Introduction
As the fourth leading cause of cancer mortality,
pancreatic cancer (PC) is an aggressive disease, and its global
prevalence has been increasing (1,2). The
5-year overall survival rate of patients diagnosed with PC is
<5% (3). It has been reported
that obesity is one of the risk factors for PC development, and
over the past decade, the rate of obesity has risen at an
unprecedented rate (4,5). Chronic obesity changes the production
and secretion of adipokines, a type of cytokine secreted by the
adipose tissue (6–9).
The adipokine leptin has been reported to be highly
expressed in obese patients (6,7).
Leptin regulates a variety of biological processes, such as
satiety, food intake and energy expenditure, through leptin
receptor (LepR) (10–12). Glucose metabolism, controlled by
cellular ATP and metabolite levels, offers energy and required
substance for tissue anabolism and catabolism, and is essential for
the growth and development of cells and tissues (13,14).
In addition, increasing evidence showed that glucose is an
important regulator for leptin expression and secretion (15–17).
LepR is composed of several isoforms including the short isoform
(LepR-short) and long isoform (LepR-long). It has been reported
that LepR can trigger the activation of several pathways, including
PI3K/AKT and Janus-activated kinase/STAT (18–20).
LepR common variant (LepR-common) is reported to influence
gestation glycemic traits (21).
Leptin is highly expressed in subsets of cancer patients including
breast, colon and prostate cancer, and stimulates various
biological activities in cancer cells, including cell
proliferation, migration and invasion (22–25).
Leptin is a growth factor for colon epithelial cells, and it has
been demonstrated to promote motility and invasiveness in human
colon cancer cells (22,23). Leptin also promotes invasion and
migration of breast cancer cells through transactivation of
epidermal growth factor receptor (23). Pancreatic β-cells have been
demonstrated to express functional LepRs, and LepR-short and
LepR-long in PC cells (4,26). A previous study reported that
leptin signaling enhances human PC cell invasion and metastasis by
increasing matrix metalloproteinase-13 (MMP-13) (27). However, the effects of LepR on the
proliferation and glucose metabolism of human PC, as well as its
underlying mechanisms remain unclear.
In the present study, in vitro leptin
stimulation significantly promoted cell proliferation and enhanced
glucose metabolism of human PC and normal pancreas cells in a
dose-dependent manner, accompanied by an increase in the expression
levels of the glycolytic enzymes hexokinase II (HKII) and glucose
transporter 1 (GLUT1). Silencing of LepR decreased AKT
phosphorylation. Additionally, the induction of leptin stimulation
was significantly counteracted by treatment with an AKT inhibitor
(LY294002), whereas the effect of LepR silencing was counteracted
by the AKT activator insulin-like growth factor 1 (IGF-1). The
results of the present study suggested that leptin may contribute
to human PC cell proliferation and glucose metabolism, which may be
through activation of the AKT pathway.
Materials and methods
Cell culture
Two human PC cell lines, BxPC3 and Panc-1, and
normal pancreas cells HPC-Y5 were purchased from The Cell Bank of
Type Culture Collection of the Chinese Academy of Sciences. The
cells were cultured in a 37°C, 5% CO2 incubator (Thermo
Forma 3111; Thermo Fisher Scientific, Inc.) in RPMI-1640 medium
(cat. no. SH30809.01B; HyClone; GE Healthcare Life Sciences), which
contained 10% FBS (cat. no. 16000-044; Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin/streptomycin (cat. no.
P1400-100; Beijing Solarbio Science & Technology Co., Ltd.).
The medium was refreshed every two days during incubation.
Lentiviral transduction
Short hairpin RNA (shRNA)-1 and shRNA-2 targeting
two different sites of LepR (GenBank no. BC131779.1; Table I) were constructed and inserted
into the Agel I/Ecol I restriction sites of a
pLKO.1-puro vector (Addgene, Inc.). Following confirmation by DNA
sequencing (Shanghai Majorbio Pharmaceutical Technology Co., Ltd.),
LepR-shRNA-1 or LepR-shRNA-2 was co-transfected with the viral
packaging plasmids psPAX2 and pMD2G (Addgene, Inc.) into 293T (The
Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences) cells using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) in a 37°C incubator. The virus
particles were collected by ultracentrifugation 48 h
post-transfection.
 | Table I.Leptin receptor interference target
design results. |
Table I.
Leptin receptor interference target
design results.
| Name | Sequence |
|---|
| Leptin receptor
target site 1 (842–860) | Sense
5′-GGGUACUGAGGUAACCUAUUU-3′ |
|
| Antisense
5′-AUAGGUUACCUCAGUACCCUU-3′ |
| Leptin receptor
target site 2 (863–881) | Sense
5′-GGACGAAAGCCAGAGACAAUU-3′ |
|
| Antisense
5′-UUGUCUCUGGCUUUCGUCCUU-3′ |
BxPC3 cells were transduced with lentiviruses
expressing shRNA negative control (shNC), LepR-shRNA-1 and
LepR-shRNA-2; medium-treated cells were used as the untransfected
control. Following 48 h of transduction by lentivirus infection in
a 37°C, 5% CO2 incubator, the silencing efficiency of
LepR-shRNA-1 and LepR-shRNA-2 were evaluated by reverse
transcription-quantitative PCR (RT-qPCR) and western blot analysis.
Subsequently, cell proliferation, glucose uptake and lactate
production, as well as the expression of several related genes and
proteins including LepR-short, LepR-long, HKII, GLUT1, AKT and
p-AKT, were measured at 48 h of transduction.
Experimental groups
BxPC3, Panc1 or HPC-Y5 cells were treated with 0,
20, 50 or 100 ng/ml Leptin. Subsequently, cell proliferation,
glucose uptake, lactate production as well as the expression of
HKII and GLUT1 were detected. The treatment groups for BxPC3 cells
were: i) RPMI-1640 medium (control); ii) shRNA negative control
(shNC); iii) LepR-shRNA-1 (shRNA-1); iv) LepR-shRNA-2 (shRNA-2), or
i) Medium + DMSO (control); ii) 50 ng/ml leptin + DMSO; iii) medium
+ 25 µmol/l LY294002 (AKT inhibitor); and iv) 50 ng/ml leptin + 25
µmol/l LY294002. A subset of shNC and shRNA-2-transfected BxPC3
cells were further treated with DMSO or 50 ng/ml IGF-1 (AKT
activator). Following treatment, cell proliferation, glucose
uptake, lactate production, and the expression of several related
genes (HKII, GLUT1, AKT and p-AKT) were examined.
Proliferation assay
The proliferation of treated human PC cells (BxPC3
and Panc1) and normal pancreas cells HPC-Y5 were evaluated by the
Cell Counting Kit-8 assay (CCK-8; cat. no. CP002; SAB
Biotherapeutics, Inc.). Human PC cells were seeded in 96-well
plates (3×103 cells/well) and cultured in a 37°C, 5%
CO2 incubator overnight. Following treatment for 24 h,
100 µl CCK-8 solution (diluted 1:10 in serum-free medium) was added
and incubated for 1 h in a 37°C, 5% CO2 incubator.
Subsequently, the absorbance value [optical density (OD)] at 450 nm
was determined using a microplate reader (DNM-9602; Perlong Medical
Equipment Co., Ltd.).
Detection of glucose uptake and
lactate production
Human PC cells (BxPC3 and Panc1) and normal pancreas
cell HPC-Y5 in the logarithmic growth phase were inoculated in
6-well plates (5×105 cells/well) and cultured in a 37°C
incubator overnight. After treatment with a range of leptin
concentrations (0, 20, 50 and 100 ng/ml) or according to the
grouping aforementioned, the cells were cultured for 3 h in
low-glucose DMEM, followed by washing with glucose-free
Krebs-Ringer bicarbonate buffer (containing 2% BSA) at 37°C. The
cells were subsequently incubated in glucose-free DMEM containing
100 µM 2-NBDG (cat. no. 0467597-16; Cayman Chemical Company) for 45
min, and glucose uptake was evaluated using a 2-NBDG Glucose Uptake
Assay kit (Nanjing Jiancheng Bioengineering Institute). Lactate
production was evaluated using a Lactate Assay Kit (Nanjing
Jiancheng Bioengineering Institute). The supernatant of the treated
cells was prepared according to the manufacturer's protocol and the
OD was measured at 530 nm using a spectrophotometer.
RT-qPCR
Following treatment, total RNA from human PC cells
(BxPC3 and Panc1) and normal pancreas cells HPC-Y5 was extracted
using TRIzol reagent (cat. no. 1596-026; Invitrogen; Thermo Fisher
Scientific, Inc.). Following RNA quantification and confirmation of
RNA integrity by electrophoresis with 1% gel, 1 µg of RNA was
reverse transcribed into cDNA using the RevertAid First strand cDNA
synthesis kit (cat. no. K1622; Fermentas; Thermo Fisher Scientific,
Inc.). RT-qPCR was performed in triplicate using an ABI-7300
Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific,
Inc., USA) and the Maxima SYBR Green/ROX qPCR Master Mix kit (cat.
no. K0223; Thermo Fisher Scientific, Inc.). GAPDH was used as an
internal reference. mRNA expression levels of HKII, GLUT1,
LepR-common, LepR-short and LepR-long were analyzed using the
2−ΔΔCT method (28).
The primers used were as follows: HKII, forward
5′-ACGACAGCATCATTGTTAAGG-3′, reverse 5′-TTTGGCAAAGTGAGGATGTAG-3′;
GLUT1, forward 5′-TGCAGGAGATGAAGGAAG-3′, reverse
5′-CAATGGTGGCATACACAG-3′; LepR-common, forward
5′-TTGTGCCAGTAATTATTTCCTCTT-3′, reverse
5′-CACACCAAAGAATGAAAAAGCTAT-3′; LepR-short, forward
5′-TTCCTGGGCACAAGGACTTA-3′- reverse 5′-GCTCCAAAAGAAGAGGACCA-3′;
LepR-long, forward 5′-TTCCTGGGCACAAGGACTTA-3′, reverse
5′-TTTGTGTCCCTGGGTACTTGA-3′; GAPDH, forward
5′-AATCCCATCACCATCTTC-3′, reverse 5′-AGGCTGTTGTCATACTTC-3′. The
thermocycling conditions were as follows: 95°C for 10 min; followed
by 40 cycles of 95°C for 15 sec and 60°C for 45 sec (29).
Western blot analysis
Following the various treatments, total protein from
human PC cells (BxPC3 and Panc1) and normal pancreas cells HPC-Y5
were extracted using RIPA buffer supplemented with protease and
phosphatase inhibitors (cat. no. R0010; Beijing Solarbio Science
& Technology Co., Ltd.), followed by quantification by the BCA
Kit (cat. no. PICPI23223; Thermo Fisher Scientific, Inc.). Proteins
(25 µg) were separated using 10 (spacer) and 8% (separation)
SDS-PAGE, and transferred to PVDF membranes (cat. no. HATF00010;
EMD Millipore). Subsequently, the membranes were blocked in 5%
skimmed milk (cat. no. BYL40422; BD Biosciences) for 1 h at room
temperature and incubated with primary antibodies against GLUT1
(1:1,000; cat. no. ab115730; Abcam), HKII [1:1,000; cat. no. 2867;
Cell Signaling Technology (CST)], AKT (1:1,000; cat. no. 2920;
CST), phosphorylated (p)-AKT (1:2,000; cat. no. 4060; CST),
LepR-long (1:250; cat. no. sc-1835; Santa Cruz Biotechnology, Inc.,
California, USA), LepR-short (1:250; catalog no. sc-8325; Santa
Cruz Biotechnology, Inc.) and GAPDH (1:1,000; cat. no. 5174; CST)
overnight at 4°C with gentle agitation. Following 5–6 washes in TBS
+ 0.1% Tween-20, the membranes were incubated for 2 h at room
temperature with horseradish peroxidase-conjugated goat anti-rabbit
secondary antibody (1:1,000; cat. no. ZB2301, OriGene Technologies,
Inc.). The blots were visualized by ECL reagent (cat. no.
WBKLS0100; EMD Millipore) and images were captured with a
Tanon-5200 ECL imaging system (Tanon Science and Technology Co.,
Ltd.). Protein expressions levels were normalized to GAPDH and
analyzed using ImageJ 1.47v (National Institutes of Health).
Statistical analysis
All statistical analyses and calculations in this
study were carried out using GraphPad Prism 7.0 software (GraphPad
Software, Inc.). The statistical significance of differences
between the two groups was determined using two-tailed Student's
t-test, and multiple comparisons were made by one-way ANOVA
followed by Tukey's test for multiple comparisons. All experiments
were performed in triplicate, and data are expressed as the mean ±
standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results
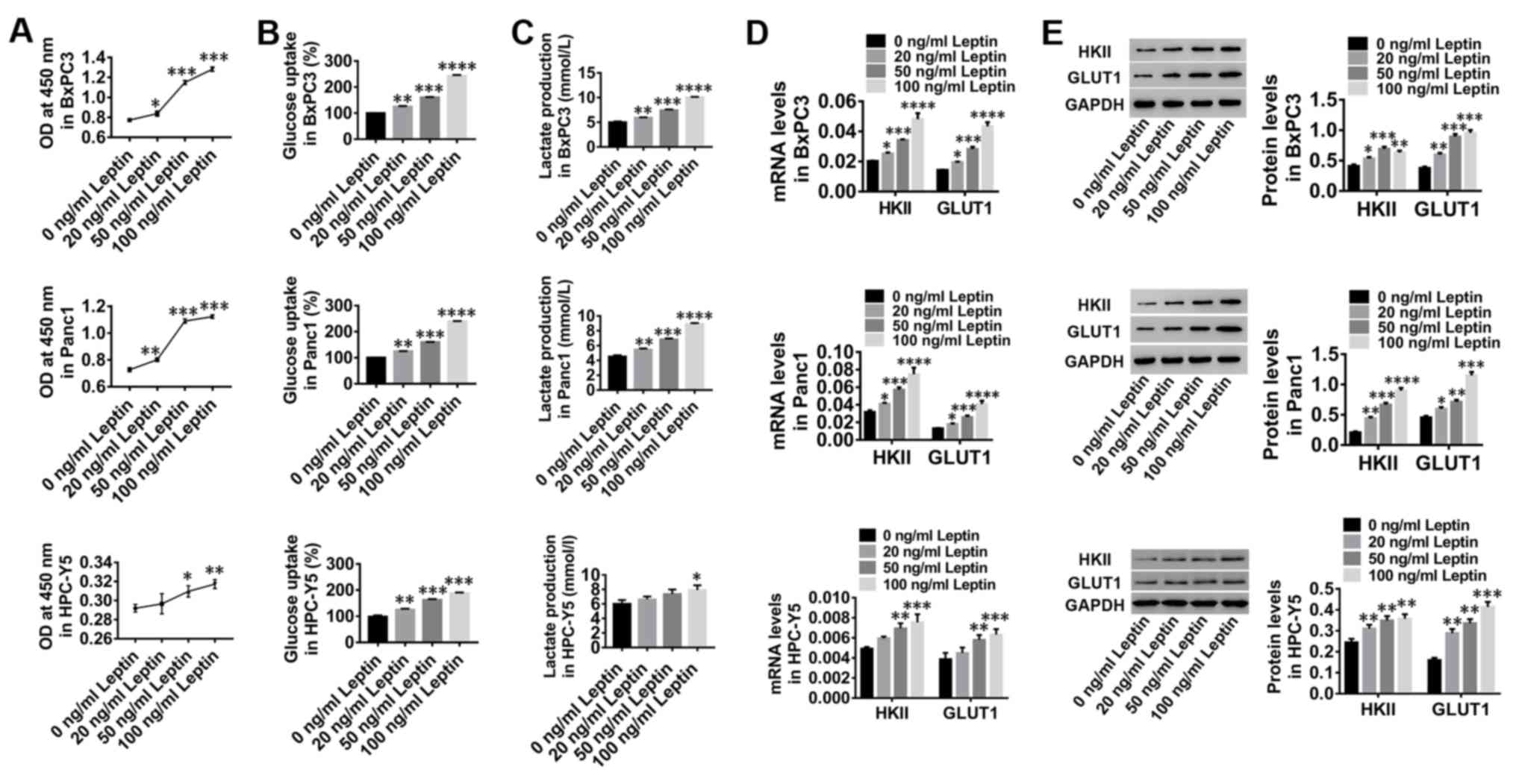
Leptin stimulates the proliferation
and glucose metabolism of human PC cells
Human PC cell lines, BxPC3 and Panc1, and normal
pancreas cells were treated with increasing concentrations of
leptin (0, 20, 50 and 100 ng/ml) for 24 h. In vitro
treatment with leptin significantly stimulated the proliferation of
BxPC3 and Panc1 cells as well as pancreas cells HPC-Y5 in a
dose-dependent manner (Fig. 1A).
These results indicated that leptin stimulation may promote the
proliferation of human PC and pancreas cells. In addition, glucose
uptake (Fig. 1B) and lactate
production (Fig. 1C) of BxPC3,
Panc1 and pancreas cells were markedly increased by leptin
stimulation, accompanied by increased expression of HKII and GLUT1
(Fig. 1D and E). These data
suggested that leptin stimulation may contribute to glucose
metabolism and proliferation of human PC cells and healthy
pancreatic cells.
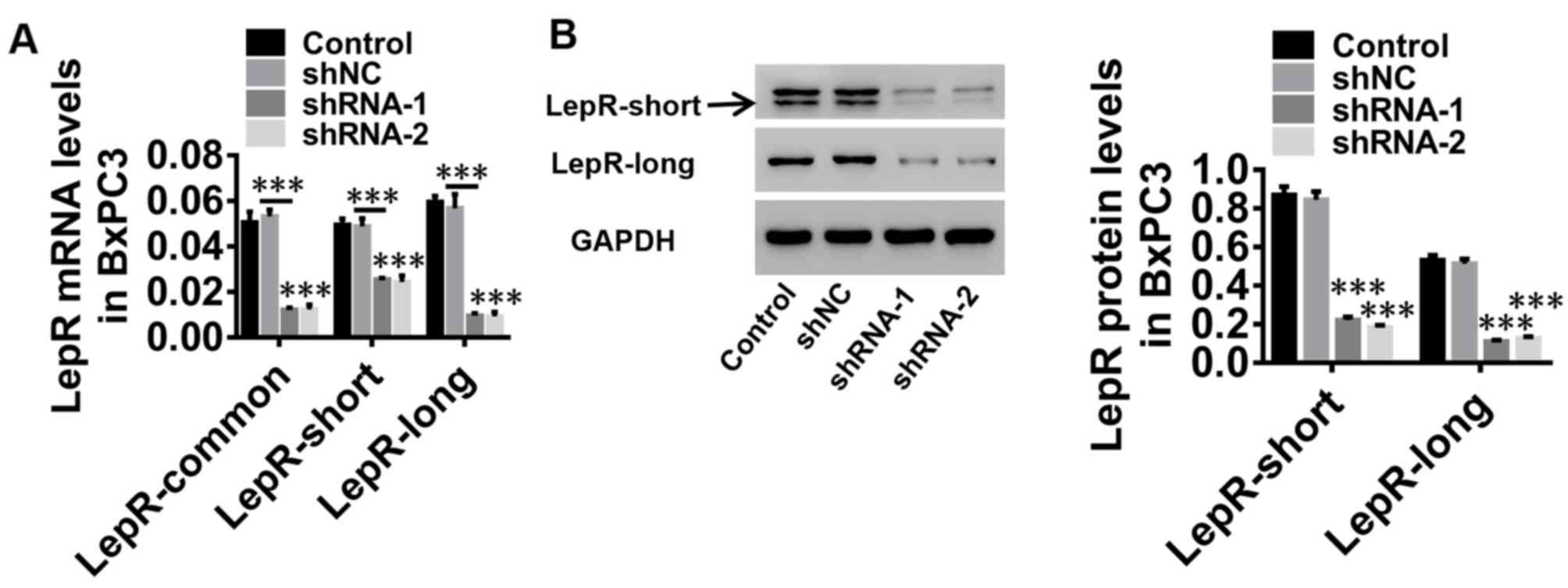
Silencing of LepR expression levels in
BxPC3 cells
Due to the significant increase of proliferation,
glucose uptake and lactate production in both BxPC3 and Panc1
cells, in the present study, one cell line, BxPC3, was selected for
further investigation. BxPC3 cells were infected with lentiviruses
expressing shNC, LepR-shRNA-1 and LepR-shRNA-2; untreated cells
were used as a control. mRNA expression of LepR-common, LepR-short
and LepR-long (Fig. 2A), as well
as the protein expression levels of LepR-short and LepR-long, were
significantly downregulated by LepR-shRNA-1 and LepR-shRNA-2
(Fig. 2B). Therefore, the
LepR-shRNA-1 and LepR-shRNA-2 vectors were used for subsequent
experiments.
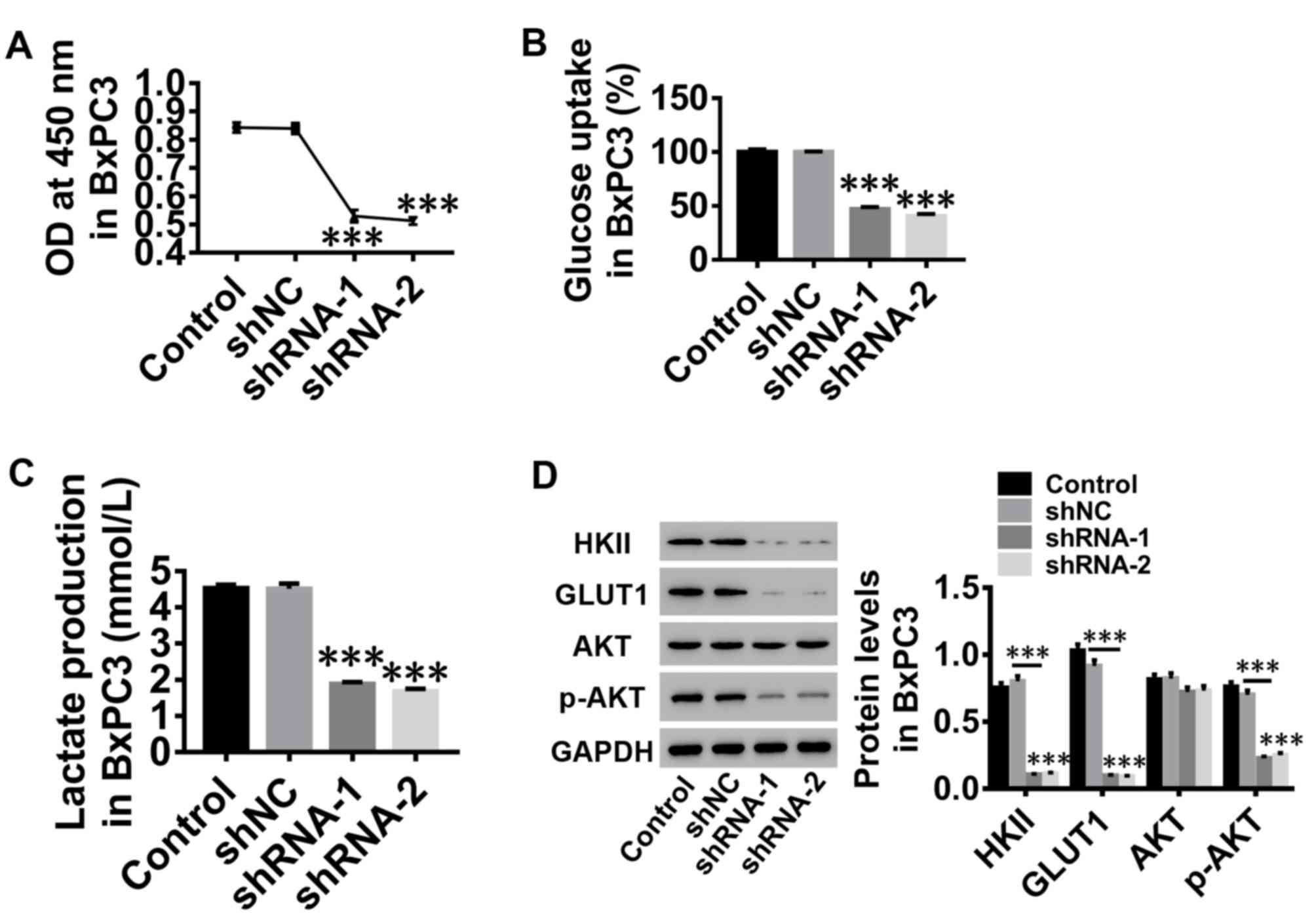
Silencing of LepR inhibits
proliferation and glucose metabolism of human PC cells
To investigate the role of leptin in human PC cells,
proliferation, glucose uptake and lactate production were assessed
in BxPC3 cells following LepR silencing. The results indicated that
the proliferation of BxPC3 cells was notably suppressed by
silencing of LepR, compared with the control groups (Fig. 3A). Glucose uptake (Fig. 3B) and lactate production (Fig. 3C) of BxPC3 cells were also
inhibited, accompanied by decreased protein expression levels of
HKII, GLUT1 and p-AKT, whereas total AKT protein expression was
unaltered (Fig. 3D), suggesting an
inhibitory effect of leptin silencing on AKT activation. These
results demonstrated the beneficial effects of leptin in glucose
metabolism and proliferation of human PC cells, which may be
involved in AKT pathway activation.
Leptin stimulates the proliferation
and glucose metabolism of human PC cells via activation of the AKT
pathway
The molecular mechanisms of leptin in mediating
glucose metabolism and proliferation of human PC cells were
explored. Leptin-stimulated proliferation, glucose uptake and
lactate production were effectively counteracted by co-treatment
with the AKT inhibitor LY294002 (Fig.
4A-C, respectively). The increased protein expression levels of
HKII, GLUT1 and p-AKT induced by leptin stimulation were also
significantly decreased (Fig. 4D),
whereas total AKT was unchanged (Fig.
4D). Leptin stimulation potently attenuated the effects of AKT
inhibitor, LY294002, suggesting that there was a rescue effect of
leptin stimulation on AKT inhibition. In addition, the inhibition
of proliferation, glucose uptake and lactate production induced by
Lep2-shRNA-2 transfection were significantly counteracted by
co-treatment with the AKT activator IGF-1 (Fig. 4E-G, respectively), accompanied by
an increased expression of GLUT1 and HKII protein (Fig. 4H). In the present study,
Lep2-shRNA-2 was used due to its better downregulation efficiency
compared with shRNA-1. These results further demonstrated that
leptin may stimulate glucose metabolism and proliferation of human
PC cells potentially through activating the AKT pathway.
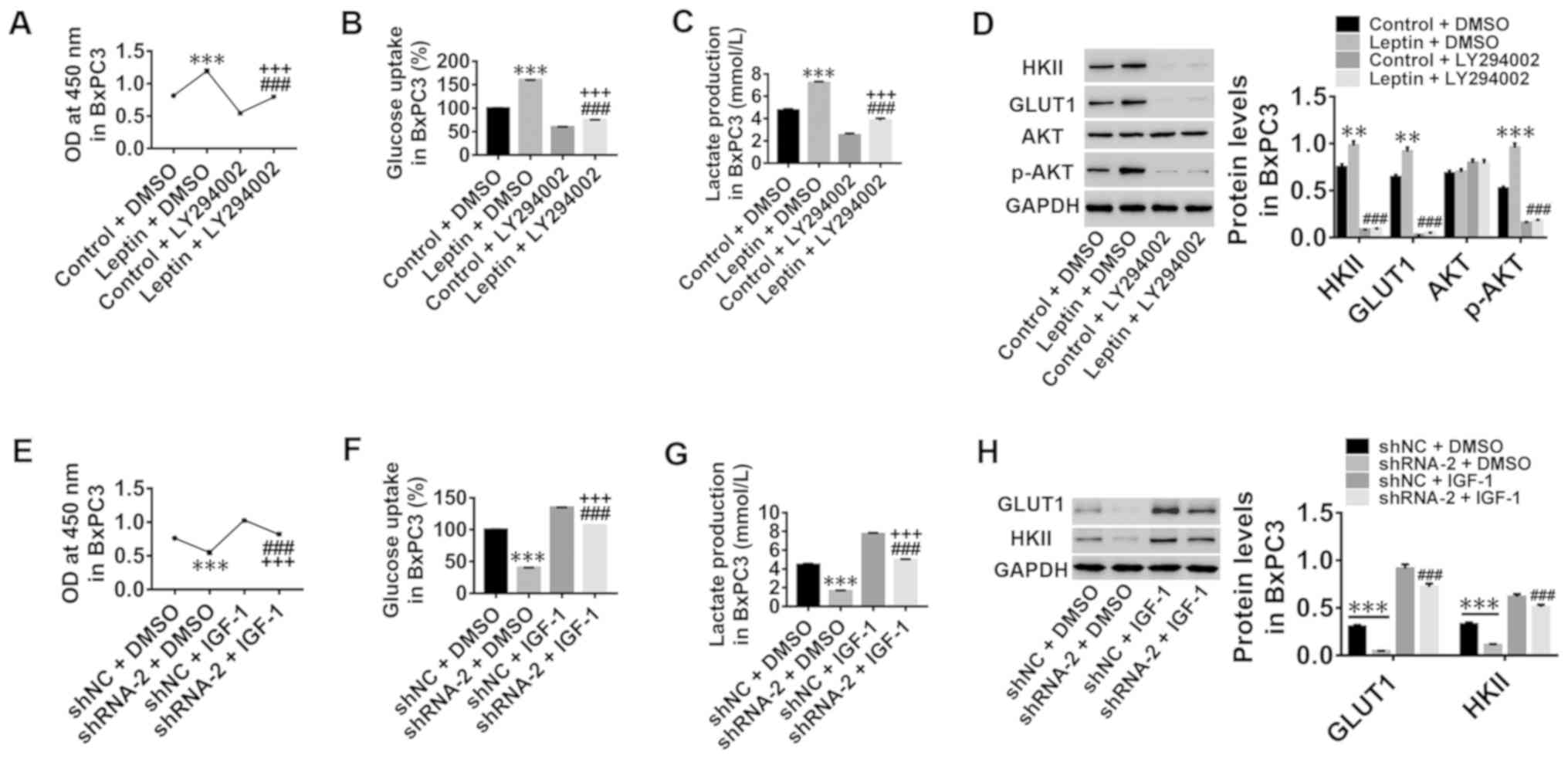 | Figure 4.Leptin stimulates the proliferation
and glucose metabolism of human PC cells via activation of the AKT
pathway. BxPC3 cells were treated with medium (control), 50 ng/ml
leptin, medium + 25 µmol/l LY294002 (an AKT inhibitor), or 50 ng/ml
leptin + 25 µmol/l LY294002. (A) Cell proliferation was assessed.
(B) Glucose uptake was calculated. (C) Lactate production was
calculated. (D) The protein levels of GLUT1, HKII and the
p-AKT/total AKT ratio were detected. BxPC3 cells were treated with
shNC, shRNA-2, shNC + 50 ng/ml IGF-1 (an AKT activator), and
shRNA-2 + 50 ng/ml IGF-1. (E) Cell proliferation was assessed. (F)
Glucose uptake was calculated. (G) Lactate production was
calculated. (H) The protein levels of GLUT1, HKII were detected.
**P<0.01 and ***P<0.001 vs. control + DMSO or shNC + DMSO;
###P<0.001 vs. leptin + DMSO or shRNA-2 + DMSO;
+++P<0.001 vs. control + LY294002 or shNC + IGF-1.
GLUT1, glucose transporter 1; HKII, hexokinase II; p-AKT,
phosphorylated AKT; shNC, negative control shRNA; shRNA, short
hairpin RNA. |
Discussion
Leptin is a hormone that is highly expressed in
overweight and obese individuals and may be involved in the
progression of many cancers. Previous studies have shown that
leptin can stimulate the proliferation of various cancer cell lines
(30–33). An increase in leptin was reported
in plasma and pancreatic tissue, suggesting that leptin may be
involved in tumor growth in the pancreas (4). The distribution and high abundance of
positive leptin or LepR in tumor tissue samples of PC patients have
been described in our previous study (27). Leptin acts as a growth factor to
promote proliferation in multiple cancers, such as breast and lung
cancer (34,35). Leptin stimulates cell proliferation
and survival in breast cancer cells through LepR (36,37).
In the current study, leptin stimulation promoted the glucose
metabolism and proliferation of human PC cells and non-cancerous
cells. Silencing of LepR by LepR-shRNAs had the opposite effect as
leptin stimulation on human PC cells, which agreed with a previous
report (38) and suggested that
LepR knockdown may attenuate human PC progression by mediating
glucose metabolism and cell proliferation.
The underlying mechanisms of LepR on human PC cell
growth were also explored. GLUT1 and HKII are two key enzymes that
regulate cellular glycolysis and energy metabolism. HKII has been
demonstrated to be the key enzyme to catalyze the first step in the
glycolytic pathway (39,40), and GLUT1 typically regulates the
first rate-limiting step of glucose metabolism when glucose flows
into cells (41,42). The results of the present study
demonstrated that GLUT1 and HKII expression in human PC cells and
healthy pancreatic cells was significantly increased by leptin
stimulation, but LepR silencing in human PC cells significantly
decreased GLUT1 and HKII expression. Studies have showed that
leptin is a cytokine with multiple biological roles including
regulation of energy metabolism and plays an important role in cell
proliferation, invasion, metastasis and survival in cancers
including breast, esophageal and endometrial cancers (43–46).
Thus, the increase of GLUT1 and HKII expression in healthy pancreas
cells may be caused by the stimulation of leptin. In our previous
study, compared with normal tissues, high abundance of positive
leptin or LepR was found in tumor tissue samples of PC patients
(27). These results demonstrated
that leptin may be a molecular switch that regulates
glycolytic-related proteins such as GLUT1 and HKII, thus regulating
glucose metabolism of human PC cells. The PI3K/AKT pathway has been
suggested to be the main signaling cascade in glucose metabolism
and cell growth regulation. It is known that activation of AKT
regulates cell growth and controls the rates of glucose uptake
through the GLUT1 transporter. It has also been found that AKT can
further affect glycolysis through HKII (47–50).
Therefore, the AKT pathway was analyzed in the present study.
GLUT1, HKII and p-AKT were significantly decreased by the silencing
of leptin receptor, whereas AKT levels were unchanged.
Additionally, high levels of GLUT1, HKII and p-AKT induced by
leptin stimulation were effectively counteracted by co-treatment
with the AKT inhibitor LY294002, whereas inhibition of GLUT1, HKII
and p-AKT by LepR silencing was counteracted by IGF-1 co-treatment.
Increased levels of GLUT1 have been reported to enhance glucose
uptake, which in turn elevates the rate of glycolysis to enhance
ATP production, ultimately enhancing the growth of tumors (51). These findings demonstrated that
LepR may contribute to the glucose metabolism and proliferation of
human PC cells in vitro, possibly through the activation of
the AKT pathway. Compared with our previous studies and other
related studies (27,52–54)
which reported that leptin enhances PC invasion through the
increase in MMP-13 production but does not affect PC cell
proliferation, the present study targeted LepR instead of leptin
and revealed that LepR had an effect on the proliferation and
metabolism of PC cells and further demonstrated that this
regulation may be through the activation of the AKT signaling
pathway. Therefore, targeting the LepR/AKT signaling pathway is a
potential therapeutic strategy for PC. However, the current results
are based solely on in vitro experiments and are therefore
not comprehensive enough. Further in vivo studies should be
conducted in the future to confirm these results.
In conclusion, the present study demonstrated that
knockdown of LepR may attenuate the development of human PC by
inhibiting glucose metabolism and cell proliferation. LepR has the
potential to be a molecular switch that regulates
glycolysis-related proteins such as GLUT1 and HKII through
activation of the AKT pathway, further regulating glucose
metabolism and proliferation in human PC cells. These findings
offer the foundation for further study of the role of LepR in the
relief or treatment of pancreatic cancer.
Acknowledgements
Not applicable.
Funding
This work was funded by The National Natural Science
Foundation of China (grant no. 81672083), Shanghai No. 6 People's
Hospital group project (grant no. 2016jy201602) and Shanghai Jiao
Tong University Medical and Engineering Cross Subject (grant no.
YG2016MS69).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
WX and HS conceived and designed the study. YX, MT,
XT, JuZ, JiZ and JC performed the experiments. WX and HS wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Harbuzariu A, Daley-Brown DS, Harmon TL,
Garrison RC, Beech DJ, Cason FD, Klug C and Gonzalez-Perez RR:
Abstract B26: Leptin affects proliferation, stem cells and
chemotherapeutic treatment outcome of pancreatic cancer: A link to
health disparity. Cancer Epidemiol Biomarkers Prev. 25:B262016.
|
|
4
|
Mendonsa AM, Chalfant MC, Gorden LD and
VanSaun MN: Modulation of the leptin receptor mediates tumor growth
and migration of pancreatic cancer cells. PLoS One.
10:e01266862015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ng M, Fleming T, Robinson M, Thomson B,
Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF,
et al: Global, regional, and national prevalence of overweight and
obesity in children and adults 1980–2013: A systematic analysis for
the Global Burden of Disease Study 2013. Lancet. 384:766–781. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Inadera H: The usefulness of circulating
adipokine levels for the assessment of obesity-related health
problems. Int J Med Sci. 5:248–262. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
de Luis DA, González Sagrado M, Conde R,
Aller R, Izaola O and Castro MJ: Circulating adipocytokines in
morbid obese patients, relation with cardiovascular risk factors
and anthropometric parameters. Nutr Hosp. 26:91–96. 2011.PubMed/NCBI
|
|
8
|
Ebert T, Roth I, Richter J, Tönjes A,
Kralisch S, Lossner U, Kratzsch J, Blüher M, Stumvoll M and
Fasshauer M: Different associations of adipokines in lean and
healthy adults. Horm Metab Res. 46:41–47. 2014.PubMed/NCBI
|
|
9
|
Balistreri CR, Caruso C and Candore G: The
role of adipose tissue and adipokines in obesity-related
inflammatory diseases. Mediators Inflamm. 2010:8020782010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Collins S, Kuhn CM, Petro AE, Swick AG,
Chrunyk BA and Surwit RS: Role of leptin in fat regulation. Nature.
380:6771996. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lindheim SR, Sauer MV, Carmina E, Chang
PL, Zimmerman R and Lobo RA: Circulating leptin levels during
ovulation induction: Relation to adiposity and ovarian morphology.
Fertil Steril. 73:493–498. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Henson M and Castracane V: Leptin in
pregnancy. Biol Reprod. 63:1219–1228. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shaw RJ: Glucose metabolism and cancer.
Curr Opin Cell Biol. 18:598–608. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang G and Zhang BB: Glucagon and
regulation of glucose metabolism. Am J Physiol Endocrinol Metab.
284:E671–E678. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mueller WM, Gregoire FM, Stanhope KL,
Mobbs CV, Mizuno TM, Warden CH, Stern JS and Havel PJ: Evidence
that glucose metabolism regulates leptin secretion from cultured
rat adipocytes. Endocrinology. 139:551–558. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Frühbeck G and Salvador J: Relation
between leptin and the regulation of glucose metabolism.
Diabetologia. 43:3–12. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Morton GJ and Schwartz MW: Leptin and the
central nervous system control of glucose metabolism. Physiol Rev.
91:389–411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ceddia RB: Direct metabolic regulation in
skeletal muscle and fat tissue by leptin: Implications for glucose
and fatty acids homeostasis. Int J Obes (Lond). 29:1175–1183. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vansaun MN: Molecular pathways:
Adiponectin and leptin signaling in cancer. Clin Cancer Res.
19:1926–1932. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cottrell EC and Mercer JG: Leptin
receptors. Handb Exp Pharmacol. 209:3–21. 2012. View Article : Google Scholar
|
|
21
|
Lin R, Ju H, Yuan Z, Zeng L, Sun Y, Su Z,
Yang Y, Wang Y and Jin L: Association of maternal and fetal LEPR
common variants with maternal glycemic traits during pregnancy. Sci
Rep. 7:31122017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hardwick JC, Van Den Brink GR, Offerhaus
GJ, Van Deventer SJ and Peppelenbosch MP: Leptin is a growth factor
for colonic epithelial cells. Gastroenterology. 121:79–90. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jaffe T and Schwartz B: Leptin promotes
motility and invasiveness in human colon cancer cells by activating
multiple signal-transduction pathways. Int J Cancer. 123:2543–2556.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Saxena NK, Taliaferro-Smith LT, Knight BB,
Merlin D, Anania FA, O'Regan RM and Sharma D: Bidirectional
crosstalk between leptin and insulin-like growth factor-I signaling
promotes invasion and migration of breast cancer cells via
transactivation of epidermal growth factor receptor. Cancer Res.
68:9712–9722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fava G, Alpini G, Rychlicki C, Saccomanno
S, DeMorrow S, Trozzi L, Candelaresi C, Venter J, Di Sario A,
Marzioni M, et al: Leptin enhances cholangiocarcinoma cell growth.
Cancer Res. 68:6752–6761. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Islam MS, Morton NM, Hansson A and
Emilsson V: Rat insulinoma-derived pancreatic beta-cells express a
functional leptin receptor that mediates a proliferative response.
Biochem Biophys Res Commun. 238:851–855. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fan Y, Gan Y, Shen Y, Cai X, Song Y, Zhao
F, Yao M, Gu J and Tu H: Leptin signaling enhances cell invasion
and promotes the metastasis of human pancreatic cancer via
increasing MMP-13 production. Oncotarget. 6:16120–16134. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hong JY, Kang B, Kim A, Hwang S, Ahn J,
Lee S, Kim J, Park JH and Cheon DS: Development of a highly
sensitive real-time one step RT-PCR combined complementary locked
primer technology and conjugated minor groove binder probe. Virol
J. 8:3302011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han G, Wang L, Zhao R, Yue Z, Zhou X, Hu
X, Cao Y, Dai D and Liu J: Leptin promotes human glioblastoma
growth through activating Signal Transducers and Activators of
Transcription 3 signaling. Brain Res Bull. 87:274–279. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu Y, Lv L, Xiao W, Gong C, Yin J, Wang D
and Sheng H: Leptin activates STAT3 and ERK1/2 pathways and induces
endometrial cancer cell proliferation. J Huazhong Univ Sci
Technolog Med Sci. 31:3652011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Endo H, Hosono K, Uchiyama T, Sakai E,
Sugiyama M, Takahashi H, Nakajima N, Wada K, Takeda K, Nakagama H
and Nakajima A: Leptin acts as a growth factor for colorectal
tumours at stages subsequent to tumour initiation in murine colon
carcinogenesis. Gut. 60:1363–1371. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Saxena NK, Vertino PM, Anania FA and
Sharma D: Leptin-induced growth stimulation of breast cancer cells
involves recruitment of histone acetyltransferases and mediator
complex to CYCLIN D1 promoter via activation of Stat3. J Biol Chem.
282:13316–13325. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Garofalo C and Surmacz E: Leptin and
cancer. J Cell Physiol. 207:12–22. 2010. View Article : Google Scholar
|
|
35
|
Somasundar P, McFadden DW, Hileman SM and
Vona-Davis L: Leptin is a growth factor in cancer. J Surg Res.
116:337–349. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hu X, Juneja SC, Maihle NJ and Cleary MP:
Leptin-A growth factor in normal and malignant breast cells and for
normal mammary gland development. J Natl Cancer Inst. 95:1704–1711.
2002. View Article : Google Scholar
|
|
37
|
Dieudonne MN, Machinal-Quelin F,
Serazin-Leroy V, Leneveu MC, Pecquery R and Giudicelli Y: Leptin
mediates a proliferative response in human MCF7 breast cancer
cells. Biochem Biophys Res Commun. 293:622–628. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Krechler T, Zeman M, Vecka M, Macasek J,
Jachymova M, Zima T and Zak A: Leptin and adiponectin in pancreatic
cancer: Connection with diabetes mellitus. Neoplasma. 58:58–64.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lv X, Yao L, Zhang J, Han P and Li C:
Inhibition of microRNA-155 sensitizes lung cancer cells to
irradiation via suppression of HK2-modulated glucose metabolism.
Mol Med Rep. 14:1332–1338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fang R, Xiao T, Fang Z, Sun Y, Li F, Gao
Y, Feng Y, Li L, Wang Y, Liu X, et al: MicroRNA-143 (miR-143)
regulates cancer glycolysis via targeting hexokinase 2 gene. J Biol
Chem. 287:23227–23235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fan R, Hou WJ, Zhao YJ, Liu SL, Qiu XS,
Wang EH and Wu GP: Overexpression of HPV16 E6/E7 mediated HIF-1α
upregulation of GLUT1 expression in lung cancer cells. Tumour Biol.
37:4655–4663. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sasaki H, Shitara M, Yokota K, Hikosaka Y,
Moriyama S, Yano M and Fujii Y: Overexpression of GLUT1 correlates
with Kras mutations in lung carcinomas. Mol Med Rep. 5:599–602.
2012.PubMed/NCBI
|
|
43
|
Huang L and Li C: Leptin: A
multifunctional hormone. Cell Res. 10:81–92. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ogunwobi O, Mutungi G and Beales IL:
Leptin stimulates proliferation and inhibits apoptosis in Barrett's
esophageal adenocarcinoma cells by cyclooxygenase-2-dependent,
prostaglandin-E2-mediated transactivation of the epidermal growth
factor receptor and c-Jun NH2-terminal kinase activation.
Endocrinology. 147:4505–4516. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Saxena NK, Sharma D, Ding X, Lin S, Marra
F, Merlin D and Anania FA: Concomitant activation of the JAK/STAT,
PI3K/AKT, and ERK signaling is involved in leptin-mediated
promotion of invasion and migration of hepatocellular carcinoma
cells. Cancer Res. 67:2497–2507. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sharma D, Saxena NK, Vertino PM and Anania
FA: Leptin promotes the proliferative response and invasiveness in
human endometrial cancer cells by activating multiple
signal-transduction pathways. Endocr Relat Cancer. 13:629–640.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jóźwiak P, Krześlak A, Bryś M and Lipińska
A: Glucose-dependent glucose transporter 1 expression and its
impact on viability of thyroid cancer cells. Oncol Rep. 33:913–920.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hong SY, Yu FX, Luo Y and Hagen T:
Oncogenic activation of the PI3K/Akt pathway promotes cellular
glucose uptake by downregulating the expression of
thioredoxin-interacting protein. Cell Signal. 28:377–383. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hou X, Liu Y, Liu H, Chen X, Liu M, Che H,
Guo F, Wang C, Zhang D, Wu J, et al: PERK silence inhibits glioma
cell growth under low glucose stress by blockage of p-AKT and
subsequent HK2's mitochondria translocation. Sci Rep. 5:90652015.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Neary CL and Pastorino JG: Akt inhibition
promotes hexokinase 2 redistribution and glucose uptake in cancer
cells. J Cell Physiol. 228:1943–1948. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Stan SD, Singh SV and Brand RE:
Chemoprevention strategies for pancreatic cancer. Nat Rev
Gastroenterol Hepatol. 7:347–356. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bloomston M, Zervos EE and Rosemurgy AS
II: Matrix metalloproteinases and their role in pancreatic cancer:
A review of preclinical studies and clinical trials. Ann Surg
Oncol. 9:668–674. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Coussens LM, Fingleton B and Matrisian LM:
Matrix metalloproteinase inhibitors and cancer-trials and
tribulations. Science. 295:2387–2392. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Vihinen P and Kähäri VM: Matrix
metalloproteinases in cancer: Prognostic markers and therapeutic
targets. Int J Cancer. 99:157–166. 2002. View Article : Google Scholar : PubMed/NCBI
|