Introduction
Microglial cells are the resident immune cells of
the central nervous system (CNS) and can quickly respond to CNS
injury; activation occurs rapidly within hours (1). Once the microglia are activated, they
can secrete and release cytokines and chemokines, such as
interleukin (IL)-1, IL-2, IL-6, transforming growth factor (TGF)
131, and matrix metalloproteinase (MMP)9, and express complement
components and their receptors (2). Traditionally, microglial cell
activity in the CNS has been considered detrimental, as they
recruit and reactivate T cells in the CNS by secreting MMPs,
disrupting the blood-brain barrier, upregulate major
histocompatibility complex II molecules and exert OPC toxicity by
producing inflammatory cytokines and reactive oxygen species
(3). Microglia have been reported
to participate in the pathogenesis of many human diseases, such as
Alzheimer's disease, multiple sclerosis (MS), autoimmune diseases
and other disorders (4).
Particularly in MS, microglia have been reported to serve dual
roles in demyelination in the CNS, including protective and
pathogenic roles (5). However,
previous studies have demonstrated that microglia may serve an
important role in the remyelination process, through phagocytosing
debris from cells and destroyed myelin, supporting myelinating
oligodendrocytes and promoting axonal regeneration (6,7).
Tuftsin (Thr-Lys-Pro-Arg) was first described by
Fridkin et al (8) and was
thought to be a phagocytosis-stimulating factor for cells of
monocytic origin. Tuftsin activates macrophages through binding to
receptors that are expressed by cells of monocytic origin,
including microglia (9).
The present study investigated the role of microglia
in EAE progression. Tuftsin was used to alter the activation of
microglia. Early administration of tuftsin altered the activation
of microglia and attenuated the humoral immune responses associated
with EAE progression to different degrees.
Materials and methods
Experimental animals
Adult (8–9 weeks old and 18–20 g) female C57BL/6
mice were purchased from North China University of Science and
Technology. All mice were housed in a temperature-controlled room
under a 12-h light/dark cycle for 4 weeks with food and water ad
libitum. The mice were randomly divided into three groups;
control group (n=12), experimental autoimmune encephalomyelitis
group (EAE group, n=12) and Tuftsin group (n=12). All protocols
were approved by the Animal Ethics Committee of the North China
University of Science and Technology.
Induction of EAE in mice
EAE mice were induced with MOG35-55 (200 µg), and
the mice were intraperitoneally injected with pertussis toxin (500
ng, List Biological Laboratories, Inc.) at 0 and 48 h following
immunization. At least two investigators weighed and evaluated the
animals for clinical scores in a blinded manner. There were two
animals which appeared to be in intolerable distress and
self-mutilated limbs; these were sacrificed using pentobarbital
sodium (150 mg/kg).
Clinical assessment
Clinical scores (10) were determined in accordance with
the following criteria: 0, healthy; 1, limp tail; 2, ataxia and/or
paresis of the hind limbs; 3, paralysis of the hind limbs; 4,
paresis and/or paralysis of the forelimbs; 5, moribund or dead.
Time-controlled drug delivery
The present study used ALZET mini-osmotic pumps to
control drug delivery over time. The mice were injected with either
PBS or 500 mM tuftsin [Gen Script (Nanjing) Co., Ltd.] at a rate of
0.25 ml/h (total volume was 100 ml). Pumps were implanted
subcutaneously in the backs of anaesthetized mice on day 1
following immunization. On day 15, the pumps were replaced with
fresh pumps and maintained thus until day 28.
Histological staining and
immunohistochemistry
Spinal cords were obtained from anaesthetized mice,
which were perfused intracardially with 4% paraformaldehyde. The
samples underwent a dehydration in graded ethanol (70% ethanol 3–5
min; 80% ethanol 3–5 min; 90% ethanol 3–5 min; 95% ethanol 3–5
min). Paraffin-embedded tissue sections were cut in the coronal
plane at a thickness of 5 µm. Histological staining, including LFB
staining, was performed to identify demyelination. The sections
were left in LFB solution (Beijing Solarbio Science &
Technology Co., Ltd.) at 56°C overnight, excess stain rinsed off
with 95% ethyl alcohol and distilled water, differentiated in
lithium carbonate solution for 30 sec and 70% ethyl alcohol for 30
sec, counterstained in cresyl violet solution (Guidechem) for 30–40
sec, rinsed in distilled water, differentiated in 95% ethyl alcohol
for 5 min then placed in 100% alcohol for 5 min (twice) and finally
two baths in xylene for 5 min each. Immunohistochemistry was
performed with anti-myelin basic protein (MBP) antibodies to
identify MBP (1:100; sc-271524, Santa Cruz Biotechnology, Inc.).
Hematoxylin was used to stain cell morphology. The sections were
observed under light microscope (magnification, ×40) (11) and analyzed by Image 2 Pro plus 5.0
(Media Cybernetics, Inc.).
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted from the brain and spinal
cord in all groups using the RNAeasy Micro kit purchased from OMEGA
Company following the manufacturer's instructions. Reverse
transcription was performed on 1 µg of total RNA with an RT-PCR kit
(Life Technologies; Thermo Fisher Scientific, Inc.), Purity
quantification, cDNA synthesis and qPCR were performed according to
the manufacturer's protocols. Reaction procedures were as follows:
An initial step at 95°C for 5 min, 40 cycles of 94°C for 15 sec,
and 60°C for 34 sec; reaction volume 20 µl. The primer sequences
were: GAPDH: Forward: 5′-TTCACCACCATGGAGAAGGC-3′, Reverse:
5′-GGCATGGACTGTGGTCATGA-3′; tumor necrosis factor (TNF)-α: Forward:
5′-CATCTTCTCAAAATTCGAGTGACAA-3′, Reverse:
5′-TGGG-AGTAGACAAGGTACAACCC-3′; IL-10: Forward:
5′-TGGCCACACTTGA-GAGCTGC-3′, Reverse: 5′-TTCAGGGATGAAGCGGCTGG-3′;
TGF-β: Forward: 5′-CCGCAACAACGCAATCTATG-3′, Reverse:
5′-AGCCCTGTATTCCGTCTCCTT-3′.
qPCR was carried out using SYBR green mix (Roche,
USA) and analyzed using the 2−ΔΔCq method (11). The relative expression levels of
the mRNAs were reported as fold changes vs. control.
Western blot analysis
Protein was extracted from the brain and spinal
cord. The samples were lysed in Tissue Protein Lysis Solution
(Thermo Fisher Scientific, Inc.). The protein concentration was
determined using a BCA protein assay kit (OriGene Technologies,
Inc.) and 25 µg of protein was loaded per lane. The primary
antibodies used were specific for β-actin (1:1,000; cat. no.
AB8227, Abcam), for the detection of ionized calcium binding
adaptor molecule 1 (iba-1; 1:1,000; no. AB5076, Abcam), and for the
detection of MBP (1:500, no. sc-271524, Santa Cruz Biotechnology,
Inc.). Protein extracts were separated by electrophoresis on 12%
SDS-PAGE gels and transferred onto polyvinylidene fluoride
membranes. The membranes were blocked in 5% non-fat milk for 2 h at
room temperature and washed three times in PBS with Tween-20
(0.05%). The membranes were sequentially incubated at 4°C with
primary antibodies (2 h) and secondary antibodies (1 h; 1:1,000;
no. AB205718, Abcam) and enhanced chemiluminescence (ECL) solution.
Finally, the images were captured and analyzed by using ImageJ
v1.41 software (National Institutes of Health).
Statistical analysis
Data from experiments performed in triplicate were
analyzed by One-way ANOVA, and multiple comparisons between groups
were performed using the Student-Newman-Keuls method. The results
are shown as the mean ± standard error of mean. P<0.05,
**P<0.01 or ***P<0.001 was considered to indicate a
statistically significant difference.
Results
Effect of tuftsin on the activation of
microglia in EAE
To explore the function of microglia in EAE, the
expression of microglia in mice induced with tuftsin or PBS was
first examined. EAE mice were induced with the MOG35-55 peptide.
The expression levels of all groups were compared using western
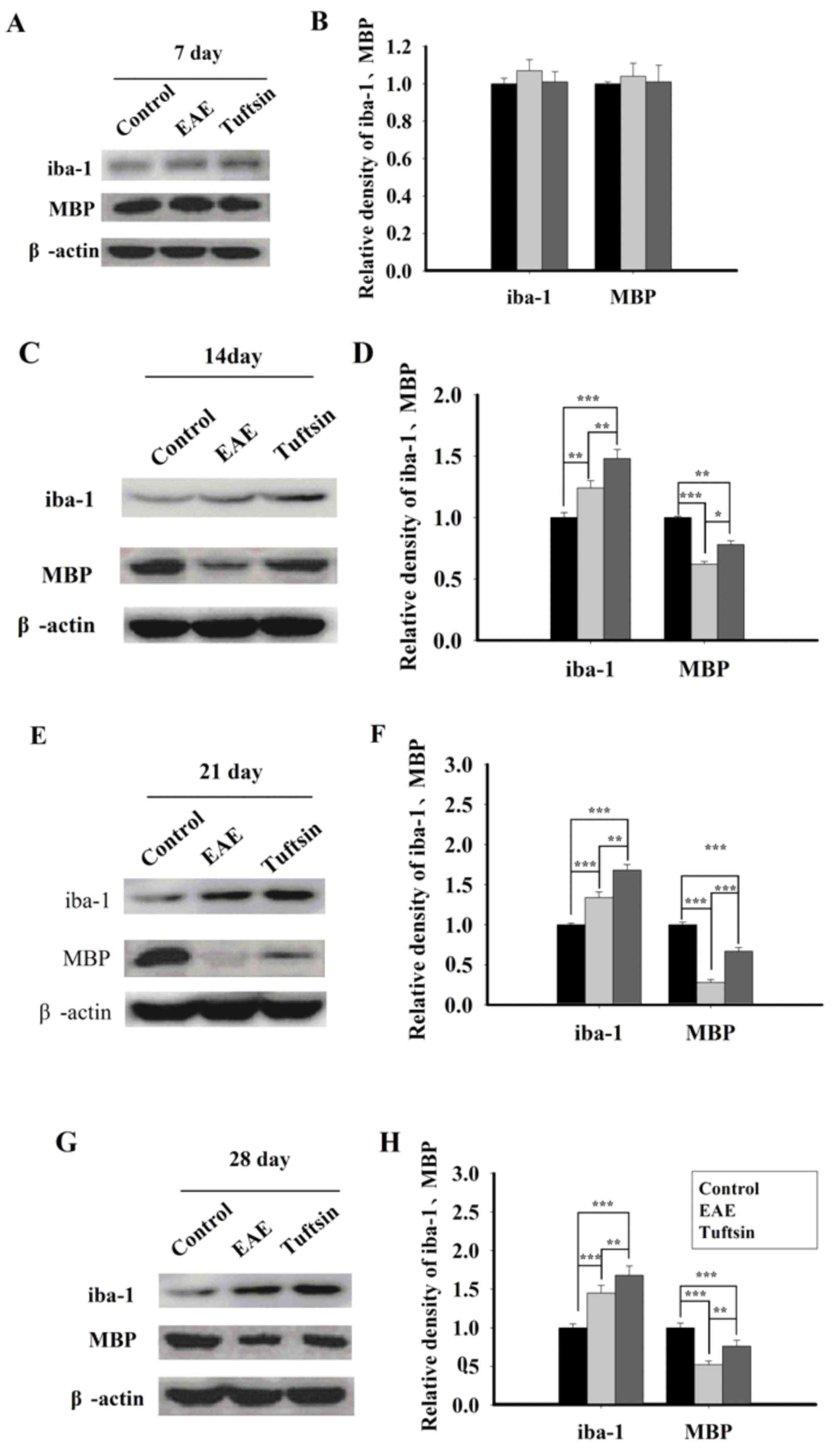
blotting at different time points of EAE. As shown in Fig. 1, western blots indicated increases
in iba-1 and MBP expression in the spinal cords of tuftsin-treated
mice compared with those of the EAE mice on day 7 (P>0.05), day
14 (P<0.01), day 21 (P<0.01), and day 28 (P<0.01). In a
comparison of the EAE group and the control group, expression of
iba-1 in EAE group demonstrated notably higher expression than the
control group on day 14 (P<0.01), day 21 (P<0.001), and day
28 (P<0.001); the EAE group demonstrated significantly higher
expression of MBP compared with the control group on day 14
(P<0.001), day 21 (P<0.001) and day 28 (P<0.001).
Effect of tuftsin on EAE clinical
scores
Clinical scores are one of the validated behavioral
methods used to evaluate the symptoms of EAE in mice. Following
induction of EAE with MOG35-55 or PBS, the symptoms of EAE mice
with and without tuftsin treatment were compared by using clinical
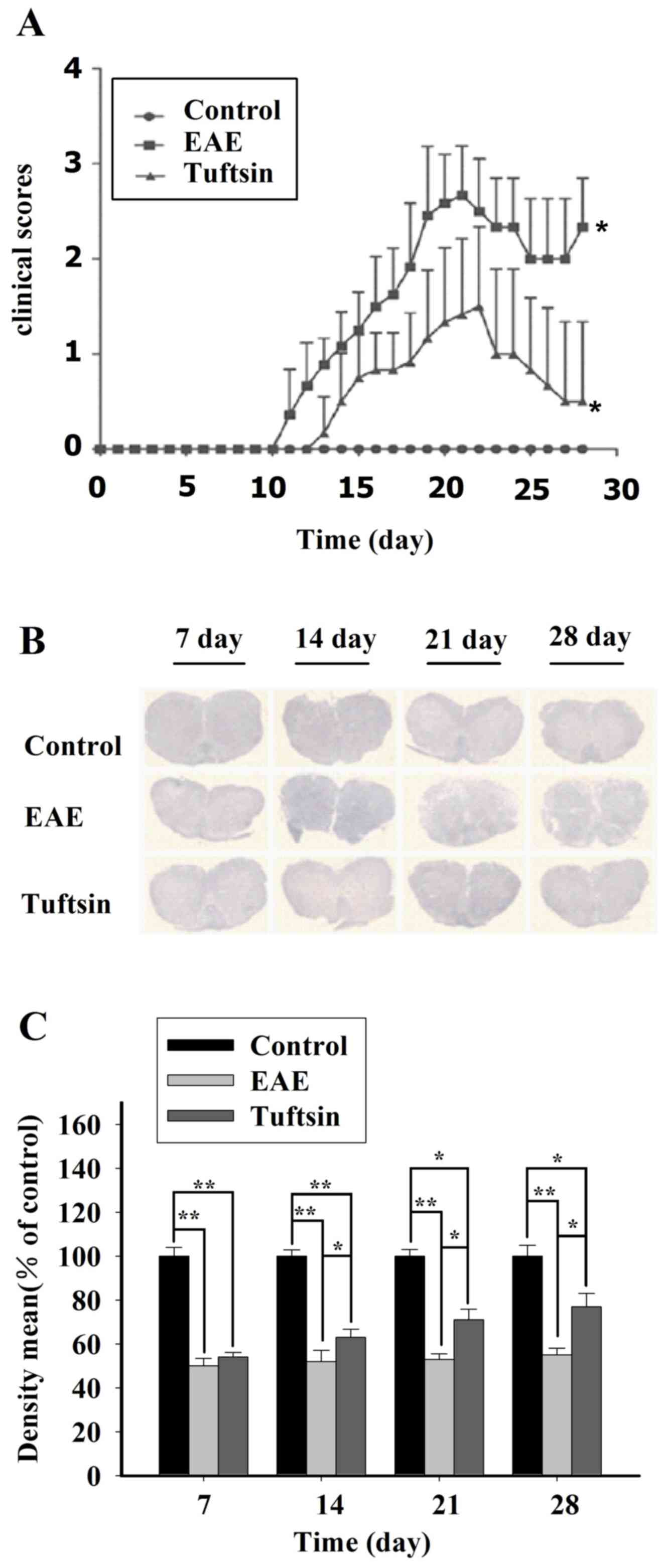
score testing. In Fig. 2A, we
observed that in the acute phase of EAE, all groups had similar
clinical scores. The clinical scores of the EAE and tuftsin groups
changed from day 11; however, the EAE and tuftsin groups achieved a
higher clinical score than the control group on day 14, day 21 and
day 28 (P<0.05). However, during the progression of EAE, the
control group had no mice without the disease. Notably, EAE mice
treated with tuftsin had a significantly lower clinical score than
did mice treated with PBS on day 14 (P<0.05), day 21 (P<0.05)
and day 28 (P<0.05). These results demonstrated that tuftsin
treatment can ameliorate the behavior impairment induced by
MOG35-55.
Remyelination was increased when the
activation of microglia was stimulated
A typical pathological change during EAE development
is demyelination, which can be evaluated utilizing LFB staining. To
investigate this aspect comprehensively, a histological examination
of spinal cord tissue isolated from mice in the control, EAE and
tuftsin groups was performed at different time points of the EAE
process. The spinal cord tissue sections were subjected to LFB
staining in which myelin is stained blue to green,
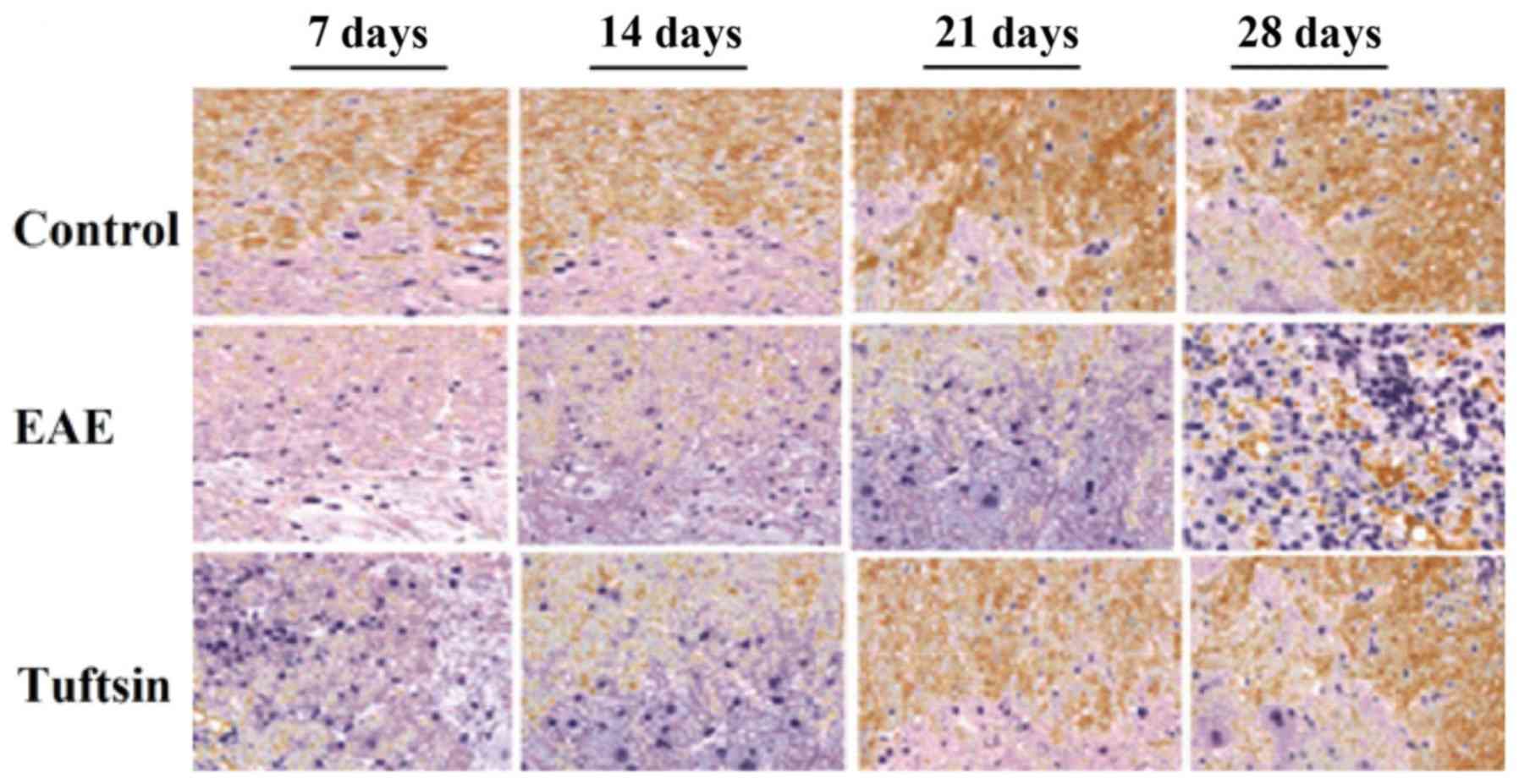
immunohistochemical examination. As shown in Figs. 2B and C, and 3, significant demyelination was not
observed in any group on day 7. At day 14, the clinical onset
point, LFB staining of the EAE group demonstrated slight
demyelination in the white matter. However, in the tuftsin group,
the demyelination was less intense at the same time point. The
structure of the myelin sheath in the control group, however,
exhibited integrity. At the peak of the EAE process, day 21, a
gradual but obvious change in the myelin sheath revealed severe
deficiency in the EAE group. In contrast, demyelination was milder
in the tuftsin group. On day 28, during the recovery stage of EAE,
the loss of myelin did not continue but remyelination began,
particularly in the tuftsin group, in which remyelination was more
obvious than in the EAE group, showing that the structure of the
myelin sheath was relatively intact. It should be noted that the
structure of myelin in the control group remained unaltered.
Microglia subset alteration alleviates
EAE symptoms
Microglia were demonstrated to have M1 and M2
subsets, which represent pro- and anti-inflammatory populations,
respectively (12). TNF-α and
nitric oxide were secreted by M1 microglia, while neuroprotective
M2 microglia produced protective cytokines, including IL-10 and
TGF-β. M2 microglia have been reported to serve a protective role
in EAE by ameliorating experimental autoimmune encephalomyelitis
(13). To test whether M1 or M2
microglia contributed to the severity of EAE during development,
the concentrations of cytokines produced by M1 or M2 microglia were
detected. Total RNA was extracted the from the brains from each
group and RT-qPCR performed to determine the microglial phenotype
based on TNF-α levels for M1, and IL-10 and TGF-β levels for M2.
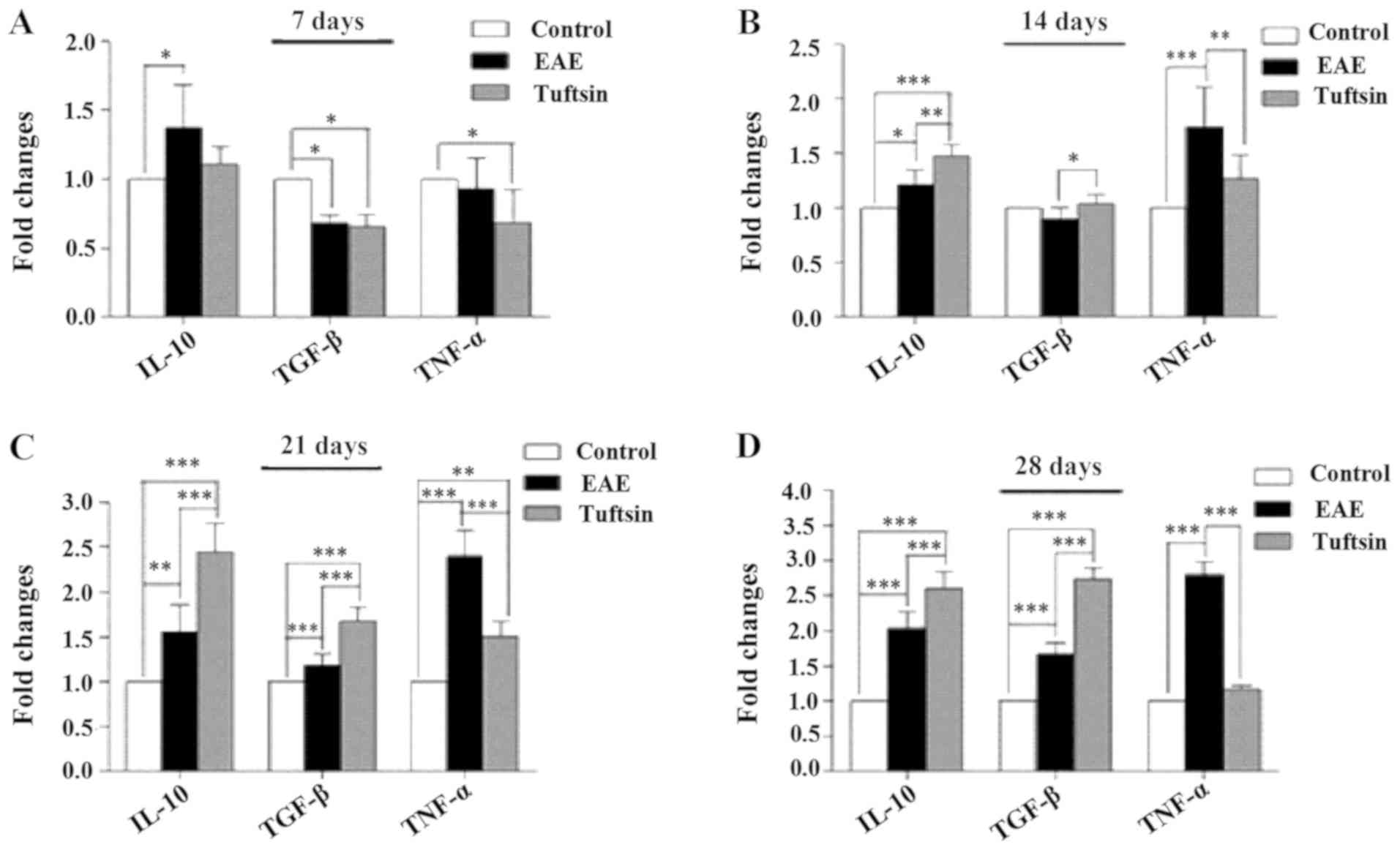
Fig. 4 shows that the amounts of
TNF-α and TGF-β were reduced in the tuftsin group and that the
level of IL-10 was increased in the EAE group. The levels of TNF-α,
IL-10 and TGF-β were increased at day 14 in the tuftsin group and
EAE group, and the levels of IL-10 and TGF-β in the
tuftsin-treatment group exceeded those in the EAE group (P<0.01
and P<0.05, respectively), but the level of TNF-α was less than
that in the EAE group (P<0.01). At days 21 and 28, a comparison
of the levels of IL-10 and TGF-β in the tuftsin treatment group
with those in the EAE group revealed that the former increased more
than the latter (P<0.001, P<0.001); however, the level of
TNF-α demonstrated the opposite pattern. The control group
generated little TNF-α, IL-10 and TGF-β. Collectively, these data
indicated that microglial activation did not contribute to the
development of EAE but rather the secretion of protective
cytokines, which ameliorated the progression of clinical EAE. Thus,
these experiments indicated that microglia released
anti-inflammatory cytokines, improving remyelination.
Discussion
The present study observed the role of microglia in
EAE progression and the findings demonstrated that demyelination
increased during EAE progression before peaking. By increasing
microglial activation, the disease severity of EAE could be
suppressed in the chronic phase. However, the recovery of EAE mice
occurred earlier in the tuftsin-treated group than in the EAE
group, correlating with microglial activation. Generally,
activation of microglia reduced EAE severity and advanced the
recovery of EAE in C57BL/6 mice.
To investigate the implications of microglia for MS,
EAE was induced with MOG35-55 in C57B6/L mice, which is the
commonest animal model used to study MS (14). Then, clinical score assessment, a
widely accepted behavioral method for evaluating EAE symptoms, was
used to observe the variation among the mice. In the present study,
mice subjected to MOG35-55 exhibited weakness in the tail and hind
limbs. The results of subsequent clinical score testing
demonstrated that the mice treated with tuftsin achieved markedly
better scores relating to limb weakness and performance compared
with their counterparts which did not receive tuftsin, especially
during the recovery phase.
Subsequently, the variation of pathology, including
LFB staining, was assessed in in the spinal cord because LFB is
considered an index of demyelination, which is a widely accepted
model for assessing the degree of demyelination at the pathology
level (15). LFB staining revealed
not only intense mitochondrial staining but also staining for
demyelinating fibers both peripherally and in the CNS (16), which was widespread in myelin
staining. An increase in myelination in the EAE group was detected
compared with the control group and the tuftsin group, which could
promote microglia; this effect decreased in the spinal cord
compared with the EAE group, especially during the recovery phase.
These data indicated that in the recovery phase, microglia could
improve remyelination and attenuate the severity of EAE
symptoms.
The role of microglia in demyelinating
neurodegenerative diseases, MS and its animal model EAE is still
controversial (6). Previous data
have shown that microglia serve a detrimental role and encompass a
wide range of harmful factors, such as serving a toxic role in
neurons and oligodendrocyte precursor cells, and releasing
proteases, inflammatory cytokines and free radicals (17), as well as recruiting and promoting
T lymphocyte reactivity in the CNS (18,19).
Emerging evidence, however, has demonstrated that microglia serve
beneficial roles in the brain during neurodegenerative disease
(5), particularly in the recovery
of EAE progression. The precise mechanisms involved in the
protective role of microglia remain unclear, but evidence indicates
that microglia serve a vital role, including axonal regeneration,
assistance with remyelination, efficient removal of injured myelin
debris and release of trophic support factors (6). By phagocytosing cellular debris
(20) and releasing trophic
support factors and anti-inflammatory cytokines, microglia can
protect neurons from injury and repair the injured brain and spinal
cord (21). Furthermore, all of
these effects contribute to myelin regeneration in MS and EAE, in
which remyelination occurs spontaneously (22).
One mechanism of remyelination is the efficient
clearance of myelin debris that contains inhibitory factors for
axonal growth, which is a critical step during the remyelination
progress (23). Microglia, the
resident immune cells in the CNS, could serve a phagocytic role
similar to that of other cells of monocytic origin. Microglial
phagocytosis has been suggested to remove myelin debris and
apoptotic cell debris during demyelination and is beneficial to
promoting regeneration (21).
Independent studies have shown that microglia function through
lectin-, integrin-, and phosphatidylserine-mediated recognition of
apoptotic neurons, followed by phagocytosis and clearance of
apoptotic neurons (24). In the
present study, administering tuftsin promoted phagocytosis for
microglia expressing tuftsin receptors and enhanced the
phagocytosis of microglia. Therefore, tuftsin treatment may
upregulate the role of phagocytosis by combining with its receptor
on microglia, which could efficiently clear the debris from
disrupted myelin sheaths and injured neurons and may maintain an
environment favorable for remyelination.
One important factor in remyelination is
oligodendrocyte precursor cells (25), which migrate to the demyelination
lesion, differentiate into myelinating oligodendrocytes and
synthesize myelin (26). A
previous study demonstrated that remyelination occurs during
demyelination progression, in which endogenous oligodendrocyte
precursor cells (OPCs) intend to remyelinate, but this process
fails (25). In addition, blocking
the differentiation of oligodendroglial progenitor cells causes
remyelination failure during MS progression (27). A study indicated that different
states of microglia were related to oligodendrogenesis by producing
IGF-1 and downregulating TNF-α (28). Microglia have been demonstrated to
present a cytokine and chemokine repertoire, which enables them to
activate and recruit endogenous OPCs to the MS lesion and transmit
trophic support during remyelination.
Inflammatory autoimmune attack against myelin in the
CNS leads to myelin destruction and demyelinated lesion formation
(plaques) in MS (29). Thus,
inhibition of the inflammatory response, instead of improving
anti-inflammation, contributes to remyelination. Microglia serve an
important role in the pathology and pathogenesis of MS. A previous
study suggested that microglia might express immune receptors,
nod-like receptors (NLRs), which are intracellular receptors
capable of suppressing inflammation and ameliorating EAE symptoms
(30). In addition, deregulation
of cytokines (M1 vs. M2) has been reported to be involved in the
pathogenesis of remyelination (31). An previous study demonstrated that
microglia/macrophages contribute to driving oligodendrocyte
differentiation during CNS remyelination by shifting the phenotype
from proinflammatory M1 to anti-inflammatory M2, which triggers
remyelination (32). The M2
phenotype of microglia secrete IL-10 and TGF-β, providing evidence
for the induction of an anti-inflammatory M2 phenotype to suppress
innate immunity in affected brain tissue (31). TGF-β is a signature of microglia
and exerts a protective effect against various neuronal insults
(33). Therefore, the M1/M2
balance in EAE was determined by measuring TNF-α as an M1 cytokine
and IL-10 and TGF-β as M2 cytokines. In the present study, EAE
induced a decrease in IL-10 and TGF-β but an increase in TNF-α in
the spinal cord; however, tuftsin treatment normalized these
cytokine levels. On the other hand, a previous study indicated that
inflammation can convert a non-remyelinating context to a
remyelinating context (34).
Together, the results the present study illustrated
the possibility that microglia may be activated towards an
anti-inflammatory state by shifting to the M2 mode, generating a
properly regulated immunosuppressive response. The findings
indicated that microglial activation could significantly enhance
remyelination induced by anti-immunization in mice with EAE.
Diminishing the response to inflammation and enhancing white matter
lesions may result from the neuroprotective effect of microglia.
More studies are required to detail the mechanisms of microglial
activation at the molecular level by ameliorating neurologic
deficits. The results of the present study provide useful
information for considering the intervention of microglial
activation as an alternative treatment choice for multiple
sclerosis.
As for the concentration of the tuftsin, it was
thought to be the optimal concentration. At this concentration,
there were very few adverse reactions and no mortality in the
animals, and this concentration has a good therapeutic effect.
However, there were two animals which appeared to be in intolerable
distress and self-mutilated limbs and culled for this reason. Of
course, in future experiments, we will continue to explore the
concentration of the tuftsin to find the more appropriate
concentration.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZB and FF designed the research study. YL performed
the experiments and research. JH analyzed the data and wrote the
manuscript. All authors contributed to editorial changes in the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All protocols were approved by the Animal Ethics
Committee of the North China University of Science and Technology,
and animal handling was carried out according to the guidelines
from the National Institutes of Health.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xu L, He D and Bai Y: Microglia-mediated
inflammation and neurodegenerative disease. Mol Neurobiol.
53:6709–6715. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kreutzberg GW: Microglia: A sensor for
pathological events in the CNS. Trends Neurosci. 19:312–318. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Suzumura A: Neurotoxicity by microglia:
The mechanisms and potential therapeutic strategy. Fukuoka Igaku
Zasshi. 100:243–247. 2009.(In Japanese). PubMed/NCBI
|
|
4
|
Kim SU and de Vellis J: Microglia in
health and disease. J Neurosci Res. 81:302–313. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rawji KS and Yong VW: The benefits and
detriments of macrophages/microglia in models of multiple
sclerosis. Clin Dev Immunol. 2013:9489762013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Napoli I and Neumann H: Protective effects
of microglia in multiple sclerosis. Exp Neurol. 225:24–28. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rus H, Cudrici C, Niculescu F and Shin ML:
Complement activation in autoimmune demyelination: Dual role in
neuroinflammation and neuroprotection. J Neuroimmunol. 180:9–16.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fridkin M and Najjar VA: Tuftsin its
chemistry, biology, and clinical potential. Crit Rev Biochem Mol
Biol. 24:1–40. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bump NJ and Najjar VA: Tuftsin
(Thr-Lys-Pro-Arg), a natural modulator of macrophage activity:
Further studies. Mol Cell Biochem. 63:137–142. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Terry RL, Ifergan I and Miller SD:
Experimental autoimmune encephalomyelitis in mice. Methods Mol
Biol. 1304:145–160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kigerl KA, Gensel JC, Ankeny DP, Alexander
JK, Donnelly DJ and Popovich PG: Identification of two distinct
macrophage subsets with divergent effects causingeither
neurotoxicity or regeneration in the injured mouse spinal cord. J
Neurosci. 29:13435–13444. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu M, Nissen JC, Chen EI and Tsirka SE:
Tuftsin promotes an Anti-Inflammatory switch and attenuates
symptoms in experimental autoimmune encephalomyelitis. PLoS One.
7:e349332012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baker D and Amor S: Experimental
autoimmune encephalomyelitis is a good model of multiple sclerosis
if used wisely. Mult Scler Relat Disord. 3:555–564. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Salthouse TN: Luxol fast blue G as a
myelin stain. Stain Technol. 39:1231964.PubMed/NCBI
|
|
16
|
Snodgress AB, Dorsey CH and Lacey LB:
Luxol fast blue staining of degenerating myelinated fibers. Anat
Rec. 140:83–90. 1961. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Benveniste EN: Role of
macrophages/microglia in multiple sclerosis and experimental
allergic encephalomyelitis. J Mol Med (Berl). 75:165–173. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bilimoria PM and Stevens B: Microglia
function during brain development: New insights from animal models.
Brain Res. 1617:7–17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rothhammer V and Quintana FJ: Role of
astrocytes and microglia in central nervous system inflammation
Introduction. Semin Immunopathol. 37:575–576. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lampron A, Larochelle A, Laflamme N,
Préfontaine P, Plante MM, Sánchez MG, Yong VW, Stys PK, Tremblay MÈ
and Rivest S: Inefficient clearance of myelin debris by microglia
impairs remyelinating processes. J Exp Med. 212:481–495. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Program and Abstracts of the 5th Joint
Italian-German Purine Club Meeting, . ‘Fostering translational
research on Purines by Italian-German joint efforts’. Purinergic
Signal. 10:369–417. 2014. View Article : Google Scholar
|
|
22
|
Piaton G, Williams A, Seilhean D and
Lubetzki C: Remyelination in multiple sclerosis. Prog Brain Res.
175:453–464. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang L, Johnson D and Johnson JA:
Deletion of Nrf2 impairs functional recovery, reduces clearance of
myelin debris and decreases axonal remyelination after peripheral
nerve injury. Neurobiol Dis. 54:329–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Witting A, Muller P, Herrmann A,
Kettenmann H and Nolte C: Phagocytic clearance of apoptotic neurons
by microglia/brain macrophages in vitro: Involvement of lectin-,
integrin-, and phosphatidylserine-mediated recognition. J
Neurochem. 75:1060–1070. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bramow S, Frischer JM, Lassmann H,
Koch-Henriksen N, Lucchinetti CF, Sørensen PS and Laursen H:
Demyelination versus remyelination in progressive multiple
sclerosis. Brain. 133:2983–2998. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tanaka T and Yoshida S: Mechanisms of
remyelination: Recent insight from experimental models. Biomol
Concepts. 5:289–298. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kuhlmann T, Miron V, Cuo Q, Wegner C,
Antel J and Brück W: Differentiation block of oligodendroglial
progenitor cells as a cause for remyelination failure in chronic
multiple sclerosis. Brain. 131:1749–1758. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Butovsky O, Landa G, Kunis G, Ziv Y,
Avidan H, Greenberg N, Schwartz A, Smirnov I, Pollack A, Jung S and
Schwartz M: Induction and blockage of oligodendrogenesis by
differently activated microglia in an animal model of multiple
sclerosis. J Clin Invest. 116:905–915. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dutta R and Trapp BD: Mechanisms of
neuronal dysfunction and degeneration in multiple sclerosis. Prog
Neurobiol. 93:1–12. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gharagozloo M, Mahvelati TM, Imbeault E,
Gris P, Zerif E, Bobbala D, Ilangumaran S, Amrani A and Gris D: The
nod-like receptor, Nlrp12, serves an anti-inflammatory role in
experimental autoimmune encephalomyelitis. J Neuroinflamm.
12:1982005. View Article : Google Scholar
|
|
31
|
Mikita J, Dubourdieu-Cassagno N, Deloire
MS, Vekris A, Biran M, Raffard G, Brochet B, Canron MH, Franconi
JM, Boiziau C and Petry KG: Altered M1/M2 activation patterns of
monocytes in severe relapsing experimental rat model of multiple
sclerosis. Amelioration of clinical status by M2 activated monocyte
administration. Mult Scler. 17:2–15. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh
JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin
RJM and Ffrench-Constant C: M2 microglia and macrophages drive
oligodendrocyte differentiation during CNS remyelination. Nat
Neurosci. 16:1211–1275. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Butovsky O, Jedrychowski MP, Moore CS,
Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM,
Doykan CE, et al: Identification of a unique TGF-β dependent
molecular and functional signature in microglia. Nat Neurosci.
17:131–143. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Foote AK and Blakemore WF: Inflammation
stimulates remyelination in areas of chronic demyelination. Brain.
128:528–539. 2005. View Article : Google Scholar : PubMed/NCBI
|