Introduction
Albinism is a rare hereditary disease associated
with an absence of pigment in the eyes, skin and hair, due to
congenital defects in melanocytes. Oculocutaneous albinism and
ocular albinism type 1 (OA1) are the two main subtypes of albinism.
The birth prevalence of OA1 is ~1 in 60,000 (1); however, reports on Chinese patients
with OA1 are rare and epidemiological data are lacking. Compared
with other types of albinism, patients with OA1 appear to be almost
exclusively males and present with only ocular abnormalities. OA1
is characterized by varying degrees of iris depigmentation,
nystagmus, loss of stereoscopic vision, foveal hypoplasia, fundus
hypopigmentation and reduced visual acuity (2–6).
Female carriers do not exhibit clinical symptoms but have spotty or
patchy pigment loss in the fundus, which is also considered a
characteristic of OA1 (7).
OA1 has a Mendelian inheritance pattern and the G
protein-coupled receptor 143 gene (GPR143; OMIM: #300808, NCBI gene
ID: 4935) has been identified as the disease-causing gene. The
GPR143 gene, which has also been reported to cause congenital
nystagmus, possesses nine exons and spans ~40 kb of genomic DNA
(8). GPR143 encodes proteins that
are important for the development and maturation of melanosomes,
and is only localized in the lysosomes and melanosomes of cells
(9). A previous study reported
that OA1 is a disorder involving protein misfolding as a pathogenic
mechanism, rather than the lack of melanin synthesis (10). The typical clinical features of OA1
are iris and fundus depigmentation, nystagmus, foveal hypoplasia,
and normally pigmented skin and hair. In addition, OA1 is often
associated with reduced visual acuity.
Since iris or fundus depigmentation are very slight
and insidious among Asian patients with OA1, it can easily be
misdiagnosed as other congenital eye diseases, such as congenital
idiopathic nystagmus (CIN), although the treatment principle for
OA1 and CIN is the same (11–16).
Molecular diagnosis combined with detailed eye examinations,
including optical coherence tomography (OCT) and detailed slit lamp
examination, are effective tools for differential diagnosis
(17–20). In the present study, the clinical
manifestations were described and a molecular genetic analysis was
performed on a Chinese family with X-linked OA1. Reports of
X-linked OA1 in Asian populations are relatively rare (7–16);
therefore, the present results may enrich the mutation spectrum of
GPR143 in the Asian population.
Materials and methods
Recruitment of subjects
A total of 18 members of a family affected by OA1
(nine affected patients and nine normal subjects), from Sanya
(Hainan, China), were recruited to the present study in December
2017 at the Department of Ophthalmology, Chinese PLA General
Hospital. This study was approved by the Hospital Ethics Committee
and strictly followed the Helsinki Declaration. All participants
provided written informed consent. Additionally, 100 healthy normal
people (50 men and 50 women) between the age of 18–65 years were
recruited as controls in the present study.
Patient assessment
Detailed family and medical histories were
collected. All participants underwent careful ophthalmologic
testing, which covered the first occurrence of nystagmus,
intraocular pressure, anterior segment of the eyes, best corrected
visual acuity (BCVA), indirect ophthalmoscopy and fundus
photography for vitreous and fundus examination, OCT for retinal
structure examination and electrophysiological assessment.
Genetic mutation screening
Blood samples (8–10 ml) were collected from six
affected patients and seven normal individuals within the 18 family
members. Genomic DNA was extracted from blood lymphocytes using a
SeqCap EZ MedExome target enrichment kit (Tiangen Biotech Co.,
Ltd.) according to the manufacturer's protocol. A high-throughput
sequencer Illumina HiSeq2500 Analyzer (Illumina, Inc.) was used for
continuous bidirectional sequencing for 90 cycles and the raw
sequencing numbers were read using Illumina Pipeline software
(version 1.3.4; Illumina, Inc.). SOAPsnp software
(SOAPsnp-v1.03.tar.gz; http://soap.genomics.org.cn) and Samtools software
(Samtools version 0.1.19; http://samtools.sourceforge.net/) were used to perform
single nucleotide variant and insertion and deletion analysis, in
order to generate the target region base polymorphism results. The
primer and Illumina sequencing reaction conditions were performed
by Beijing Huada Gene Technology, Ltd. The coding exon and intron
sequences of the GPR143 gene were amplified by polymerase chain
reaction (PCR). The PCR reaction conditions were as follow: Initial
denaturation at 95°C for 5 min, followed by 35 cycles at 94°C for
30 sec, annealing at 58°C for 30 sec and 72°C extension for 30 sec,
and a final extension cycle at 72°C for 5 min. The reaction
products were purified using the Purification kit (Qiagen GmbH) and
BigDye Terminator v3.1 Cycle Sequencing kit (Thermo Fisher
Scientific, Inc.) was used for Sanger Sequencing, according to the
manufacturer's protocol. The genes were read using an ABl3130
sequencer (Thermo Fisher Scientific, Inc.), in accordance with the
manufacturer's protocol. The sequencing outcomes were analyzed
using Chromas 2.0 (Technelysium Pty Ltd.) and DNAStar 8.0 software
(http://www.dnastar.com). The sequencing results
were compared with the Reference Sequence (RefSeq; release 34;
http://www.ncbi.nlm.nih.gov/LocusLink/refseq.html)
database, and the identified novel mutation was named according to
the nomenclature recommended by Human Genome Variation Society
(http://www.hgvs.org/). Sanger sequencing was also
performed on the GPR143 gene in samples collected from 100 healthy
individuals.
Results
Clinical phenotype
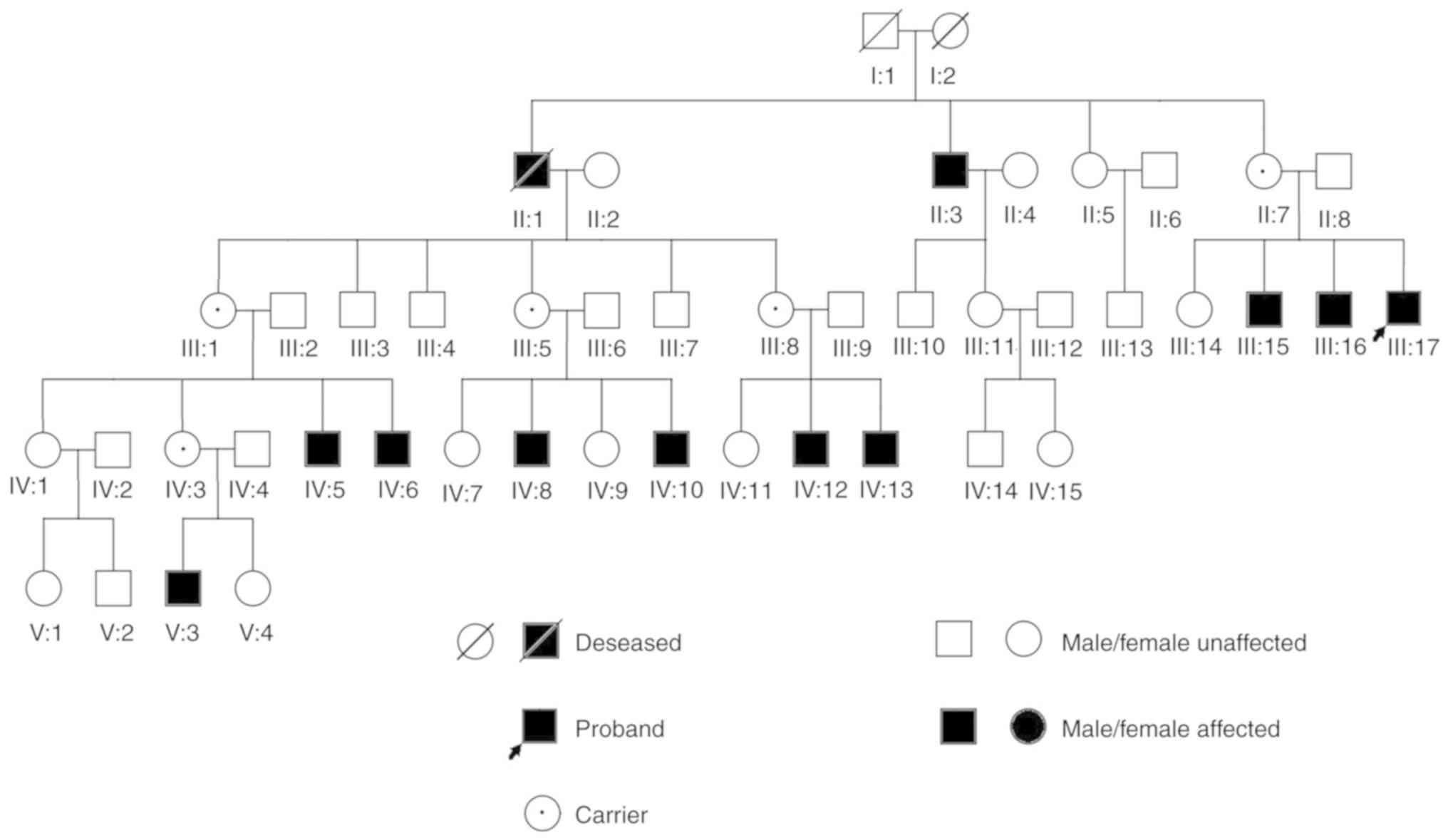
All of the affected patients in the recruited family
were male and were the offspring of female carriers of the
mutation; men with the mutation develop the disorder, whereas women
can be carriers or healthy individuals. This is in line with
X-linked recessive inheritance (Fig.
1). All patients with OA1 exhibited different degrees of
horizontal nystagmus and low BCVA. The patients exhibited nystagmus
from birth to 6 months of age, and the BCVA data ranged between 0.1
and 0.4. All carriers presented normal BCVA and no symptoms
(Tables I and II).
 | Table I.Clinical features of patients with
ocular albinism type 1 in a Chinese family. |
Table I.
Clinical features of patients with
ocular albinism type 1 in a Chinese family.
| ID | Sex | Age (years) | BCVA
(right/left) | Iris
hypopigmentation | Fundus
hypopigmentation | CN | Macular
hypoplasia | Coloboma
chorioideae |
|---|
| II:3 | Male | 65 | 0.12/0.2 | Yes | Yes | Yes | Yes | No |
| III:15 | Male | 29 | 0.15/0.25 | Yes | Yes | Yes | Yes | No |
| III:16 | Male | 27 | 0.12/0.12 | No | Yes | Yes | Yes | Yes |
| III:17 | Male | 23 | 0.25/0.25 | Yes | Yes | Yes | Yes | No |
| IV:6 | Male | 22 | 0.3/0.25 | Yes | Yes | Yes | Yes | No |
| IV:8 | Male | 19 | 0.25/0.3 | Yes | Yes | Yes | Yes | No |
| IV:10 | Male | 12 | 0.25/0.3 | Yes | Yes | Yes | Yes | No |
| IV:12 | Male | 15 | 0.25/0.3 | Yes | Yes | Yes | Yes | No |
| IV:13 | Male | 10 | 0.2/0.25 | Yes | Yes | Yes | Yes | No |
| Table II.Clinical features of carriers of
ocular albinism type 1 in a Chinese family. |
Table II.
Clinical features of carriers of
ocular albinism type 1 in a Chinese family.
| ID | Sex | Age (years) | BCVA
(right/left) | Iris
hypopigmentation | Fundus
hypopigmentation | CN | Macular
hypoplasia | Coloboma
chorioideae |
|---|
| II:7 | Female | 58 | 0.5/0.6 | Slight | Slight | No | No | No |
| III:1 | Female | 51 | 0.8/0.8 | Slight | Slight | No | No | No |
| III:5 | Female | 47 | 1.0/1.0 | Slight | Slight | No | No | No |
| III:8 | Female | 41 | 1.0/1.0 | Slight | Slight | No | No | No |
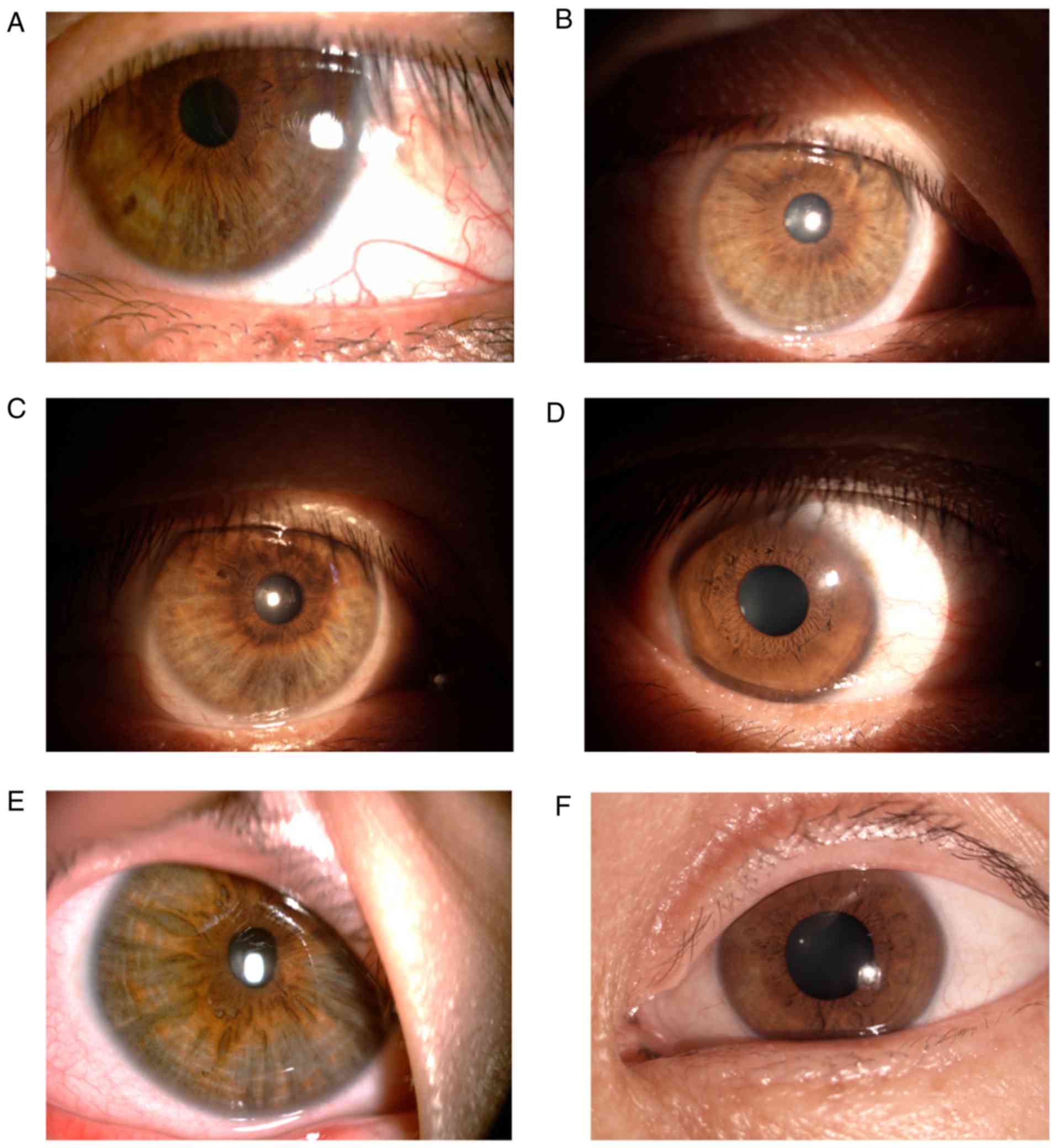
Using slit lamp examination, nine patients were
observed to have different degrees of abnormal iris pigmentation.
Some patients exhibited peripheral iris depigmentation in a ring or
fan and there was only one patient who did not present
depigmentation of the iris. In addition, a slight depigmentation of
the iris was also observed in all the carriers (Fig. 2).
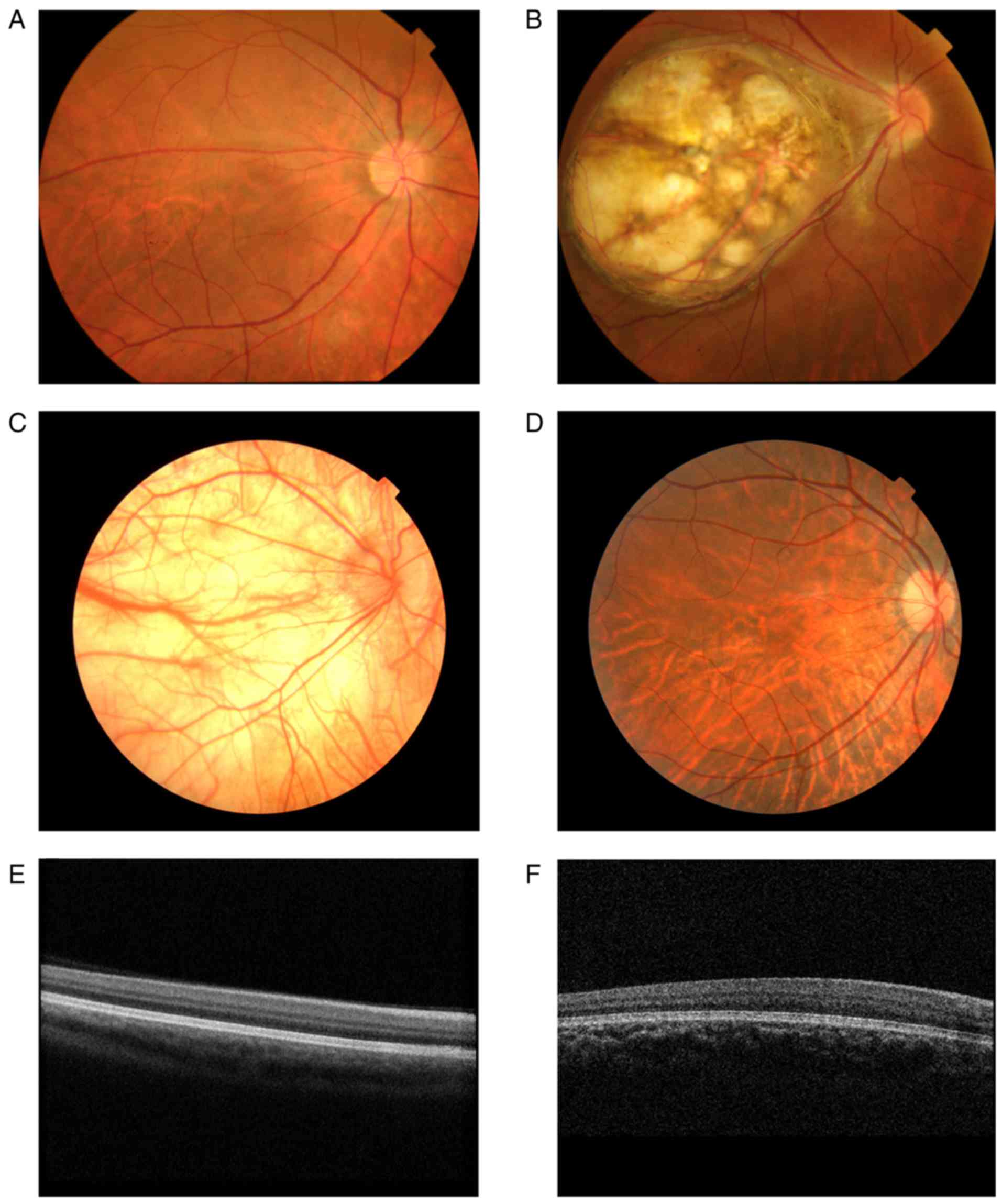
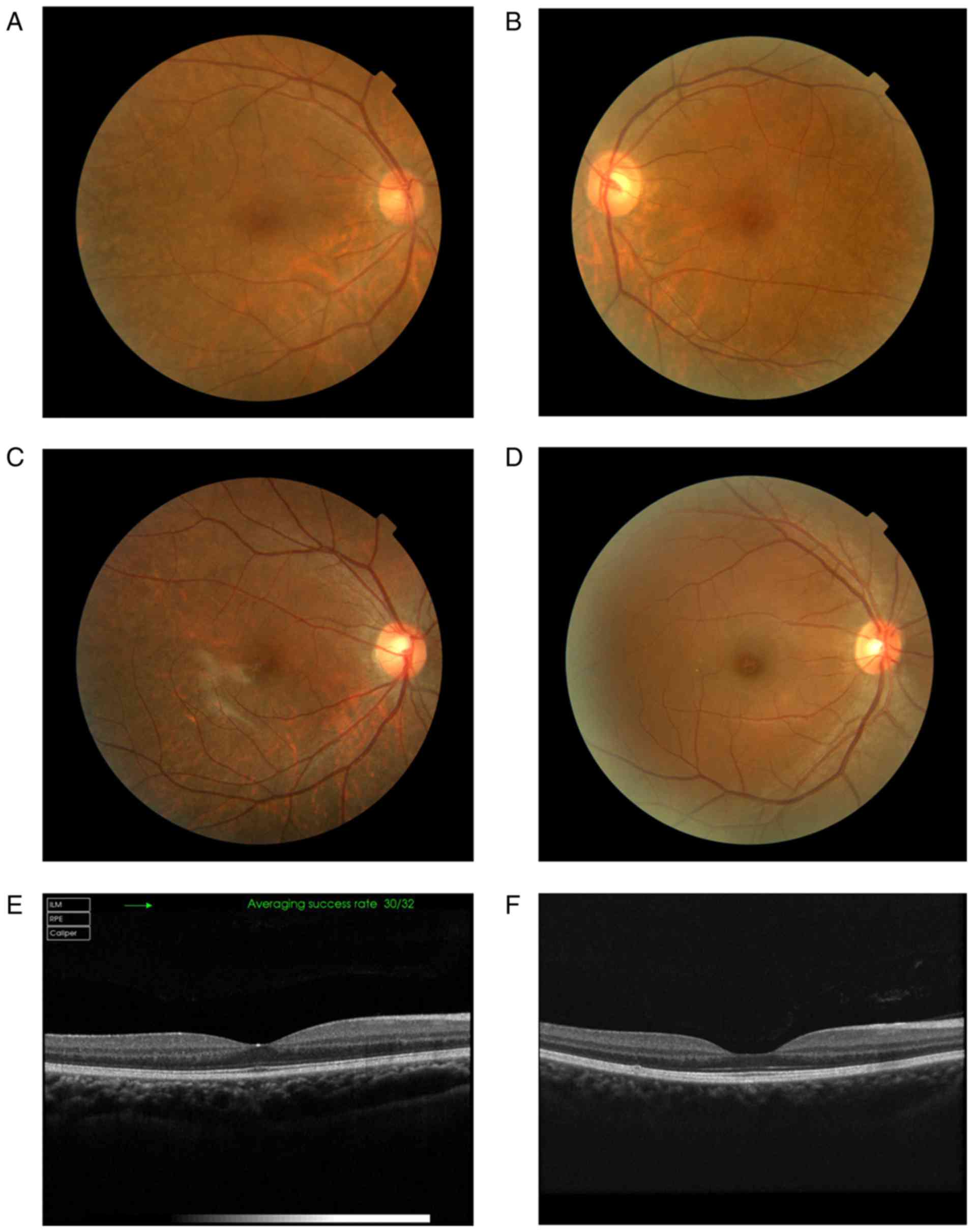
According to the fundus examination, nine patients
presented different degrees of retinal hypopigmentation and foveal
hypoplasia. The choroidal blood vessels of some patients were
clearly visible due to retinal depigmentation (Fig. 3A). In addition, one patient
(III:16) had a normal iris with a large choroid membrane coloboma
at the fundus of the eye (Fig.
3B). The fundi of some patients presented irregular retinal
alternating shades of pigment, which resembled highly myopic eyes
(Fig. 3D). OCT examination could
not visualize the macular foveal structures in nine patients
(Fig. 3E and F). The participants
in this study presented with normal skin and hair color. Only one
patient (IV:13) exhibited light brown hair with complete
depigmentation at the fundus (Fig.
3C). In addition, the carriers with normal foveae exhibited
slight spotty depigmentaton in the peripheral fundus of the eyes
(Figs. 3 and 4).
Mutation analysis
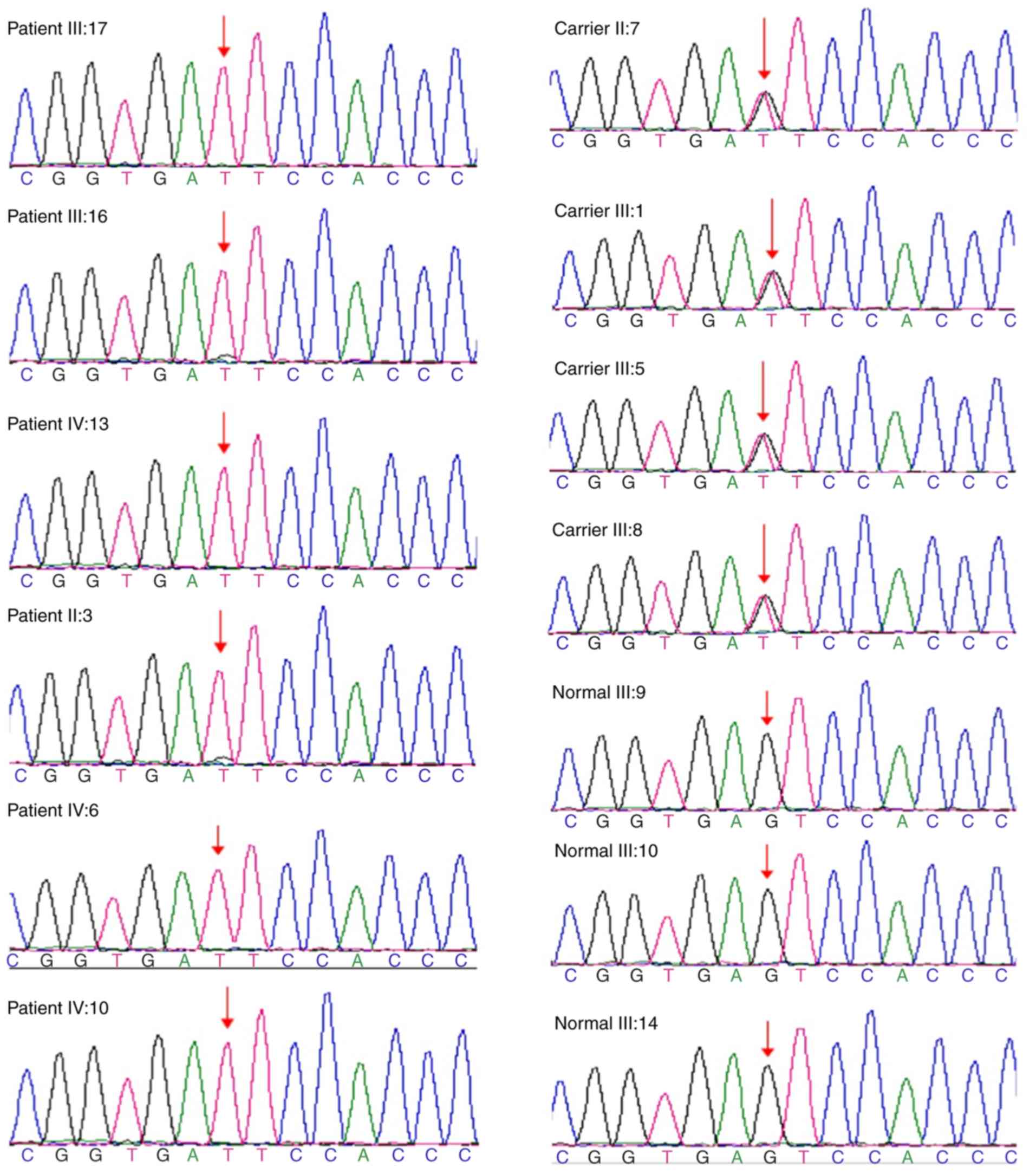
Following Illumina sequence analysis of the GPR143
gene, the proband III:17 and 5 patients with OA1 underwent mutation
analysis; the hemizygous mutation c.360+5G>T in GPR143 gene was
detected on II:3, III:16, III:17, IV:6, IV:10 and IV:13 in this
family (Fig. 5). However, the
clinical significance of this mutation was not clear.
Validation
Sanger sequencing of the GPR143 gene c.360+5G>T
site was performed on 12 participants of this family, with the
exception of the proband III:17 (sequencing was completed by
Beijing Huada Gene Technology, Ltd.). The results revealed that
four cases (II:7, III:1, III:5 and III:8 carriers) exhibited
heterozygous mutations and three cases (III:9, III:10, and III:14
normal individuals) did not possess the mutation (Fig. 5).
The mutation c.360+5G>T described was absent in
the 100 normal controls. These findings indicated that the
c.360+5G>T mutation in the GPR143 gene was a novel mutation site
that leads to OA1. This mutation may be the pathogenic mutation
site of the recruited family.
Discussion
The present study described the clinical features of
a Chinese family with OA1; a novel mutation in the GPR143 gene
(c.360+5G>T) was detected in this family. All patients had
normally pigmented skin and hair but presented with different
degrees of iris depigmentation, horizontal jerk nystagmus, low BCVA
and foveal hypoplasia.
There have been many reports in Caucasian
individuals regarding the clinical characteristics and pathogenic
mechanism of OA1 (21,22). In addition, it has been reported
that OA1 in African-American patients presents as non-albinotic,
with no translucency of the iris and a moderately pigmented fundus;
however, during ophthalmoscopic examination, these patients always
present foveal hypoplasia (23).
It has been reported that Japanese patients with OA1 exhibit fundus
pigmentation to a degree somewhere between Caucasian patients and
patients of African descent (24).
In 2008, Fang et al (13)
first reported clinical studies of OA1 in the Chinese population.
It has been demonstrated that regardless of race, all patients with
OA1 consistently exhibit signs of foveal hypoplasia (14,20–24).
It is thought that ~80% of heterozygous female carriers exhibit an
alternating pattern of streaks of pigment with streaks of low
pigment in the fundus (25). This
is consistent with the present study. This study revealed that even
in female carriers, one X chromosome carried the disease gene,
whereas the other X chromosome carried the normal GPR143 gene,
which can induce the expression of the normal OA1 protein.
The GPR143 gene, which spans ~40 kb and encodes a
404 amino acid membrane glycoprotein, is located on chromosomal
region Xp22.3. The GPR143 protein is mainly expressed in the iris,
retinal pigment epithelium and melanocytes (19,26).
As a receptor of tyrosine, levodopa and dopamine, the GPR143
protein has been reported to regulate the early stages of
melanosome biogenesis, organization and signal transduction
(27,28). Until now, the Human Gene Mutations
Database (http://www.hgmd.cf.ac.uk) has
described >100 mutations in the GPR143 gene that have been
reported to be responsible for OA1.
OA1 is easily misdiagnosed as other diseases,
particularly in East Asian patients, as patients with brown irises
usually have no typical iris hypopigmentation or albinotic type of
retinal pigment. In the present study, patients were initially
misdiagnosed as having CIN, as CIN exhibits similar features to
those of OA1. Therefore molecular testing combined with
comprehensive clinical analysis is a good method for accurate
diagnosis, particularly when clinical symptoms are conflicting. The
patients within the present family exhibited congenital nystagmus
between birth and 6 months of age; the symptoms included horizontal
pendular nystagmus, which was accompanied by head tremor, amblyopia
and poor lateral vision. All patients presented different degrees
of retinal hypopigmentation in the fundus as well as severe foveal
hypoplasia. It was speculated that there may be possible linkage
inheritance of albinism with nystagmus. It has been confirmed that
the GPR143 gene is both the pathogenic gene of OA1 and the
pathogenic candidate gene of congenital nystagmus (16–19).
However, how the two diseases interact with each other requires
further study. In the examination of the anterior segment, all
patients with OA1, with the exception of one, exhibited iris
depigmentation in the shape of a ring or fan. This is consistent
with previous reports in Chinese OA1 families (14,29).
This study confirmed the GPR143 gene mutation
through Sanger sequencing and detected a hemizygous mutation
(c.360+5G>T) in the affected family; this mutation is a splicing
mutation. The mutation was detected in six patients in this family
and was later verified to be absent in 100 healthy people who did
not have the disease and had no family history of OA1. Therefore,
the novel splicing mutation in the GPR143 gene, c.360+5G>T, was
identified as the pathogenic mutation of this OA1 family. However,
the prevalence of this GPR143 mutation in Chinese patients is
unclear.
The novel mutation c.360+5G>T is located in the
shearing area after exon 4 of the GPR143 gene, changing the shear
of RNA and affecting the stability and translation of RNA. Splicing
mutations are a type of mutation that changes the splicing mode of
RNA precursors due to a mutation of the splicing donor, receptor
site or its side conservative sequence, which results in mature RNA
containing a class of mutations that contain intron or missing exon
sequences. In the present study, the fifth intron in the shearing
area after exon 4 was mutated from the original G base to a T base,
thereby resulting in a change from AGT-serine to ATT-isoleucine;
this alteration may lead to abnormal functional or structural
characteristics of terminal protein products. Therefore, this may
be the ultimate cause of the disease in this affected family. It
has been speculated that transcriptional mutations may lead to a
reduction in the function of the nonsense-mediated mRNA degradation
pathway, thus leading to the generation of truncated proteins that
affect function (30,31); however, the specific pathogenesis
requires further study and confirmation.
In conclusion, the present study identified a novel
mutation in the GPR143 gene in a Chinese family affected by OA1;
the mutation c.360+5G>T was successfully located. This study
expanded the mutation spectrum of the GPR143 gene, particularly
enriching our current knowledge on the GPR143-associated OA1,
thereby supporting future genetic diagnosis and treatment of OA1.
The results of this study, combined with other novel mutations
affecting the GPR143 protein, may provide a basis for further
proteomics research. Genetic analysis, as well as careful clinical
examination, may contribute to the accurate diagnosis of disorders
and may inform genetic counseling, providing information about
prognosis and avoiding unnecessary and inappropriate
interventions.
Acknowledgements
The authors would like to thank Miss Hong Jiang and
Mr Bing Chen of Chinese PLA General Hospital, for contributing to
fundus photography and optical coherence tomography used in the
present study.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
TCL made substantial contributions to the conception
and design of the current study. MNZ performed test method
guidance. LW collected blood specimen. AD collected the clinical
data. XC analyzed and interpreted the patient data. RPL analyzed
the sequencing results. XHG interpreted the data and wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by The Hospital
Ethics Committee and strictly followed the Helsinki Declaration.
All participants provided written informed consent.
Patient consent for publication
All participants provided written informed consent
for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rosenberg T and Schwartz M: X-linked
ocular albinism: Prevalence and mutations-a national study. Eur J
Hum Genet. 6:570–577. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Preising M, Op de Laak JP and Lorenz B:
Deletion in the OA1 gene in a family with congenital X linked
nystagmus. Br J Ophthalmol. 85:1098–1103. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sallmann GB, Bray PJ, Rogers S, Quince A,
Cotton RG and Carden SM: Scanning the ocular albinism 1 (OA1) gene
for polymorphisms in congenital nystagmus by DHPLC. Ophthalmic
Genet. 27:43–49. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lam BL, Fingert JH, Shutt BC, Singleton
EM, Merin LM, Brown HH, Sheffield VC and Stone EM: Clinical and
molecular characterization of a family affected with X-linked
ocular albi-nism (OA1). Ophthalmic Genet. 18:175–184. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
O'Donnell FE Jr, King RA, Green WR and
Witkop CJ Jr: Autosomal recessively inherited ocular albinism. A
new form of ocular albinism affecting females as severely as males.
Arch Ophthalmol. 96:1621–1625. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cortin P, Tremblay M and Lemagne JM:
X-linked ocular albinism: Relative value of skin biopsy, iris
transillumination and funduscopy in identifying affected males and
carriers. Can J Ophthalmol. 16:121–123. 1981.PubMed/NCBI
|
|
7
|
Zou X, Li H, Yang L, Sun Z, Yuan Z, Li H
and Sui R: Molecular genetic and clinical evaluation of three
Chinese families with X-linked ocular albinism. Sci Rep.
6:337132017. View Article : Google Scholar
|
|
8
|
Bassi MT, Schiaffino MV, Renieri A, De
Nigris F, Galli L, Bruttini M, Gebbia M, Bergen AA, Lewis RA and
Ballabio A: Cloning of the gene for ocular albinism type 1 from the
distal short arm of the X chromosome. Nat Genet. 10:13–19. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Palmisano I, Bagnato P, Palmigiano A,
Innamorati G, Rotondo G, Altimare D, Venturi C, Sviderskaya EV,
Piccirillo R, Coppola M, et al: The ocular albinism type 1 protein,
an intracellular G protein-coupled receptor, regulates melanosome
transport in pigment cells. Hum Mol Genet. 17:3487–3501. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
d'Addio M, Pizzigoni A, Bassi MT,
Baschirotto C, Valetti C, Incerti B, Clementi M, De Luca M,
Ballabio A and Schiaffino MV: Defective intracellular transport and
processing of OA1 is a major cause of ocular albinism type 1. Hum
Mol Genet. 9:3011–3018. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu JY, Ren X, Yang X, Guo T, Yao Q, Li L,
Dai X, Zhang M, Wang L, Liu M and Wang QK: Identification of a
novel GPR143 mutation in a large Chinese family with congenital
nystagmus as the most prominent and consistent manifestation. J Hum
Genet. 52:565–570. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Guo X, Wei A, Zhu W, Li W and Lian
S: Identification of a novel mutation in a Chinese family with
X-linked ocular albinism. Eur J Ophthalmol. 19:124–128. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fang S, Guo X, Jia X, Xiao X, Li S and
Zhang Q: Novel GPR143 mutations and clinical characteristics in six
Chinese families with X-linked ocular albinism. Mol Vis.
14:1974–1982. 2008.PubMed/NCBI
|
|
14
|
Xiao X and Zhang Q: Iris hyperpigmentation
in a Chinese family with ocular albinism and the GPR143 mutation.
Am J Med Genet A 149A. 1786–1788. 2009. View Article : Google Scholar
|
|
15
|
Yan N, Liao X, Cai SP, Lan C, Wang Y, Zhou
X, Yin Y, Yu W and Liu X: A novel nonsense mutation of the GPR143
gene identified in a Chinese pedigree with ocular albinism. PLoS
One. 7:e431772012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cai CY, Zhu H, Shi W, Su L, Shi O, Cai CQ,
Ling C and Li WD: A novel splicing site mutation of the GPR143 gene
in a Chinese X-linked ocular albinism pedigree. Genet Mol Res.
12:5673–5679. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pan Q, Yi C, Xu T, Liu J, Jing X, Hu B and
Wang Y: A novel mutation, c.494C>A (p.Ala165Asp), in the GPR143
gene causes a mild phenotype in a Chinese X-linked ocular albinism
patient. Acta Ophthalmol. 94:417–418. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu J, Liang D, Xue J, Liu J and Wu L: A
novel GPR143 splicing mutation in a Chinese family with X-linked
congenital nystagmus. Mol Vis. 17:715–722. 2011.PubMed/NCBI
|
|
19
|
Han R, Wang X, Wang D, Wang L, Yuan Z,
Ying M and Li N: GPR143 gene mutations in five chinese families
with X-linked congenital nystagmus. Sci Rep. 5:120312015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kinnear PE, Jay B and Witkop CJ Jr:
Albinism. Surv Ophthalmol. 30:75–101. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Charles SJ, Green JS, Grant JW, Yates JR
and Moore AT: Clinical features of affected males with X linked
ocular albinism. Br J Ophthalmol. 77:222–227. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schnur RE, Wick PA, Bailey C, Rebbeck T,
Weleber RG, Wagstaff J, Grix AW, Pagon RA, Hockey A and Edwards MJ:
Phenotypic variability in X-linked ocular albinism: Relationship to
linkage genotypes. Am J Hum Genet. 55:484–496. 1994.PubMed/NCBI
|
|
23
|
O'Donnell FE Jr, Green WR, Fleischman JA
and Hambrick GW: X-linked ocular albinism in Blacks. Ocular
albinism cum pigmento. Arch Ophthalmol. 96:1189–1192. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hayakawa M, Kanai A, Kato K, Nakajima A
and Takamori K: A Japanese family of nettleship falls X-linked
ocular albinism. Nippon Ganka Gakkai Zasshi (Japanese).
94:1181–1187. 1990.
|
|
25
|
Oetting WS: New insights into ocular
albinism type 1 (OA1): Mutations and polymorphisms of the OA1 gene.
Hum Mutat. 19:85–92. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sone M and Orlow SJ: The ocular albinism
type 1 gene product, OA1, spans intracellular membranes 7 times.
Exp Eye Res. 85:806–816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schiaffino MV, d'Addio M, Alloni A,
Baschirotto C, Valetti C, Cortese K, Puri C, Bassi MT, Colla C, De
Luca M, et al: Ocular albinism: Evidence for a defect in an
intracellular signal transduction system. Nat Genet. 23:108–112.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Giordano F, Bonetti C, Surace EM, Marigo V
and Raposo G: The ocular albinism type 1 (OA1) G-protein-coupled
receptor functions with MART-1 at early stages of melanogenesis to
control melanosome identity and composition. Hum Mol Genet.
18:4530–4545. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jia X, Yuan J, Jia X, Ling S, Li S and Guo
X: GPR143 mutations in Chinese patients with ocular albinism type
1. Mol Med Rep. 15:3069–3075. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Inoue K, Khajavi M, Ohyama T, Hirabayashi
S, Wilson J, Reggin JD, Mancia P, Butler IJ, Wilkison MF, Wegner M
and Lupski JR: Molecular mechnism for distinct neurological
phenotypes conveyed by allelic truncating mutations. Nat Genet.
36:361–369. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Holbrook JA, Neu-Yilik G, Hentze MW and
Kulozik AE: Nonsense-mediated decay approaches the clinic. Nat
Genet. 36:801–808. 2004. View
Article : Google Scholar : PubMed/NCBI
|