulted in a significant augmentation of autophagy accompanied by
increased mdr1/P-gp expression in K562/ADM cells. In addition, the
autophagy inhibitor HCQ markedly inhibited the ADM-induced increase
in autophagy, as well as enhancing mdr1/P-gp expression in the
cells, and sensitised the drug-resistant cells to ADM. This may
mean that inhibition of autophagy attenuates the expression of
P-gp, and that P-gp may be involved in autophagic regulation in
drug-resistant cells. The true co-regulation and interaction
between autophagy and P-gp in modifying multidrug resistance
requires confirmation in future investigations. In addition, Liu
et al (51) suggested that
nelfinavir could reverse ADM resistance in K562/ADR cells by
inhibiting the P-gp efflux function without affecting P-gp protein
and mRNA expression levels. However, the results of the present
study indicated that the autophagy inhibitor HCQ might sensitise
the drug-resistant cells to ADM by reducing mdr1/P-gp expression at
the transcriptional level in cells.
Collectively, the present findings demonstrated that
HCQ can reverse ADM resistance in K562/ADM cells by inhibiting
autophagy, increasing K562/ADM cell apoptosis and decreasing P-gp
expression. It is hypothesised that P-gp may potentially sustain
drug resistance in leukaemia cells by participating in the
regulation of autophagic activity. The present study revealed a
potential novel target for leukaemia intervention and provides
further insight into understanding the role of autophagy in the
drug resistance of leukaemia cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China grant no. 81541025 (to HLW),
the Fundamental Research Funds for the Central Universities grant
no. lzujbky-2016-174 (to HLW) and the Natural Science Fund of Gansu
grant no. 1208RJZA183 (to HLW). This work was also supported by the
National Natural Science Foundation of China grant no. 31571196 (to
LW).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FFW performed the examinations, and was a major
contributor in writing the manuscript. LW and HLW made substantial
contributions to conception and design. WTL interpreted data,
translated and revised the manuscript critically for important
intellectual content. ZWZ, JC, JY, CMY, MMY, MYW, NZ and XMQ helped
to analyze and interpret data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wouters BJ and Delwel R: Epigenetics and
approaches to targeted epigenetic therapy in acute myeloid
leukemia. Blood. 127:42–52. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Burnett A, Wetzler M and Lowenberg B:
Therapeutic advances in acute myeloid leukemia. J Clin Oncol.
29:487–494. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gao W and Estey E: Moving toward targeted
therapies in acute myeloid leukemia. Clin Adv Hematol Oncol.
13:748–754. 2015.PubMed/NCBI
|
|
4
|
Ramos P and Bentires-Alj M:
Mechanism-based cancer therapy: Resistance to therapy, therapy for
resistance. Oncogene. 34:3617–3626. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jones PM and George AM: The ABC
transporter structure and mechanism: Perspectives on recent
research. Cell Mol Life Sci. 61:682–699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Borst P, Evers R, Kool M and Wijnholds J:
A family of drug transporters: The multidrug resistance-associated
proteins. J Natl Cancer Inst. 92:1295–1302. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bodor M, Kelly EJ and Ho RJ:
Characterization of the human MDR1 gene. AAPS J. 7:E1–E5. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ambudkar SV, Dey S, Hrycyna CA,
Ramachandra M, Pastan I and Gottesman MM: Biochemical, cellular,
and pharmacological aspects of the multidrug transporter. Annu Rev
Pharmacol Toxicol. 39:361–398. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abraham J, Salama NN and Azab AK: The role
of P-glycoprotein in drug resistance in multiple myeloma. Leuk
Lymphoma. 56:26–33. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Danial NN and Korsmeyer SJ: Cell death:
Critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Delbridge AR and Strasser A: The BCL-2
protein family, BH3-mimetics and cancer therapy. Cell Death Differ.
22:1071–1080. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng JH, Viacava Follis A, Kriwacki RW
and Moldoveanu T: Discoveries and controversies in BCL-2
protein-mediated apoptosis. FEBS J. 283:2690–2700. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ghibelli L and Diederich M: Multistep and
multitask bax activation. Mitochondrion. 10:604–613. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kvansakul M and Hinds MG: The Bcl-2
family: Structures, interactions and targets for drug discovery.
Apoptosis. 20:136–150. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Indran IR, Tufo G, Pervaiz S and Brenner
C: Recent advances in apoptosis, mitochondria and drug resistance
in cancer cells. Biochim Biophys Acta. 1807:735–745. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhi X, Feng W, Rong Y and Liu R: Anatomy
of autophagy: From the beginning to the end. Cell Mol Life Sci.
75:815–831. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Levy JMM, Towers CG and Thorburn A:
Targeting autophagy in cancer. Nat Rev Cancer. 17:528–542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Budini M, Buratti E, Morselli E and
Criollo A: Autophagy and its impact on neurodegenerative diseases:
New roles for TDP-43 and C9orf72. Front Mol Neurosci. 10:1702017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Morandi E, Jagessar SA, t Hart BA and Gran
B: EBV infection empowers human b cells for autoimmunity: Role of
autophagy and relevance to multiple sclerosis. J Immunol.
199:435–448. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cosin-Roger J, Simmen S, Melhem H, Atrott
K, Frey-Wagner I, Hausmann M, de Vallière C, Spalinger MR,
Spielmann P, Wenger RH, et al: Hypoxia ameliorates intestinal
inflammation through NLRP3/mTOR downregulation and autophagy
activation. Nat Commun. 8:982017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fulda S: Autophagy in cancer therapy.
Front Oncol. 7:1282017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gewirtz DA: The four faces of autophagy:
Implications for cancer therapy. Cancer Res. 74:647–651. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Puissant A, Robert G, Fenouille N, Luciano
F, Cassuto JP, Raynaud S and Auberger P: Resveratrol promotes
autophagic cell death in chronic myelogenous leukemia cells via
JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res.
70:1042–1052. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang F, Chen J, Zhang Z, Yi J, Yuan M,
Wang M, Zhang N, Qiu X, Wei H and Wang L: Differences of basic and
induced autophagic activity between K562 and K562/ADM cells.
Intractable Rare Dis Res. 6:281–290. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu K, Liu PC, Liu R and Wu X: Dual AO/EB
staining to detect apoptosis in osteosarcoma cells compared with
flow cytometry. Med Sci Monit Basic Res. 21:15–20. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Daver N, Cortes J, Kantarjian H and
Ravandi F: Acute myeloid leukemia: Advancing clinical trials and
promising therapeutics. Expert Rev Hematol. 9:433–445. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Plasschaert SL, de Bont ES, Boezen M,
vander Kolk DM, Daenen SM, Faber KN, Kamps WA, de Vries EG and
Vellenga E: Expression of multidrug resistance-associated proteins
predicts prognosis in childhood and adult acute lymphoblastic
leukemia. Clin Cancer Res. 11:8661–8668. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen J, Wei H, Xie B, Wang B and Cheng J
and Cheng J: Endoplasmic reticulum stress contributes to arsenic
trioxide-induced apoptosis in drug-sensitive and -resistant
leukemia cells. Leuk Res. 36:1526–1535. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li CL, Wei HL, Chen J, Wang B, Xie B, Fan
LL and Li LJ: Arsenic trioxide induces autophagy and antitumor
effects in burkitt's lymphoma raji cells. Oncol Rep. 32:1557–1563.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Han W, Sun J, Feng L, Wang K, Li D, Pan Q,
Chen Y, Jin W, Wang X, Pan H and Jin H: Autophagy inhibition
enhances daunorubicin-induced apoptosis in K562 cells. PLoS One.
6:e284912011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Riz I, Hawley TS and Hawley RG:
KLF4-SQSTM1/p62-associated prosurvival autophagy contributes to
carfilzomib resistance in multiple myeloma models. Oncotarget.
6:14814–14831. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun Q, Liu T, Yuan Y, Guo Z, Xie G, Du S,
Lin X, Xu Z, Liu M, Wang W, et al: MiR-200c inhibits autophagy and
enhances radiosensitivity in breast cancer cells by targeting
UBQLN1. Int J Cancer. 136:1003–1012. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang H, Tang J, Li C, Kong J, Wang J, Wu
Y, Xu E and Lai M: MiR-22 regulates 5-FU sensitivity by inhibiting
autophagy and promoting apoptosis in colorectal cancer cells.
Cancer Lett. 356:781–790. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim SH, Kim KY, Yu SN, Park SK, Choi HD,
Ji JH and Ahn SC: Autophagy inhibition enhances silibinin-induced
apoptosis by regulating reactive oxygen species production in human
prostate cancer PC-3 cells. Biochem Biophys Res Commun.
468:151–156. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li CL, Wei HL, Chen J, Wang B, Xie B, Fan
LL and Li LJ: Ebb-and-flow of macroautophagy and chaperone-mediated
autophagy in raji cells induced by starvation and arsenic trioxide.
Asian Pac J Cancer Prev. 15:5715–5719. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Caminal-Montero L and Suárez-Díaz S:
Hydroxychloroquine and antimalarials. J Rheumatol.
15:jrheum.190559. 2019.
|
|
41
|
Zhang H, Han D, Lv T, Liu K, Yang Y, Xu X
and Chen Y: Novel peptide myristoly-CM4 induces selective
cytotoxicity in leukemia K562/MDR and Jurkat cells by necrosis
and/or apoptosis pathway. Drug Des Devel Ther. 13:2153–2167. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: P38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Oral O, Akkoc Y, Bayraktar O and Gozuacik
D: Physiological and pathological significance of the molecular
cross-talk between autophagy and apoptosis. Histol Histopathol.
31:479–498. 2016.PubMed/NCBI
|
|
44
|
Zhou Y, Sun K, Ma Y, Yang H, Zhang Y, Kong
X and Wei L: Autophagy inhibits chemotherapy-induced apoptosis
through downregulating bad and bim in hepatocellular carcinoma
cells. Sci Rep. 4:53822014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yuan N, Song L, Zhang S, Lin W, Cao Y, Xu
F, Fang Y, Wang Z, Zhang H, Li X, et al: Bafilomycin A1 targets
both autophagy and apoptosis pathways in pediatric B-cell acute
lymphoblastic leukemia. Haematologica. 100:345–356. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rees DC, Johnson E and Lewinson O: ABC
transporters: The power to change. Nat Rev Mol Cell Biol.
10:218–227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Breier A, Gibalova L, Seres M, Barancik M
and Sulova Z: New insight into p-glycoprotein as a drug target.
Anticancer Agents Med Chem. 13:159–170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Silva R, Vilas-Boas V, Carmo H,
Dinis-Oliveira RJ, Carvalho F, de Lourdes Bastos M and Remiao F:
Modulation of P-glycoprotein efflux pump: Induction and activation
as a therapeutic strategy. Pharmacol Ther. 149:1–123. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang LH, Yang AJ, Wang M, Liu W, Wang CY,
Xie XF, Chen X, Dong JF and Li M: Enhanced autophagy reveals
vulnerability of P-gp mediated epirubicin resistance in triple
negative breast cancer cells. Apoptosis. 21:473–488. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sun WL, Lan D, Gan TQ and Cai ZW:
Autophagy facilitates multidrug resistance development through
inhibition of apoptosis in breast cancer cells. Neoplasma.
62:199–208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu W, Meng Q, Sun Y, Wang C, Huo X, Liu
Z, Sun P, Sun H, Ma X and Liu K: Targeting P-Glycoprotein:
Nelfinavir reverses adriamycin resistance in K562/ADR cells. Cell
Physiol Biochem. 51:1616–1631. 2018. View Article : Google Scholar : PubMed/NCBI
|