Introduction
C-type natriuretic peptide (CNP) is the third member
of the family of natriuretic peptides (NPs) originally identified
in porcine brains (1). It is also
widely expressed in the vasculature endothelium, where it regulates
vascular tone (2,3), and in the myocardium (4). Cardiac production of CNP, and its
possible autocrine and paracrine functions, have been demonstrated
in patients with heart failure (5,6) and
endogenous CNP secreted from cardiomyocytes and fibroblasts reduces
the deleterious pathological changes occurring during heart failure
(7). In addition, activation of
the CNP/NP receptor type B (NPR-B) pathway following myocardial
infarction has been reported induce antiproliferative and
antihypertrophic effects in cardiac cells (8,9). CNP
administration improved cardiac function and attenuated cardiac
remodeling after myocardial infarction in rats in vivo, and
these effects have been attributed to its antifibrotic and
antihypertrophic actions (9,10).
The cardiac gap junction is the most important
intercellular communication structure in cardiomyocytes, and is
indispensable for effective function of the heart (11). Among various gap junctional
proteins, connexin (Cx)43 is abundant in ventricular as well as
atrial myocytes, and plays a major role in inter-myocyte
connections (12). Cx40, another
connexin isoform in the heart, has a more limited expression
pattern and is normally restricted to the atrium (13). In the infarct border zone and the
failing or hypertrophied heart, myocardial Cx43 demonstrated
decreased or non-anisotropic expression patterns, and these altered
expression profiles have been designated ‘gap junction remodeling’
(14,15). Gap junction remodeling is
considered to impair intercellular communication and myocardial
function. As a novel antifibrotic and antihypertrophic agent, CNP
and its specific receptor NPR-B may be important for regulation of
cardiac hypertrophy and remodeling. However, the effects of CNP on
atrial Cx40 and Cx43 dysregulation remain unclear. Angiotensin
(Ang) II may play a central role in the etiology and
pathophysiology of cardiovascular diseases, including cardiac
hypertrophy and remodeling in humans (16). In a previous study, it was observed
that excessive Ang II induced significant dysregulation of Cx40 and
Cx43 expression in isolated perfused beating rat atria (17). Therefore, the present study
investigated the effects of CNP on Ang II-induced Cx40 and Cx43
dysregulation in isolated perfused beating rat left atria.
Materials and methods
Preparation of cultured atrial
fibroblasts
All experimental procedures were approved by the
Yanbian University Animal Care and Use Committee and were in
accordance with the Guide for the Care and Use of Laboratory
Animals published by the National Institutes of Health (18). In total, 60 Sprague-Dawley rats
(weight, 250–300 g; age, 18 weeks; female to male ratio, 3:7) were
obtained from Yanbian University. The rats were acclimatized for
one week in the Animal Experimental Center of Yanbian University
[animal license no. SCXK(ji)2012-006] with 45–65% humidity, at a
constant temperature 24–2°C and under a 12-h light/dark cycle. Rats
were given a free access to food and water. The rats were
anesthetized with pentobarbital sodium via intraperitoneal
injection (90 mg/kg), then decapitated and sterilized with alcohol.
The hearts were dissected under aseptic conditions, and the atria
were separated and placed in phosphate-buffered saline (PBS). The
left atria were digested with 0.1% collagenase type II (Gibco;
Thermo Fisher Scientific, Inc.) in a 37°C water bath. The isolated
cells were cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
containing 20% fetal bovine serum (HyClone; GE Healthcare Life
Sciences). Cells were identified using vimentin staining as
described below to confirm that >90% of cells were atrial
fibroblasts. When the cell growth approached 95% confluency, the
cells were passaged at 1:3 culture:fresh medium. Cells at the
second to fourth passages were used in experiments.
Fibroblasts were seeded in 6-well plates and
cultured for 24 h. The cells were divided into two groups: Control
group and Ang II [100 nmol/l, as previously described (19)] group. After 24 h, the cells were
collected for western blotting analysis.
Identification of atrial fibroblasts
by immunofluorescence staining
Adherent cells were identified as atrial fibroblasts
via inverted microscopy. Second-generation atrial fibroblasts
(1×105) were inoculated into glass cover slips wells (24
mm), and sequentially incubated with 4% paraformaldehyde for 20 min
and 0.5% Triton X-100 for 20 min at room temperature. Cell slides
were then blocked with 5% BSA for 2 h at 37°C, and incubated
overnight with anti-vimentin antibody (1:200; cat. no. ab8069;
Abcam) at 4°C. Subsequently, the slides were incubated with
anti-mouse IgG-FITC (1:50; cat. no. ZF-0312; ZSGB-BIO) at room
temperature for 1 h under light-proof conditions. The cell nuclei
were stained with DAPI for 20 min at room temperature. Finally,
fluorescence images were obtained with a fluorescence microscope
(U-RFL-T; Olympus Corporation).
Preparation of perfused beating rat
atria
Sprague-Dawley rats of both sexes with a weight
range of 250–300 g were anesthetized with pentobarbital sodium by
intraperitoneal injection (90 mg/kg), then decapitated and
sterilized with alcohol. Isolated perfused beating left atria were
prepared as previously described (20). After preparation of each atrium,
transmural electrical field stimulation was applied with a luminal
electrode at 1.5 Hz (0.3 ms, 30–40 V) and the atrium was perfused
with HEPES buffer using a peristaltic pump (1.0 ml/min) to enable
atrial pacing for measurement of pulse pressure changes. Oxygen was
continuously supplied and the temperature of the atrium was
maintained at 36°C. The HEPES buffer contained (in mmol/l) 118
NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 25
NaHCO3, 10.0 glucose and 10.0 HEPES (pH 7.4 with NaOH),
together with 0.1% BSA.
Experimental protocols
The rats were randomly divided into eight groups
(n=5/group) as follows: Control group; Ang II group; CNP group; Ang
II + CNP group; Ang II + cANP4-23 (NPR-C activator)
group; Ang II + CNP + KT5823 [protein kinase G (PKG) inhibitor]
group; and Ang II + CNP + pertussis toxin (inhibitor of NPR-C)
group; Ang II + CNP + KT5823 + pertussis toxin.
Each atrium was perfused for 60 min to stabilize the
atrial dynamics. After a control cycle (12 min as an experimental
cycle), the treatment cycle was followed by seven cycles of
treatment agent infusion. The treatment agents were as follows: i)
Control group, each atrium was perfused with normal buffer for
seven cycles; ii) Ang II group, three cycles of normal buffer were
followed by five cycles of Ang II [5.0 µmol/l, as used in our
previous study (17)]; iii) Ang II
+ CNP group, three different doses of CNP (0.03, 0.1 or 0.3 µmol/l)
were used, and each atrium was perfused for one cycle of CNP after
two cycles of normal buffer, and then perfused with CNP + Ang II
for five cycles. In addition, two cycles of normal buffer were
followed by six cycles of CNP alone (0.1 µmol/l); iv) Ang II +
cANP4-23 group, after two cycles of normal buffer, one
cycle of cANP4-23 (3.0 µmol/l) was followed by five
cycles of cANP4-23 + Ang II; v and vi) Ang II + CNP +
KT5823 and Ang II + CNP + pertussis toxin groups, one cycle of
KT5823 (0.3 µmol/l) or pertussis toxin (0.06 µmol/l) after one
cycle of normal buffer, one cycle of KT5823 or pertussis toxin +
CNP was followed by five cycles of KT5823 or pertussis toxin + CNP
+ Ang II; effects of KT5823 + pertussis toxin + CNP + Ang II were
also tested. Immediately after perfusion, the atrial tissues were
collected, frozen in liquid nitrogen, and stored at −80°C for
subsequent western blot analysis.
Western blot analysis
Proteins extracted from fibroblasts and left atrial
tissue samples were analyzed via western blotting. The proteins
were lysed at 4°C in RIPA buffer (cat. no. R0010; Beijing Solarbio
Science & Technology Co., Ltd.) mixed with protease and
phosphatase inhibitors. Protein concentration was determined using
the bicinchoninic acid assay (cat. no. P0010; Beyotime Institute of
Biotechnology) Solubilized proteins (mass of protein loaded per
lane, 40 µg) were separated via 8–10% SDS-PAGE, and the protein
bands were transferred to polyvinylidene difluoride filter
membranes (Beyotime Institute of Biotechnology). The membranes were
blocked with 5% skimmed milk powder in PBS at room temperature for
2 h. Subsequently, the membranes were incubated at 4°C overnight
with a rabbit anti-CNP polyclonal antibody (1:500; cat. no.
E-AB-30982; Elabscience), rabbit anti-NPR-B antibody (1:2,000; cat.
no. ab139188; Abcam), rabbit anti-NPR-C antibody (1:2,000; cat. no.
ab177954; Abcam), rabbit anti-Cx40 polyclonal antibody (1:1,000;
cat. no. ab101929; Abcam), rabbit anti-Cx43 polyclonal antibody
(1:1,000; cat. no. ab11370; Abcam), rabbit anti-transforming growth
factor-β1 (TGF-β1) polyclonal antibody (1:1,000; cat. no. ENT4632;
Elabscience), rabbit anti-collagen I monoclonal antibody (1:1,000;
cat. no. ab138492; Abcam), rabbit anti-matrix metalloproteinase 2
(MMP2) monoclonal antibody (1:1,000; cat. no. ab92536; Abcam) or
rabbit anti-p-AMP-activated protein kinase a1 polyclonal antibody
(anti-p-AMPK; 1:500; cat. no. PA5-17831; Thermo Fisher Scientific,
Inc.). All membranes were also incubated with a rabbit anti-β-actin
monoclonal antibody (1:1,000; cat. no. ENM0028; Elabscience
Biotechnology Co., Ltd.) as a loading control. The membranes were
then incubated with appropriate horseradish peroxidase-conjugated
goat anti-rabbit IgG antibody (1:1,000; cat. no. AEP003;
Elabscience Biotechnology Co., Ltd.) for 2 h at room temperature.
After thorough washing of the membranes with phosphate-buffered
saline containing 0.1% Tween-20, the antibody-bound bands were
visualized with an ECL Plus western blotting detection system (ECL
Western Blot Kit; Beijing CoWin Biotech Co., Ltd.) and the band
densities were quantified using ImageJ software (version 1.48;
National Institutes of Health).
Histopathological analysis
Immediately after perfusion in the control, Ang II
and Ang II + CNP groups, each atrium was placed in a pre-dosed
fixative (10% formalin, Bouin's fixative) overnight at room
temperature, dehydrated with ethanol, embedded in paraffin,
sectioned at 3–4-µm thickness and subjected to Masson's three-color
dye staining using a Masson's staining kit (Beijing Solarbio
Science & Technology Co. Ltd.) for 10 min at room temperature.
The stained sections were examined via optical microscopy (five
fields analyzed/sample), and images acquired at ×100 magnification.
The collagen volume fraction (collagen area/total area ×100%) was
determined using Image-Pro Plus 6.0 software (Media Cybernetics
Inc.).
Statistical analysis
Data were analyzed with Prism 5.0 software (GraphPad
Software, Inc.). Significant differences were determined by an
unpaired t-test or two-way ANOVA followed by a Bonferroni
post hoc tests. Data were presented as means ± standard error of
the mean. P<0.05 was considered to indicate a statistically
significant difference.
Results
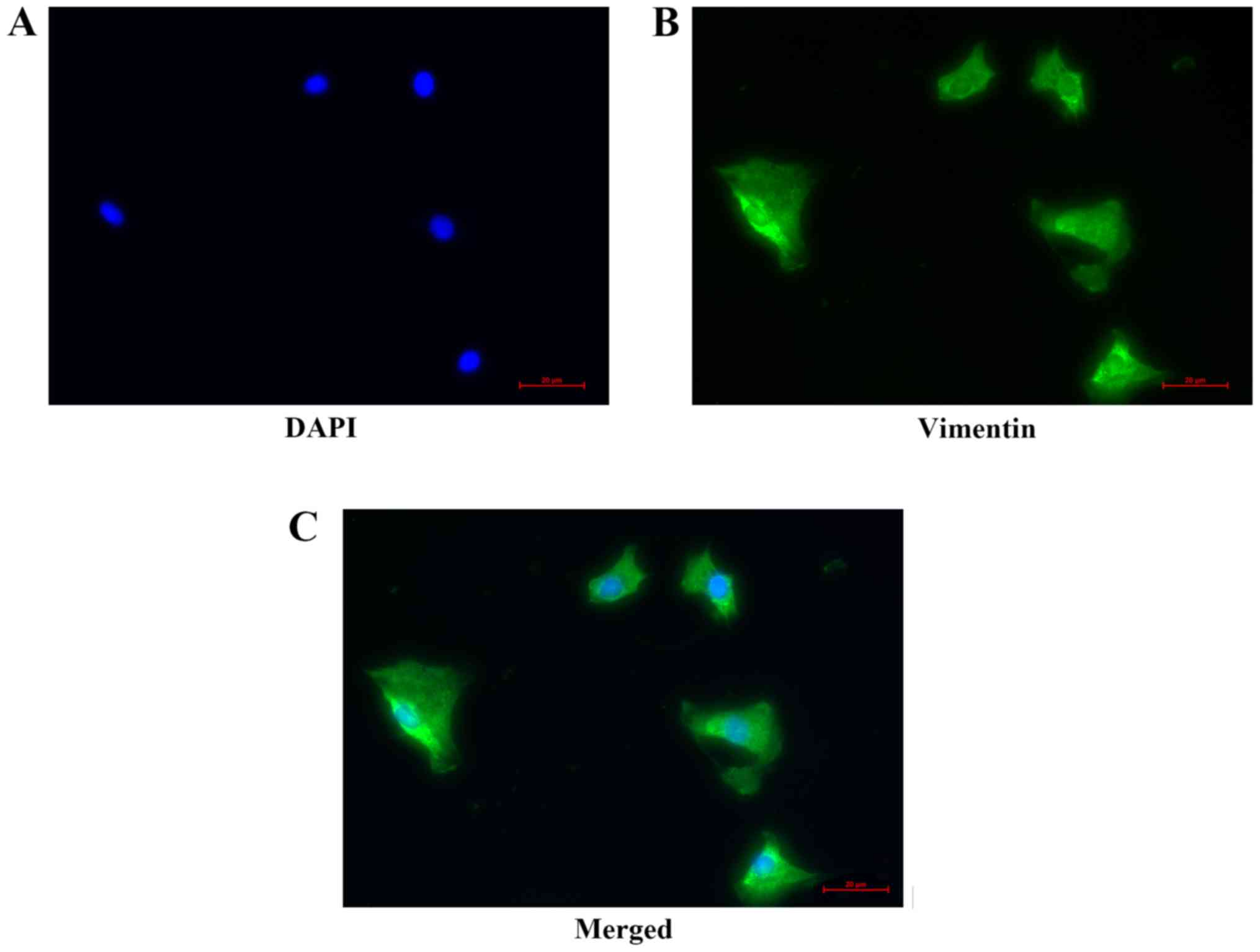
Identification of atrial
fibroblasts
Vimentin is an intermediate fiber in interstitial
cells that forms part of the cytoskeleton. It has been reported
that cardiac fibroblasts can be identified by positive expression
of vimentin (21). The results
revealed green fluorescence for vimentin-positive cells in the
highly purified atrial fibroblasts (Fig. 1).
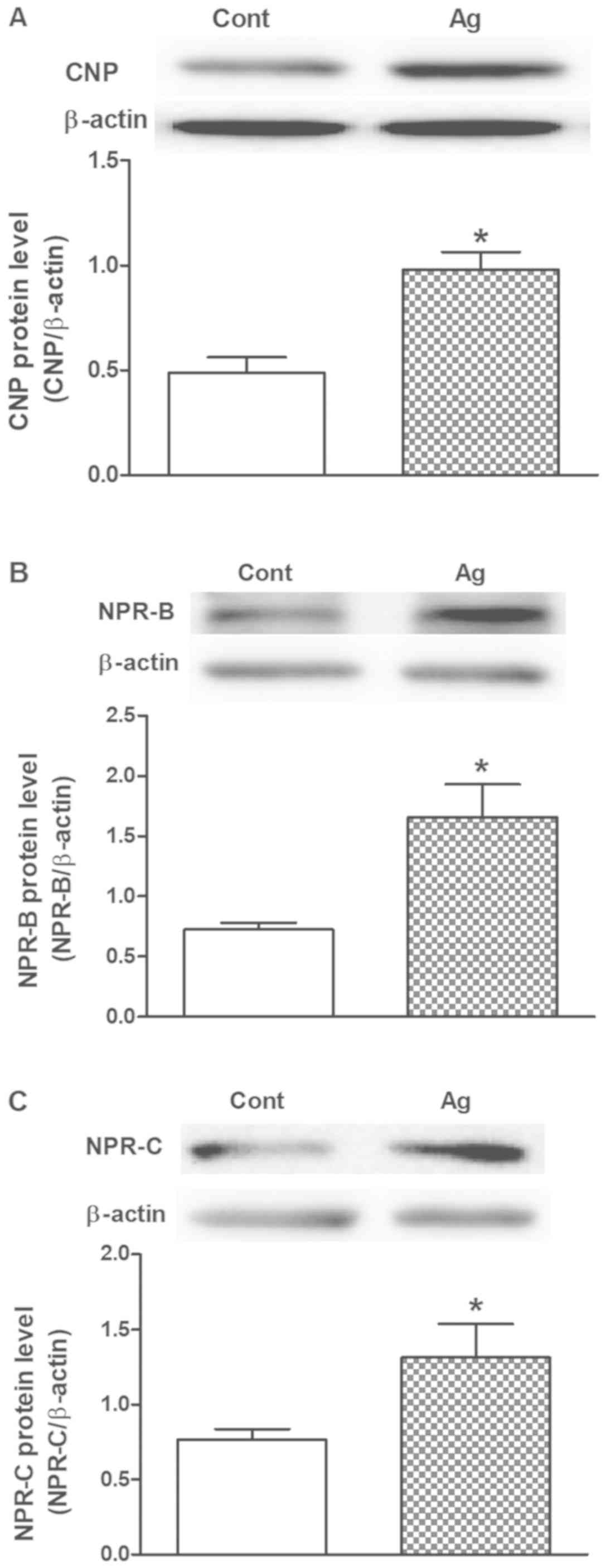
Effects of Ang II on CNP, NPR-B and
NPR-C levels in cultured atrial fibroblasts
To determine the effects of Ang II on the regulation
of CNP, NPR-B and NPR-C protein expression, a series of experiments
were performed on cultured rat atrial fibroblasts. Ang II
significantly increased CNP protein expression (P<0.05 vs.
control; Fig. 2A), as well as
NPR-B and NPR-C protein expression (P<0.05 vs. control; Fig. 2B and C) in rat cultured atrial
fibroblasts. These results suggest that Ang II can promote the
production of CNP and lead to upregulation of its receptors in rat
atrial fibroblasts.
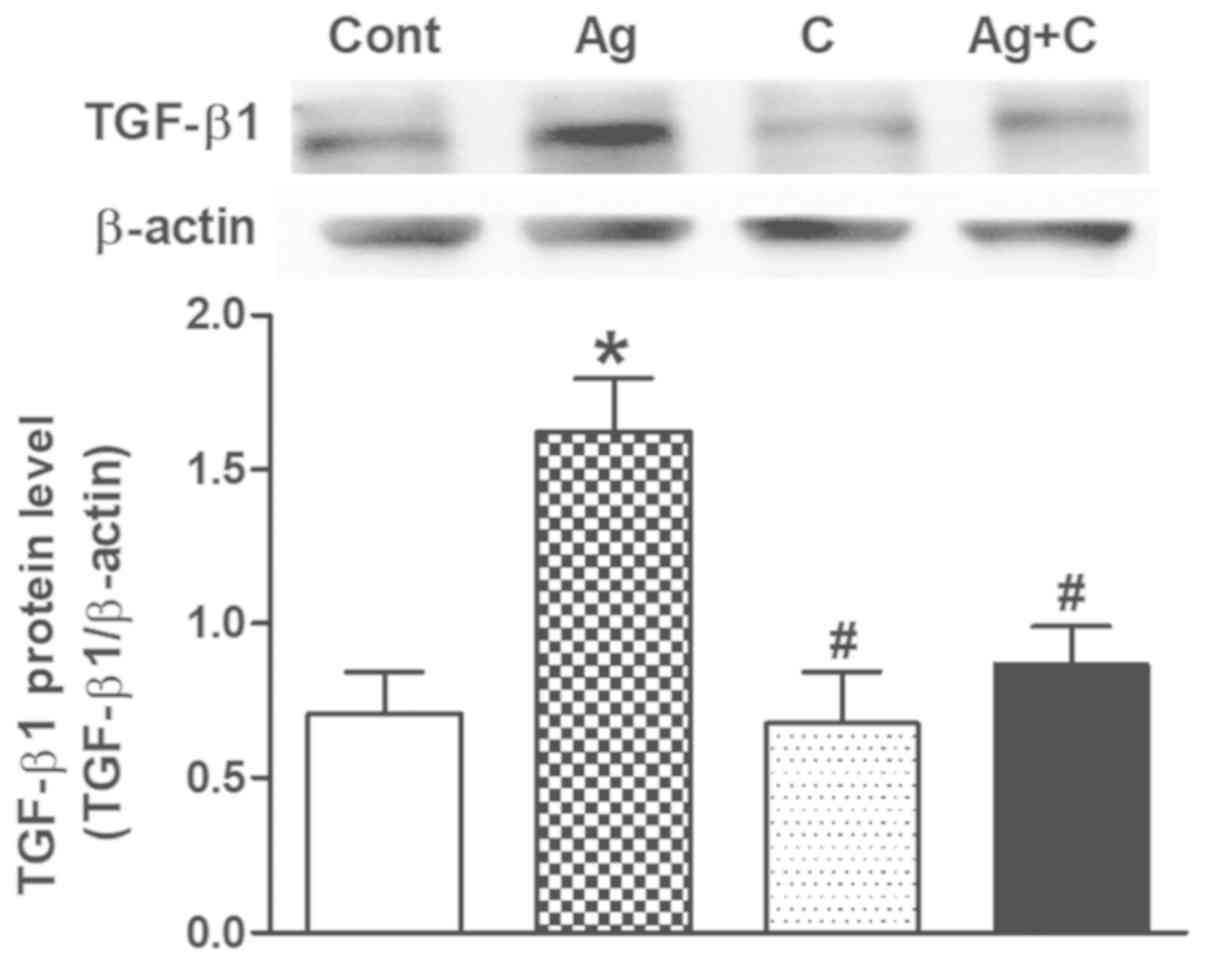
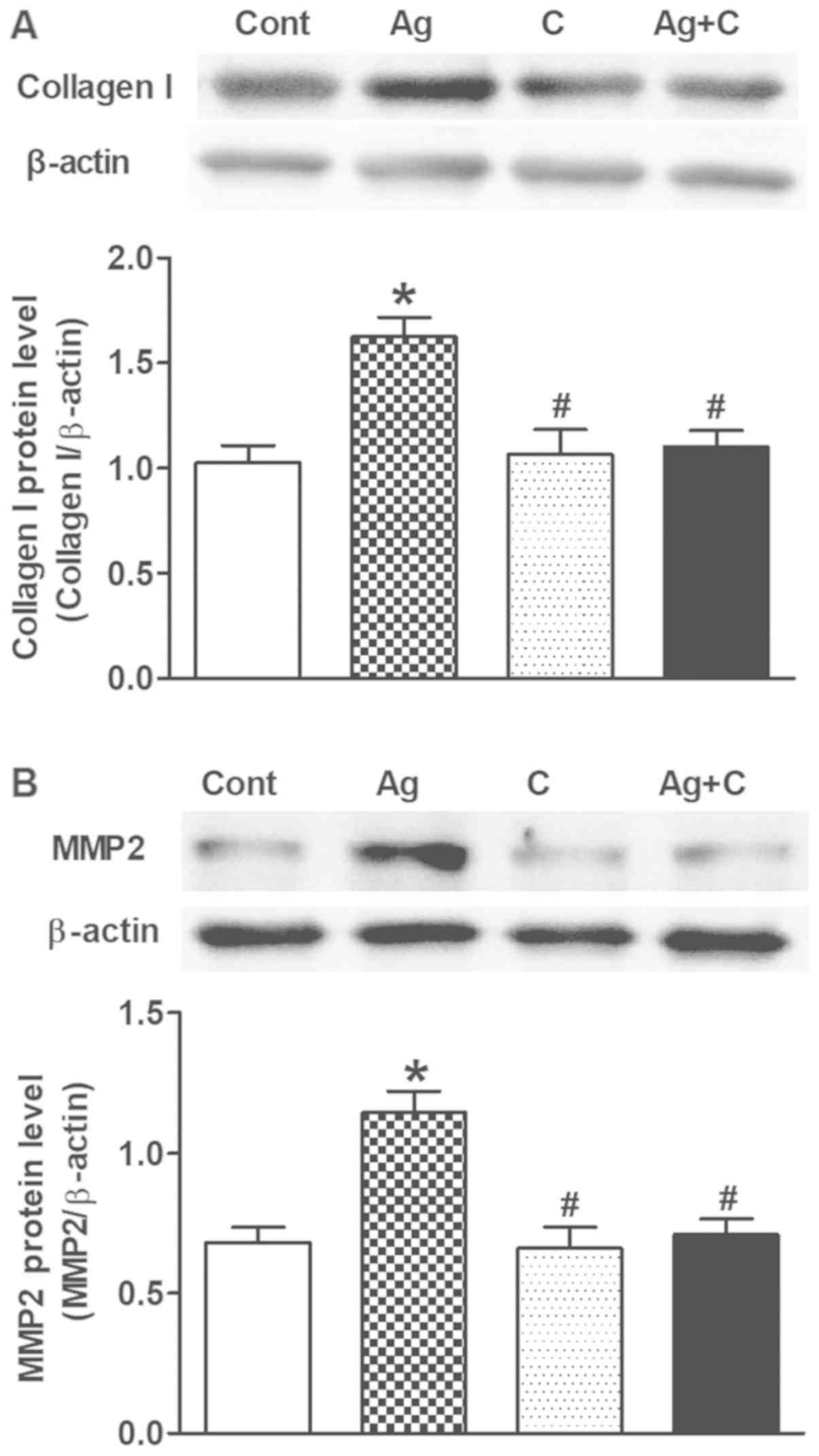
Effects of CNP on Ang II-induced
TGF-β1 expression and atrial fibrosis
Excessive Ang II-induced TGF-β1 can lead to cardiac
fibrosis and remodeling. Therefore, another series of experiments
were performed to examine the effects of CNP on Ang II-induced
TGF-β1 expression and atrial fibrosis. Ang II significantly
increased TGF-β1 expression (P<0.05 vs. control; Fig. 3) concomitantly with upregulation of
collagen I and MMP2 levels (P<0.05 vs. control; Fig. 4A and B); these effects were
completely blocked by CNP pretreatment (P<0.05 vs. control;
P<0.05 vs. Ang II; Figs. 3 and
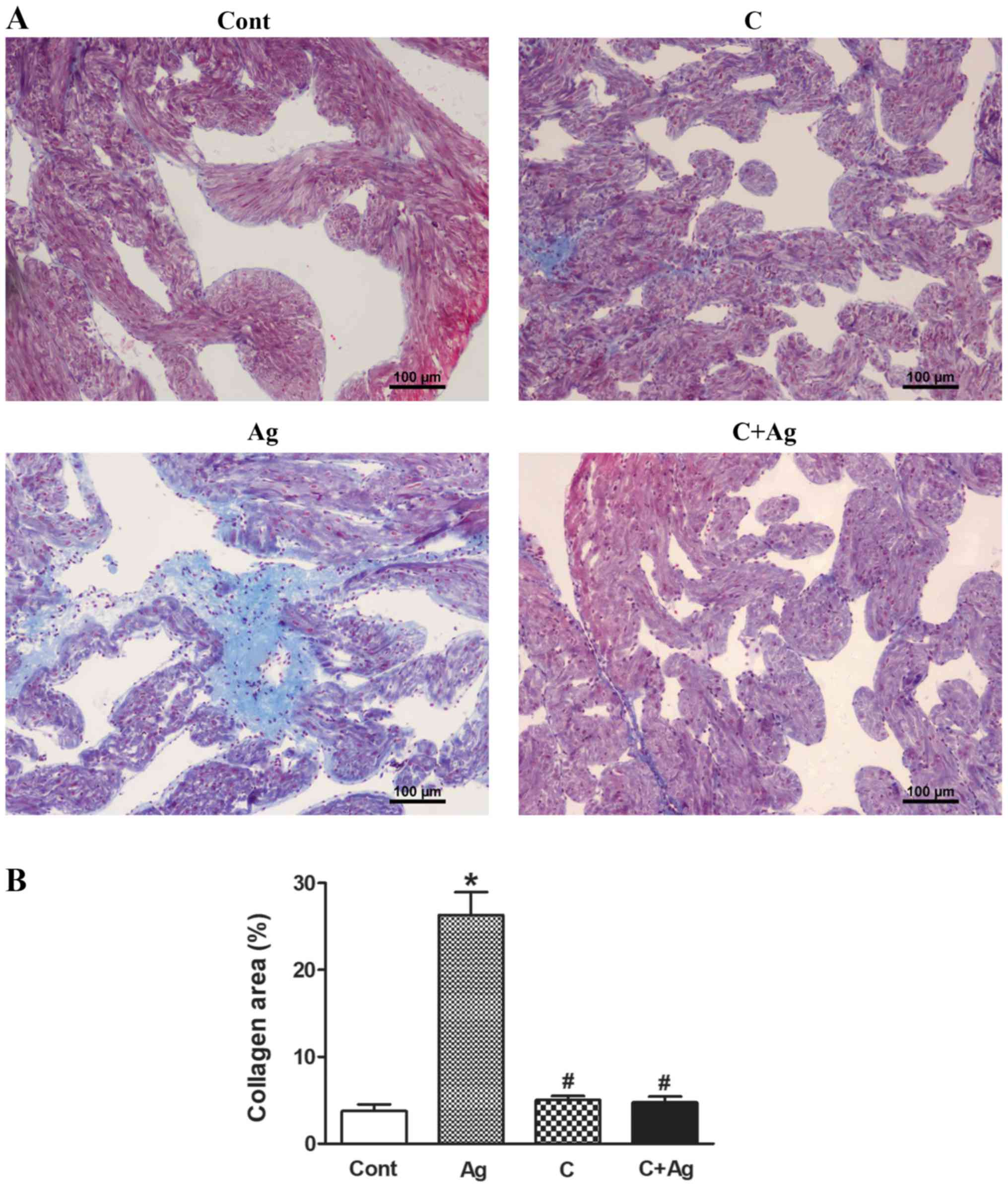
4). In addition, Masson's staining
revealed that Ang II induced widespread fibrous tissue in
interstitial areas compared with the control (collagenous fibers
were stained blue; Fig. 5A and B),
and this effect was also abolished by CNP pretreatment (Fig. 5A and B). These results suggested
that CNP induced inhibitory effects on Ang II-induced activation of
TGF-β1 and fibrosis in beating rat atria.
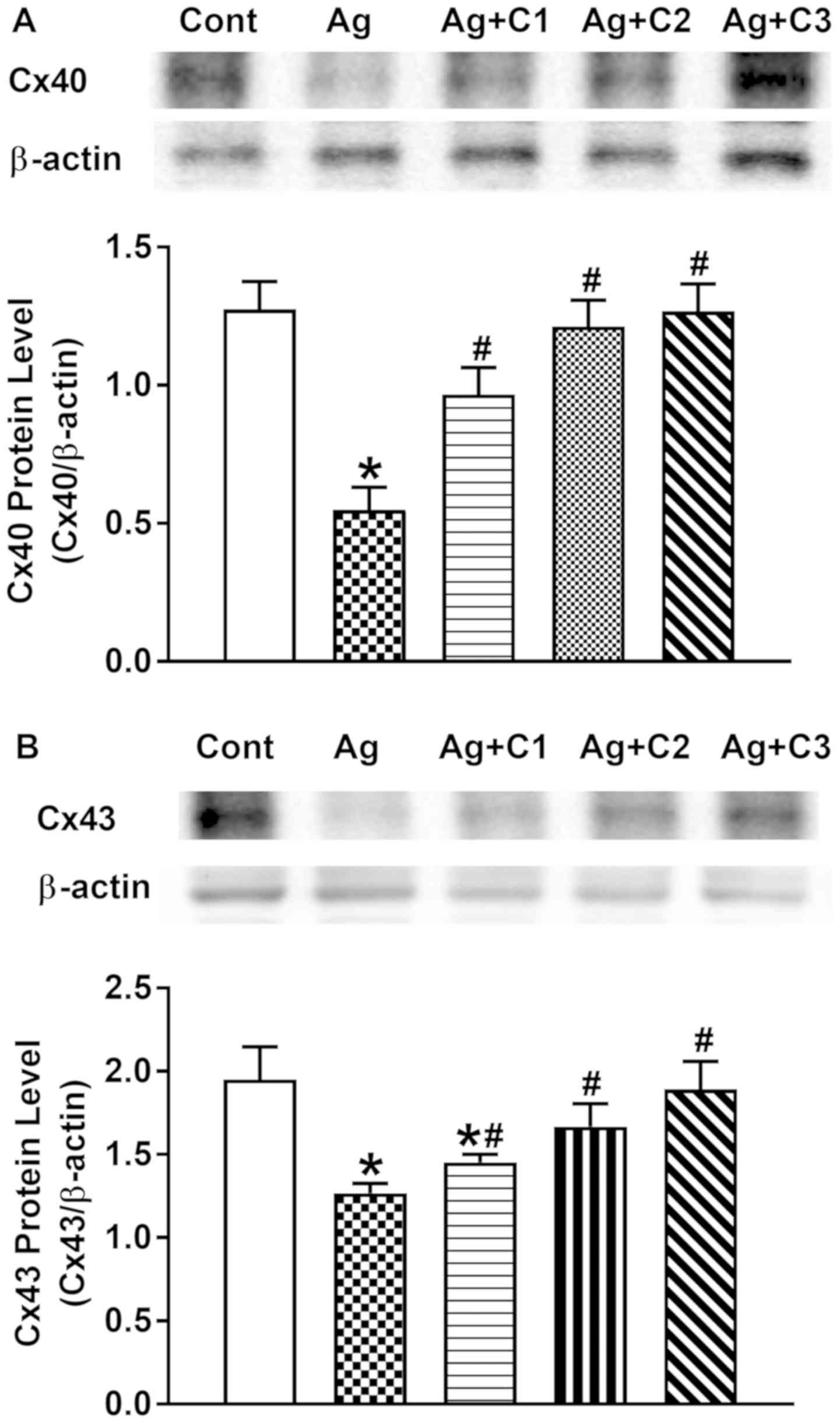
Effect of CNP on Ang II-induced
dysregulation of Cx40 and Cx43
It was demonstrated that Ang II can upregulate
TGF-β1, leading to cardiac fibrosis and subsequent cardiac
remodeling. Therefore, to examine whether Ang II induces
dysregulation of atrial connexin proteins, and the effects of CNP
on this process, the levels of Cx40 and Cx43 were analyzed. Ang II
significantly decreased atrial expression of Cx40 and Cx43, and the
effects were markedly inhibited by CNP in a dose-dependent manner
(P<0.05 vs. control; P<0.05 vs. Ang II; Fig. 6A and B). These results indicated
that CNP prevented Ang II-induced gap junction remodeling in
beating rat atria.
Effect of CNP on p-AMPK
expression
In the light of the inhibitory effects of AMPK on
Ang II-induced cardiac hypertrophy and fibrosis, p-AMPK expression
was analyzed. CNP significantly increased p-AMPK expression
(P<0.05 vs. control, P<0.05 vs. Ang II; Fig. 7). Furthermore, the NPR-C activator
cANP4-23 mimicked the effect of CNP on atrial p-AMPK
expression (P<0.05 vs. control, P<0.05 vs. Ang II; Fig. 7) in beating rat atria. Activation
of AMPK expression by CNP, as well as by cANP4-23, was
almost completely abolished by KT5823 and pertussis toxin,
inhibitors of PKG and NPR-C, respectively (P<0.05 vs. CNP,
P<0.05 vs. cANP4-23; Fig. 7). Meanwhile, Ang II had no effect
on atrial AMPK activity (P>0.05 vs. control; Fig. 7). These results demonstrated that
CNP activated atrial AMPK signaling via PKG and NPR-C.
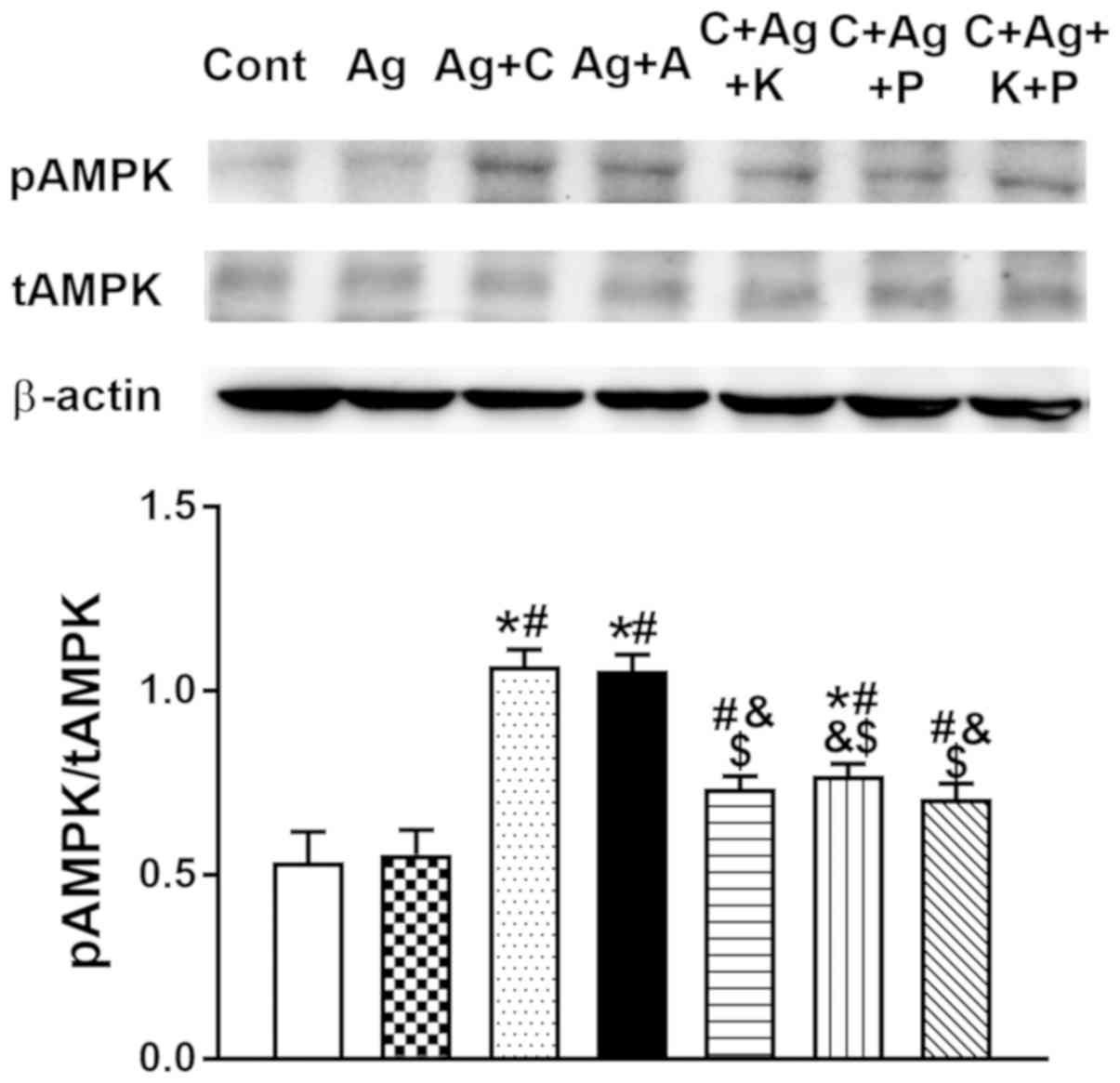 | Figure 7.Effects of CNP and
cANP4-23, an agonist of NPR-C, on the regulation of
atrial p-AMPK expression. Data were expressed as mean ± standard
error of the mean; n=5. *P<0.05 vs. Cont; #P<0.05
vs. Ag; &P<0.05 vs. Ag + C; $P<0.05
vs. Ag + A. CNP, C-type natriuretic peptide; NPR-C, natriuretic
peptide receptor type C; Cont, control; Ag, angiotensin II; C, CNP;
A, cANP4-23; KT, KT5823; P, pertussis toxin; p-,
phosphorylated; t-, total; AMPK, AMP-activated protein kinase. |
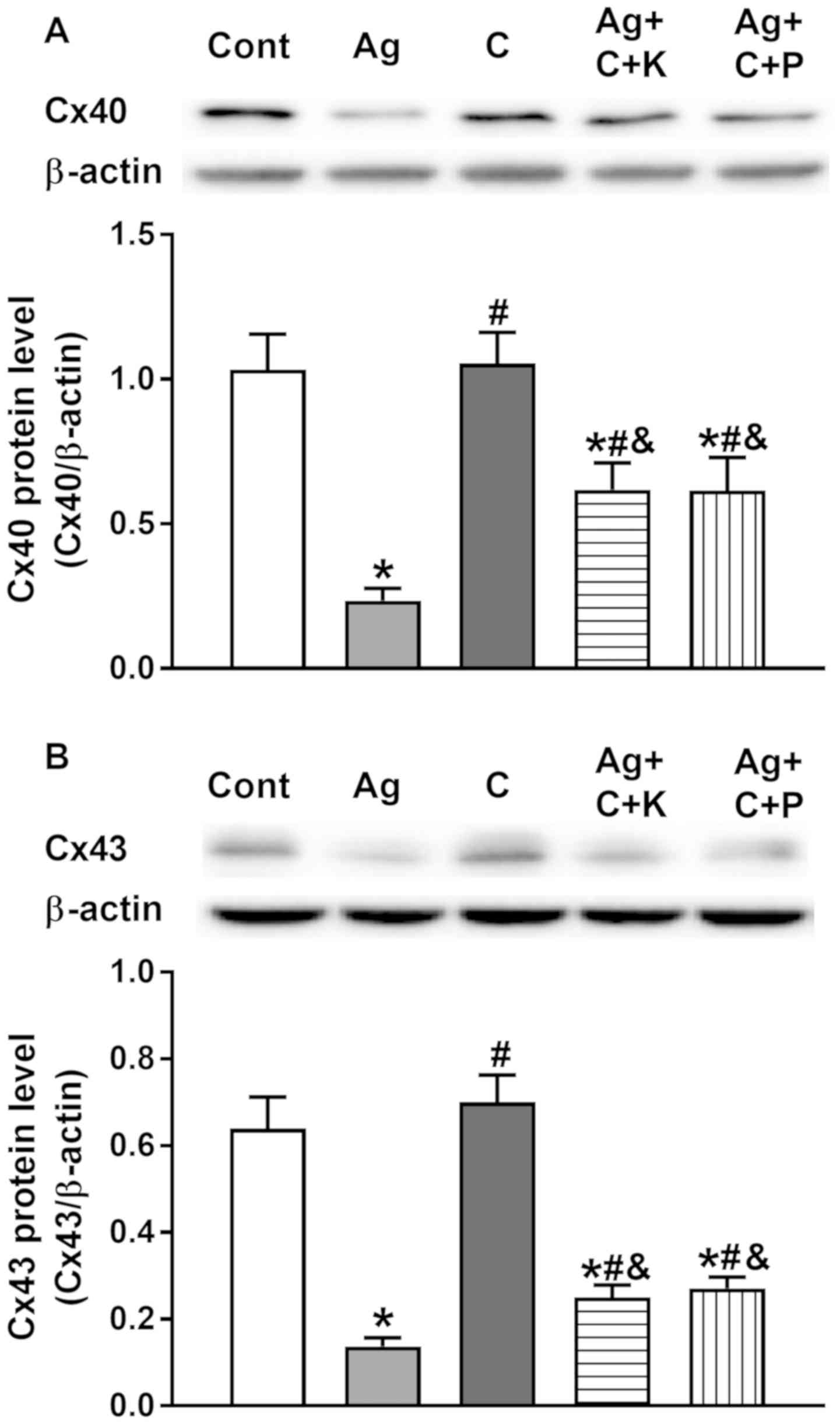
Effects of CNP receptor pathways on
Ang II-induced TGF-β1, and dysregulation of Cx40 and Cx43
expression
To confirm that CNP receptor pathways are involved
in the inhibition of Ang II-induced atrial TGF-β1 and Cx
expression, experiments were performed with PKG and NPR-C
inhibitors. As presented in Fig.
8, CNP and cANP4-23 almost completely abolished Ang
II-induced upregulation of TGF-β1 (P<0.05 vs. Ang II), and the
effect of CNP on Ang II-induced upregulation of TGF-β1 was
attenuated by KT5823 and pertussis toxin, respectively (P<0.05
vs. CNP and cANP4-23, respectively). In addition, the
preventive effects of CNP on Ang II-induced dysregulation of Cx40
and Cx43 were clearly attenuated by KT5823 and pertussis toxin
(P<0.05 vs. control, P<0.05 vs. Ang II, P<0.05 vs. CNP;
Fig. 9A and B). These results
suggested that CNP prevented Ang II-induced dysregulation of
connexin expression by inhibiting TGF-β1 activity through CNP-PKG
and CNP-NPR-C signaling in beating rat atria.
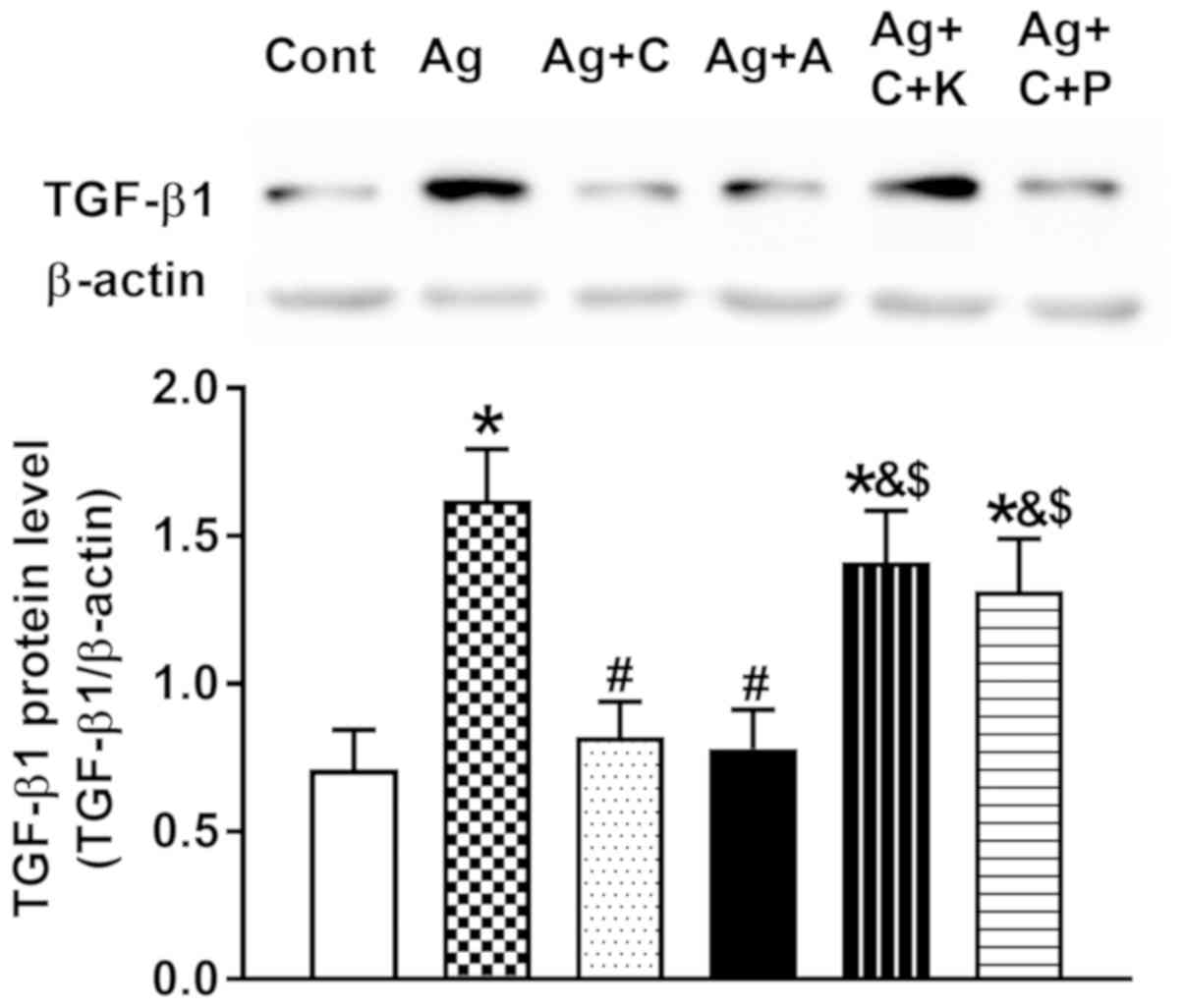 | Figure 8.Effects of CNP and
cANP4-23, an agonist of natriuretic peptide receptor
type C, on the regulation of Ang II-induced atrial TGF-β1
expression. Data were expressed as mean ± standard error of the
mean; n=5. *P<0.05 vs. Cont; #P<0.05 vs. Ang II;
&P<0.05 vs. Ag + C; $P<0.05 vs. Ag
+ A. CNP, C-type natriuretic peptide; NPR-C; Cont, control; Ag/Ang
II, angiotensin II; TGF, transforming growth factor; C, CNP; A,
cANP4-23; K, KT5823, an inhibitor of PKG; P, pertussis
toxin, an inhibitor of NPR-C. |
Discussion
The present study demonstrated that, in isolated
perfused beating rat atria, CNP prevented Ang II-induced
dysregulation of Cx40 and Cx43 protein expression via activation of
AMPK, thereby inhibiting Ang II-induced upregulation of TGF-β1 and
atrial fibrosis through both the CNP-PKG and CNP-NPR-C
pathways.
As well as being the most abundant NP in the brain,
CNP is also synthesized in cardiac fibroblasts, and may serve a
role as an autocrine regulator against excessive cardiac fibrosis
(22). Protective roles of CNP on
cardiovascular diseases have also been identified (8,23).
In the present study, Ang II significantly increased the CNP
protein level concomitantly with upregulation of NPR-B and NPR-C
expression in adult rat left atrial cultured fibroblasts. In
addition, exogenous CNP completely abolished the fibrosis induced
by Ang II in isolated beating rat atria. These findings are
consistent with the aforementioned studies.
In our previous study, it was observed that
excessive Ang II stimulated atrial TGF-β1 expression through
activation of p38-MAP kinase and nuclear transcription factors,
ultimately leading to atrial dysregulation of Cx40 and Cx43
expression (17). In the present
study, Ang II also significantly increased atrial TGF-β1 protein
expression and upregulation of collagen I as well as MMP2,
ultimately leading to atrial fibrosis. Thus, Ang II-induced
fibrosis caused downregulation of Cx40 and Cx43. These results are
consistent with previous studies (14,15,24).
In addition, it was found that CNP completely abolished Ang
II-induced upregulation of atrial TGF-β1, extracellular matrix
proteins and downregulation of Cx40 as well as Cx43. These results
demonstrate that CNP can play a protective role against Ang
II-induced connexin protein dysregulation by suppressing TGF-β1
expression in perfused beating rat atria. The present data are
consistent with previous studies that CNP prevents cardiac
hypertrophy, fibrosis, and remodeling (10,24–26).
A previous study demonstrated that stimulation of
AMPK phosphorylation led to inhibition of TGF-β/Smad3-mediated
myofibroblast differentiation (27), and prevented Ang II-induced cardiac
hypertrophy and fibrosis (28,29).
Similarly, in the present study, CNP dramatically increased atrial
AMPK phosphorylation, thereby suppressing Ang II-induced
upregulation of TGF-β1 and consequently preventing Ang II-induced
dysregulation of Cx40 and Cx43 expression. In addition, CNP-induced
upregulation of AMPK phosphorylation and downregulation of TGF-β1
were abolished by not only KT5823, a PKG pathway inhibitor, but
also by pertussis toxin, an NPR-C inhibitor. These results
suggested that CNP can stimulate AMPK phosphorylation and prevent
Ang II-induced dysregulation of Cx40 and Cx43 expression via PKG as
well as NPR-C signaling in beating rat atria. These results are
consistent with previous findings that CNP acts via the CNP-NPR-B
and CNP-NPR-C pathways to play an important protective role against
cardiac remodeling (4).
In conclusion, CNP prevented Ang II-induced
dysregulation of Cx40 and Cx43 expression through upregulation of
p-AMPK and downregulation of TGF-β1 via the CNP-PKG and CNP-NPR-C
pathways in isolated perfused beating rat atria. These findings
suggest that CNP is potentially useful for clinical applications
involving cardiac dysregulation of connexin expression-related
diseases.
Acknowledgements
Not applicable.
Funding
The present study work was supported by The National
Natural Science Foundation of China (grant nos. 81360061 and
81660089).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DD performed atrial perfusion experiments and cell
culture experiments. BZ performed immunofluorescence staining. YJ
and CG performed western blot analysis. SZ performed
histopathological analysis. XL and XC designed the experiments and
wrote the manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Yanbian University Animal Care and Use Committee and were in
accordance with the Guide for the Care and Use of Laboratory
Animals published by the National Institutes of Health.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sudoh T, Minamino N, Kangawa K and Matsuo
H: C-type natriuretic peptide (CNP): A new member of natriuretic
peptide family identified in porcine brain. Biochem Biophys Res
Commun. 168:863–870. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Abassi Z, Karram T, Ellaham S, Winaver J
and Hoffman A: Implications of the natriuretic peptide system in
the pathogenesis of heart failure: Diagnostic and therapeutic
importance. Pharm Ther. 102:223–241. 2004. View Article : Google Scholar
|
|
3
|
Scotland RS, Ahluwalia A and Hobbs AJ:
C-type natriuretic peptide in vascular physiology and disease.
Pharm Ther. 105:85–93. 2005. View Article : Google Scholar
|
|
4
|
Del Ry S, Cabiati M, Vozzi F, Battolla B,
Caselli C, Forini F, Segnani C, Prescimone T, Giannessi D and
Mattii L: Expression of C-type natriuretic peptide and its receptor
NPR-B in cardiomyocytes. Peptides. 32:1713–1718. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Del Ry S, Maltinti M, Piacenti M, Passino
C, Emdin M and Giannessi D: Cardiac production of C-type
natriuretic peptide in heart failure. J Cardiovasc Med
(Hagerstown). 7:397–399. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kalra PR, Clague JR, Bolger AP, Anker SD,
Poole-Wilson PA, Struthers AD and Coats AJ: Myocardial production
of C-type natriuretic peptide in chronic heart failure.
Circulation. 107:571–573. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moyes AJ, Chu SM, Aubdool AA, Dukinfield
MS, Margulies KB, Bedi KC, Hodivala-Dilke K, Baliga RS and Hobbs
AJ: C-type natriuretic peptide co-ordinates cardiac structure and
function. Eur Heart J. (pii): ehz093Mar 21–2019.(Epub ahead of
print). PubMed/NCBI
|
|
8
|
Calvieri C, Rubattu S and Volpe M:
Molecular mechanisms underlying cardiac anti-hypertrophic and
antifibrotic effects of natriuretic peptides. J Mol Med (Berl).
90:5–13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Y, de Waard MC, Sterner-Kock A,
Stepan H, Schultheiss HP, Duncker DJ and Walther T:
Cardiomyocyte-restricted over-expression of C-type natriuretic
peptide prevents cardiac hypertrophy induced by myocardial
infarction in mice. Eur J Heart Fail. 9:548–557. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Soeki T, Kishimoto I, Okumura H, Tokudome
T, Horio T, Mori K and Kangawa K: C-type natriuretic peptide, a
novel antifibrotic and antihypertrophic agent, prevents cardiac
remodeling after myocardial infarction. J Am Coll Cardiol.
45:608–616. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dhein S: Gap junction channels in the
cardiovascular system: Pharmacological and physiological
modulation. Trends Pharmacol Sci. 19:229–241. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Davis LM, Kanter HL, Beyer EC and Saffitz
JE: Distinct gap junction protein phenotypes in cardiac tissues
with disparate conduction properties. J Am Coll Cardiol.
24:1124–1132. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Davis LM, Rodefeld ME, Green K, Beyer EC
and Saffitz JE: Gap junction protein phenotypes of the human heart
and conduction system. J Cardiovasc Electrophysiol. 6:813–822.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Severs NJ, Coppen SR, Dupont E, Yeh HI, Ko
YS and Matsushita T: Gap junction alterations in human cardiac
disease. Cardiovasc Res. 62:368–377. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tanaka H, Matsuyama TA and Takamatsu T:
Towards an integrated understanding of cardiac
arrhythmogenesis-growing roles of experimental pathology. Pathol
Int. 67:8–16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim S and Iwao H: Molecular and cellular
mechanisms of angiotensin II-mediated cardiovascular and renal
diseases. Pharmacol Rev. 52:11–34. 2000.PubMed/NCBI
|
|
17
|
Zhang B, Cui X, Jin HH, Hong L, Liu X, Li
X, Zhang QG and Liu LP: Ginsenoside Re prevents angiotensin
II-induced gap-junction remodeling by activation of PPARγ in
isolated beating rat atria. Life Sci. 190:36–45. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals8th.
National Academies Press (US); Washington, DC: 2011
|
|
19
|
Qi H, Liu Y, Li S, Chen Y, Li L, Cao Y, E
M, Shi P, Song C, Li B and Sun H: Activation of AMPK attenuated
cardiac fibrosis by inhibiting CDK2 via p21/p27 and miR-29 family
pathways in ratsnd. Mol Ther Nucleic Acids. 8:277–290. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu X, Zhang Y, Hong L, Han CJ, Zhang B,
Zhou S, Wu CZ, Liu LP and Cui X: Gallic acid increases atrial
natriuretic peptide secretion and mechanical dynamics through
activation of PKC. Life Sci. 181:45–52. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gu X, Fang T, Kang P, Hu J, Yu Y, Li Z,
Cheng X and Gao Q: Effect of ALDH2 on high glucose-induced cardiac
fibroblast oxidative stress, apoptosis, and fibrosis. Oxid Med Cell
Longev. Oct 9–2017.(Epub ahead of print). View Article : Google Scholar
|
|
22
|
Horio T, Tokudome T, Maki T, Yoshihara F,
Suga S, Nishikimi T, Kojima M, Kawano Y and Kangawa K: Gene
expression, secretion, and autocrine action of C-type natriuretic
peptide in cultured adult rat cardiac fibroblasts. Endocrinology.
144:2279–2284. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Del Ry S: C-type natriuretic peptide: A
new cardiac mediator. Peptides. 40:93–98. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wenzel S, Taimor G, Piper HM and Schlüter
KD: Redox-sensitive intermediates mediate angiotensin II-induced
p38 MAP kinase activation, AP-1bindingactivity, and TGF-beta
expression in adult ventricular cardiomyocytes. FASEBJ.
15:2291–2293. 2001. View Article : Google Scholar
|
|
25
|
Izumiya Y, Araki S, Usuku H, Rokutanda T,
Hanatani S and Ogawa H: Chronic C-type natriuretic peptide infusion
attenuates angiotensin II-induced myocardial superoxide production
and cardiac remodeling. Int J Vasc Med. 2012:2460582012.PubMed/NCBI
|
|
26
|
Ichiki T, Boerrigter G, Huntley BK,
Sangaralingham SJ, McKie PM, Harty GJ, Harders GE and Burnett JC
Jr: Differential expression of the pro-natriuretic peptide
convertases corin and furin in experimental heart failure and
atrial fibrosis. Am J Physiol RegulIntegr Comp Physiol.
304:R102–R109. 2013. View Article : Google Scholar
|
|
27
|
Mishra R, Cool BL, Laderoute KR, Foretz M,
Viollet B and Simonson MS: AMP-activated protein kinase inhibits
transforming growth factor-beta-induced Smad3-dependent
transcription and myofibroblast transdifferentiation. J Biol Chem.
283:10461–10469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hernandez JS, Barreto-Torres G, Kuznetsov
AV, Khuchua Z and Javadov S: Crosstalk between AMPK activation and
angiotensin II-induced hypertrophy in cardiomyocytes: The role of
mitochondria. J Cell Mol Med. 18:709–720. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fujita K, Maeda N, Sonoda M, Ohashi K,
Hibuse T, Nishizawa H, Nishida M, Hiuge A, Kurata A, Kihara S, et
al: Adiponectin protects against angiotensin II-induced cardiac
fibrosis through activation of PPAR-alpha. Arterioscler Thromb Vasc
Biol. 28:863–870. 2008. View Article : Google Scholar : PubMed/NCBI
|