Introduction
Macrovascular complications, mainly characterize the
pathological change of atherosclerosis, are primarily associated
with cardiovascular and cerebrovascular diseases. In addition, as
the most common complications of DM, they are also the main causes
of fatality and disability in DM patients (1). Endothelial dysfunction is a crucial
pathophysiological step in the early stages of macrovascular
complications and is an initial event of atherogenesis (2). Under normal conditions, apoptosis and
the proliferation rate of vascular endothelial cells tends to be
low in order to maintain the balance and the stability of blood
vessels (3). Increased endothelial
cell apoptosis is one of the predominant characteristics of
endothelial dysfunction, which may promote smooth muscle cell
proliferation and migration, increase blood coagulation, enhance
leukocyte infiltration into the endothelium, and give rise to
endothelial dysfunction and atherothrombosis (4). Research has indicated the role of
inflammation in the pathogenesis of macrovascular complications of
DM (5–7). Furthermore, glucotoxicity and
inflammation can destroy endothelial function, leading to
endothelial dysfunction and atherogenesis (8–10).
Previous studies have demonstrated that high concentrations of
glucose promote inflammatory damage in vascular endothelial cells
in vitro (11–13). Therefore, inhibiting inflammation
and decreasing cell apoptosis, which improve vascular endothelial
dysfunction, are important to the treatment of macrovascular
complications of DM.
C-Jun NH2-terminal kinase (JNK) is a type
of serine/threonine protein kinase and a member of the family of
mitogen-activated protein kinase (MAPK) (14). JNK is closely associated with cell
differentiation, apoptosis, stress response and the occurrence and
development of various human diseases (15). Inflammatory cytokines are the
predominant activators of the JNK signaling pathway, which is a key
regulator of pro-inflammatory gene expression (16,17).
It has previously been suggested that the activated JNK pathway
contributes to vascular cell apoptosis and endothelial dysfunction
under the stimulation of inflammatory cytokines (18–22).
Furthermore, the JNK signaling pathway mediates apoptosis by
modulating the activities of mitochondrial pro-apoptotic and
anti-apoptotic proteins in the cytoplasm (23). Therefore, inhibition of JNK
signaling pathway activation is an important target for improving
vascular endothelial cell disorder in macrovascular complications
of DM.
Radix Astragali, a famous Traditional Chinese
Medicine, is the dry root of the perennial herbaceous plant,
Astragalus menabranaceus (Fisch.) Bunge. Astragaloside IV
(AS-IV) is one of the primary effective components of Astragali
Radix that has potent protective effects on cardiovascular disease,
DM and its complications (24).
Furthermore, Astragali Radix has been reported to have a series of
pharmacological actions, including anti-inflammatory and
antioxidant effects, and can improve endothelial dysfunction and
neovascularization associated with DM and its complications
(25). Previous studies have
revealed that AS-IV improves vascular endothelial dysfunction in
diabetic rats through inhibiting the inflammatory pathways
(26) and inhibiting inflammatory
gene expression in lipopolysaccharide (LPS)-treated mice (27). However, few studies have
investigated the protective effects and mechanisms of AST-IV with
regard to glucotoxicity and inflammation in endothelial dysfunction
in vitro. The present study aimed to determine whether AS-IV
could prevent high glucose-induced cell apoptosis and inflammatory
reactions through inhibition of the JNK signaling pathway in human
umbilical vein endothelial cells (HUVECs). In addition, an
accessible therapeutic strategy was suggested for the clinical
treatment of diabetic macrovascular complications.
Materials and methods
Chemicals and drugs
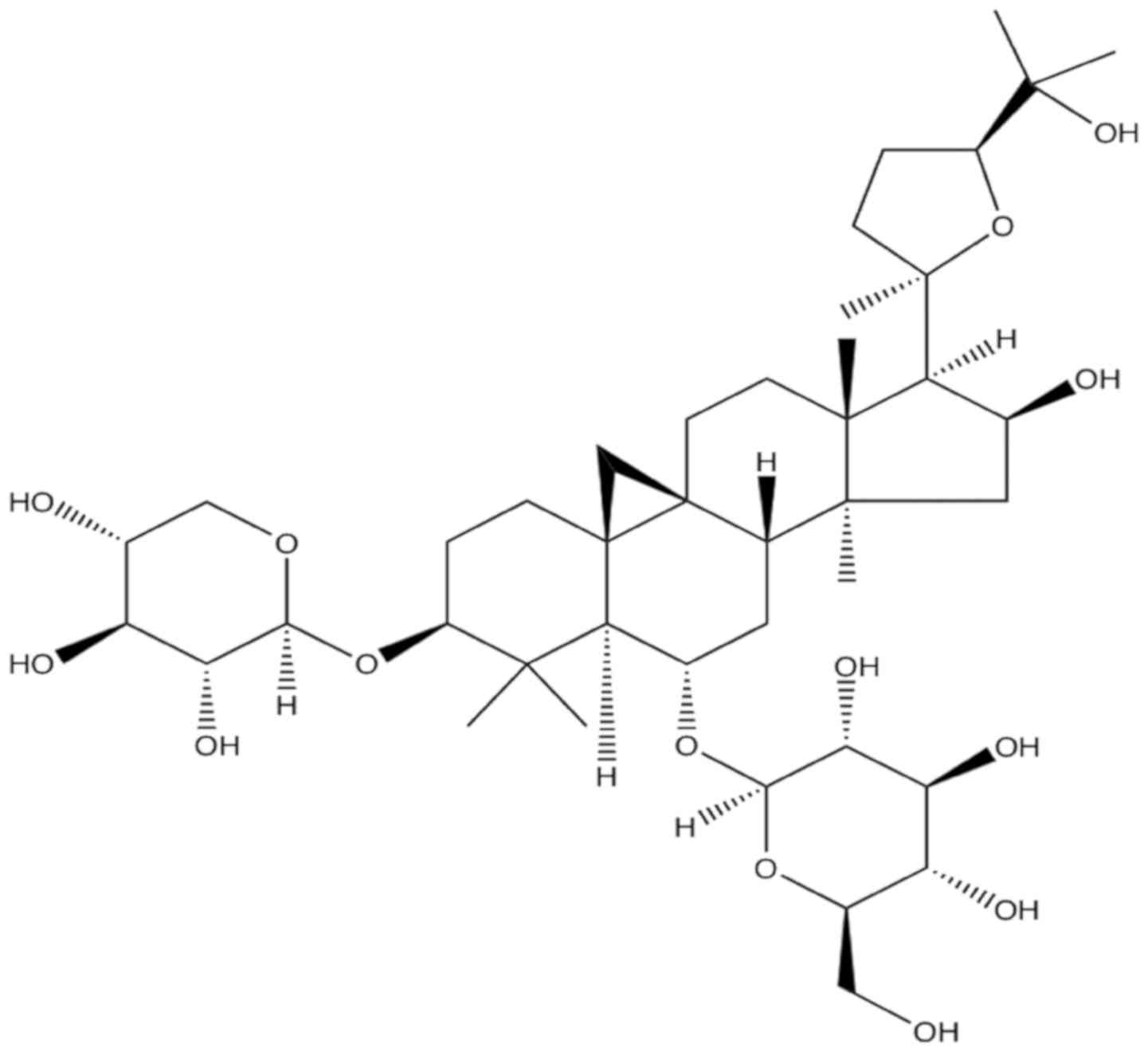
AS-IV (Fig. 1) is a
white crystalline powder and insoluble compound with >98%
purity. AS-IV has the molecular formula
C41H68O14 and a molecular weight
of 784.97 Da. AS-IV was purchased from the National Institute for
Food and Drug Control (Beijing, China). The compound was dissolved
in dimethyl sulfoxide (DMSO) to a concentration of 250 µM in a
stock solution. The stock solution was diluted with Dulbecco's
modified Eagle's medium (DMEM). The final DMSO concentration did
not exceed 0.5% (v/v).
Anti-phosphorylated (phospho/p)-JNK, anti-JNK,
anti-cleaved-caspase-3, anti-cleaved-caspase-9 and anti-β-actin
were obtained from Abcam (Cambridge, UK). Phospho-apoptosis
signal-regulating kinase-1 (ASK1), ASK1, cytochrome c (Cyt-c)
antibody, B-cell lymphoma-2-associated X protein (Bax) and B-cell
lymphoma-2 (Bcl-2) antibodies were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). SP600125,
penicillin/streptomycin solution, 0.05% trypsin-EDTA, DMSO and MTT
were purchased from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
DMEM and fetal bovine serum were purchased from HyClone (Logan, UT,
USA). Radioimmunoprecipitation assay lysis and extraction buffer,
horseradish peroxidase-conjugated AffiniPure goat anti-mouse
immunoglobulin (Ig)G and anti-rabbit IgG antibodies and D-glucose
were purchased from OriGene Technologies, Inc., (Beijing, China).
Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide
(PI) were obtained from Absin Bioscience, Inc. (Shanghai, China;
http://www.absin.cn/). A Hoechst 33258 kit was
purchased from Beijing Solarbio Science & Technology Co., Ltd.
(Beijing, China). ELISA kits for IL-1β (catalog no. SBJH0292) and
TNF-α (catalog no. SBJH0038) were purchased from the Nanjing
SenBeiJia Biological Technology Co., Ltd. (Nanjing, China).
Polymerase chain reaction (PCR) primers were obtained from Sangon
Biotech Co., Ltd. (Shanghai, China).
Cell culture and treatment
HUVECs were obtained from the Fudan Institutes of
Biomedical Sciences Cell Resource Center (The Institutes of
Biomedical Sciences, Fudan University, Shanghai, China) and
cultured in DMEM supplemented with 10% fetal bovine serum, 100
µg/ml streptomycin and 100 U/ml penicillin in a humidified
atmosphere containing 5% CO2 at 37°C. Experiments were
performed with cells at 80% confluence.
The first series of experiments were designed to
confirm the optimal glucose concentration to induce the cellular
damage model and the optimal AS-IV concentration to protect against
damaged cell. Doses of 5.55, 11.1, 22.2, 33.3 and 44.4 mM glucose
were added to HUVECs (2×105 cells/ml), which were
cultured for 24, 48 and 72 h. Doses of 0, 6.25, 12.5, 25, 50, 100,
200, 400, 800 and 1,600 µM AS-IV were added to HUVECs, which were
cultured for 48 h in the solution of 5.55 mM glucose. In addition,
doses of 25, 50, 100 µM AS-IV were added to HUVECs, which were
cultured for 48 h in a solution of 33.3 mM glucose.
The second series of experiments were designed to
examine the protective effect and possible mechanism of AS-IV.
Cells were divided into five groups at a density of
2×105 cells/ml: i) The control group, where cells were
cultured with 5.55 mM glucose and treated with 30 mM mannitol to
balance the osmotic pressure in culture medium for 48 h; ii) model
group, where cell damage was induced by incubation with 33.3 mM
glucose for 48 h; iii) the SP600125 inhibitor group, where cells
were cultured with 33.3 mM glucose for 48 h prior to the addition
of 20 µM SP600125 (JNK inhibitor); iv) AS-IV group, where cells
were cultured in 33.3 mM glucose for 48 h prior to the addition of
50 µM AS-IV; and v) the AS-IV+SP600125 group, where cells were
cultured in 33.3 mM glucose for 48 h prior to the addition of 50 µM
AS-IV and 20 µM SP600125. SP600125 was dissolved in DMSO (the final
concentration did not exceed 0.5%).
Cell viability assay
HUVECs in the logarithmic growth phase were seeded
in 96-well culture plates at 2×105 cells/well. Following
respective group treatments, 20 µl MTT (5 mg/ml) was added to 100
µl of culture medium in each well. A total of 4 h following this,
the medium was removed and 150 µl DMSO was added into each well.
The absorbance was measured at 490 nm using an automated microplate
reader (BioTek Instruments, Inc., Winooski, VT, USA). A total of
five independent experiments were repeated in each group. Cell
viability was calculated according to the following formula: Cell
viability (%)=average absorbance of the treated group/average
absorbance of the control group ×100%.
Cell apoptosis assay
Apoptosis was assessed with the Hoechst 33258
staining method, followed by photofluorography. Following the
respective group treatments, the nutrient solution was removed and
cells were washed twice with PBS. Subsequently, the cells were
fixed with 40 g/l paraformaldehyde fluid at 4°C for 10 min. Slides
were then washed twice with PBS. Once PBS was removed, the nuclear
DNA was stained with 5 mg/ml Hoechst 33258 at room temperature for
10 min and then visualized under a fluorescence microscope
(BX50-FLA; Olympus Corporation, Tokyo, Japan). The viable HUVECs
exhibited a uniform blue fluorescence throughout the nucleus,
whereas the apoptotic cells had fragmented and condensed nuclei.
The experiment was carried out three times.
Apoptosis rate was analyzed by labeling cells with
Annexin V-FITC apoptosis assay kit (cat.no. abs50001; Absin
Bioscience, Inc., Shanghai, China; www.absin.cn/). Following the treatment, cells were
collected using EDTA-free trypsin to digest the cells, followed by
centrifugation at 211 × g for 10 min at 4°C to collect the cells.
Cells were suspended with 500 µl binding buffer at a concentration
of 1×106 cells/ml and subsequently stained with 5 µl
annexin V-FITC at 4°C in the dark for 15 min followed by 10 µl PI
at 4°C in the dark for 5 min. Following this, cells were
immediately examined using a flow cytometer (Beckman Coulter, Inc.,
Brea, CA, USA) with FlowJo software (version 7.6.1; Tree Star,
Inc., Ashland, OR, USA).
ELISA analysis
The levels of IL-1β and TNF-α in culture supernatant
were determined using commercial ELISA kits according to the
manufacturers' protocols and determined using a microplate reader
at 450-nm wavelength (Multiskan FC; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from HUVECs using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. Template cDNA was prepared using a
RevertAid First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Inc.). Total RNA (5 µg) was reverse transcribed to cDNA
with 5X Reaction Buffer (4 µl), Oligo(dT)18 (1 µl), RevertAid
Reverse Transcriptase (1 µl), RiboLock RNase Inhibitor (1 µl), dNTP
Mix (2 µl) and ddH2O to supplement to a total volume of
20 µl. The synthesis temperature protocol was 60 min at 42°C, 5 min
at 70°C and 1 min at 4°C. The qPCR using the Quanti Nova SYBR Green
PCR kit (Qiagen GmbH, Hilden, Germany). qPCR was performed using an
ABI Prism 7500 Sequence Detection System (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The sequences of the primers were
as follows: 5′-TCAGCCAATCTTCATTGCTC-3′ (forward) and
5′-GCCATCAGCTTCAAAGAACA-3′ (reverse) for interleukin (IL)-1β;
5′-TTTGATCCCTGACATCTGGA-3′ (forward) and 5′-GGCCTAAGGTCCACTTGTGT-3′
(reverse) for tumor necrosis factor (TNF)-α; and
5′-GGGAAATCGTGCGTGACATTAAGG-3′ (forward) and
5′-CAGGAAGGAAGGCTGGAAGAGTG-3′ (reverse) for β-actin. The reaction
conditions included 95°C for 10 min, followed by 40 cycles of 95°C
for 30 sec, 58°C for 1 min and 72°C for 30 sec. Reaction
specificity was confirmed by melting curve analysis. Gene
expression profiles were normalized to β-actin and calculated using
the 2−ΔΔCq method (28)
for each sample.
Western blot analysis
Total proteins were extracted with a
radioimmunoprecipitation assay buffer (OriGene Technologies,
Beijing, China). Proteins from each experimental group were
quantified using the bicinchoninic acid assay (BestBio, Shanghai,
China). An equal amount of protein (30 µg) was loaded and subjected
to 10% SDS-PAGE. Gels were run at 120 V for 1.5 h and transferred
to polyvinylidene fluoride membranes at 120 V for 2 h. Non-specific
protein binding was blocked with 5% non-fat dried milk in
Tris-buffered saline with Tween 20 (TBST; 10 mM Tris-HCl, pH 7.4,
50 mM NaCl, 0.05% Tween-20) for 2 h at room temperature. Membranes
were incubated with anti-phospho-JNK (1:1,000; rabbit monoclonal
antibody, cat. no. ab124956), anti-JNK (1:1,000; cat. no.
ab179461), anti-cleaved-caspase-3 (1:1,000; cat. no. ab49822),
anti-cleaved-caspase-9 (1:1,000; cat. no. ab2324),
anti-phospho-ASK1 (1:1,000; cat. no. 3764), anti-ASK1 (1:1,000;
cat. no. 3762), anti-Cyt-c (1:1,000; cat. no. 11940), anti-Bax
(1:1,000; cat. no. 2772), anti-Bcl-2 (1:1,000; cat. no. 3498) and
anti-β-actin (1:1,000; cat. no. ab179467) overnight at 4°C. After
washing with TBST, the membranes were incubated with horseradish
peroxidase (HRP)conjugated goat anti-rabbit IgG secondary antibody
(1:20,000; cat. no. TA130015, OriGene Technologies) at room
temperature for 2 h. The signal was detected using an enhanced
chemiluminescence substrate kit (GE Healthcare, Chicago, IL, USA).
According to the manufacturer's protocol, immunoreactive bands of
proteins were scanned with a digital gel imaging and analysis
system (ProteinSimple, San Jose, CA, USA).
Statistical analysis
Data were expressed as the mean ± standard deviation
of three independent experiments and analyzed using SPSS version
13.0 (SPSS Inc., Chicago, IL, USA). The Student's ttest was used
for comparison between two groups. Multiple comparisons were
achieved using one-way analysis of variance with the
StudentNewmanKeuls test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of glucose on HUVEC
viability
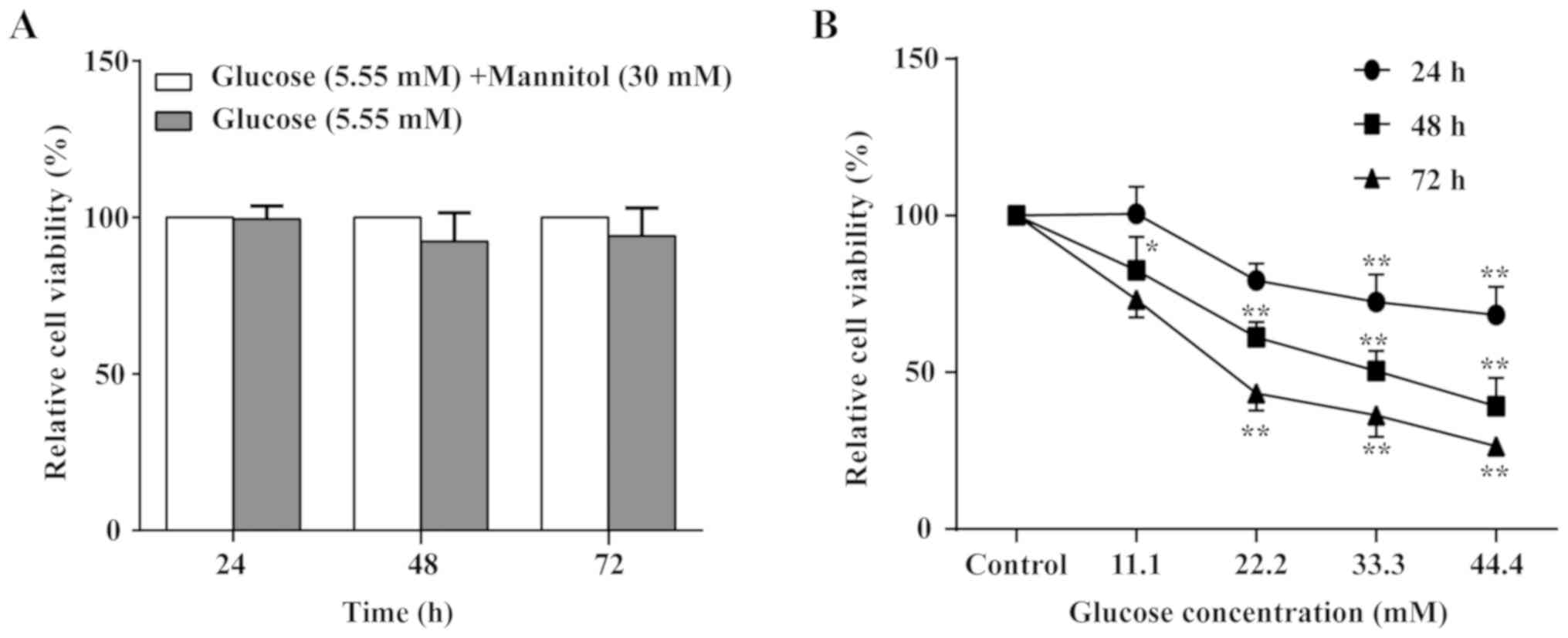
To confirm the optimal glucose concentration to
induce the cellular damage model, the MTT assay was performed to
observe HUVEC viability alterations in cells treated with glucose
for 24, 48 and 72 h at different concentrations (5.55–44.4 mM). As
indicated in Fig. 2, glucose
exhibited a time- and concentration-dependent inhibitory effect on
cell viability. The 50% inhibitory concentration (IC50)
value for the HUVECs was 66.90 mM at 24 h of glucose treatment;
however, the maximal glucose treatment still failed to reach half
of cell inhibition. The IC50 value was 21.89 mM at 72 h
of glucose treatment; however, the cells were unstable. Notably,
the IC50 value was 32.46 mM glucose treatment for 48 h
and the cells were stable. Combining the results of previous
studies (29,30) with the present study results, the
cell state was the most stable with 33.3 mM glucose treatment for
48 h. Therefore, 33.3 mM glucose for 48 h was selected to induce
the cellular damage model. In addition, there was no significant
difference in HUVEC viability between cells treated with glucose
mixed with mannitol and cells that received glucose alone (Fig. 2).
Effect of AS-IV on HUVEC
viability
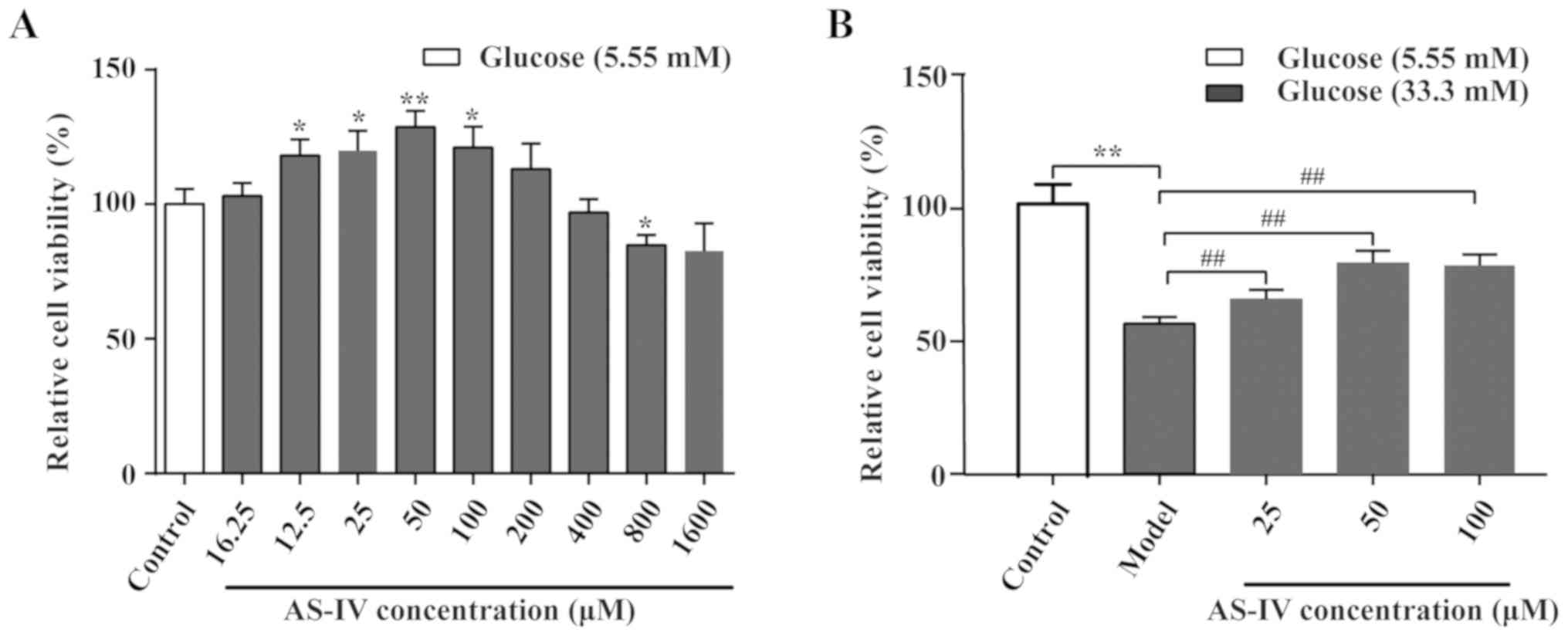
The AS-IV cell toxicity assay was performed
(Fig. 3A). Data indicated that
AS-IV could enhance cell viability over a specific concentration
range (16–200 µM). At higher concentrations (800 µM), AS-IV
significantly inhibited the cell viability compared with the
control group (P<0.05). The effects of AS-IV on HUVEC viability
under high glucose conditions were indicated (Fig. 3B). Data revealed that the cell
viability was optimal when the concentration of AS-IV was 50 µM
(P<0.01). Therefore, the 50 µM AS-IV was used in the later
experiments.
Effect of AS-IV on cell apoptosis in
HUVECs stimulated with high glucose conditions
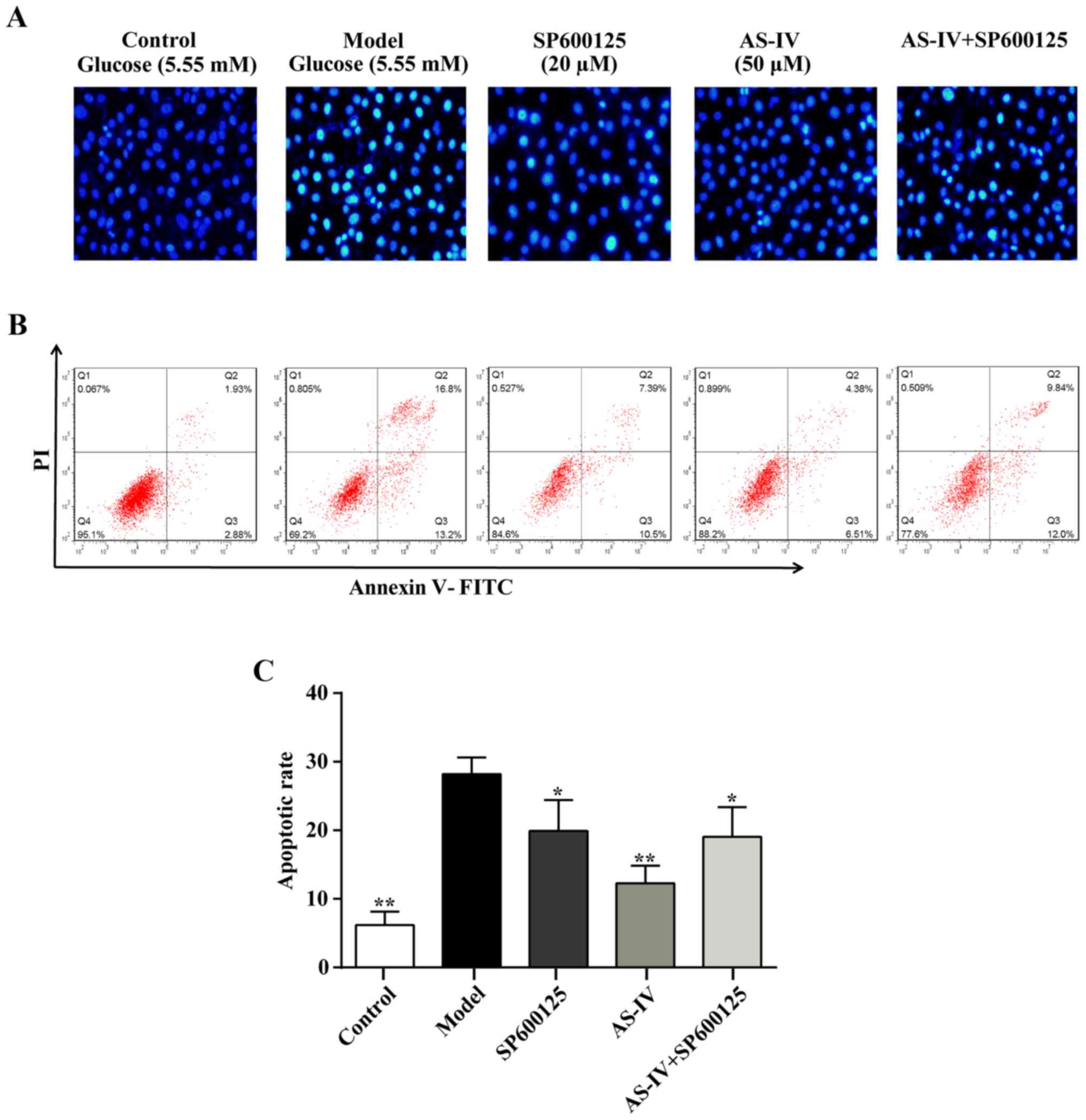
Hoechst 33258 staining was performed to identify the
nuclear morphology associated with apoptotic cell death. Exposure
to high glucose conditions for 48 h increased the number of
apoptotic cells (Fig. 4) and
nuclei pyknosis was observed. Furthermore, a large number of
chromosomal contractions could be observed in the model group.
Furthermore, specific apoptotic cell nuclei were light blue and an
apoptotic body was occasionally visible when compared with the
control group. The increased number of apoptotic cells was reversed
with AS-IV treatment (Fig. 4).
Similarly, the apoptosis rate of HUVECs was significantly decreased
with AS-IV pre-treatment according to annexin V-FITC and PI
staining analysis (P<0.05; Fig. 4B
and C).
Effect of AS-IV on high
glucose-stimulated inflammatory mediators
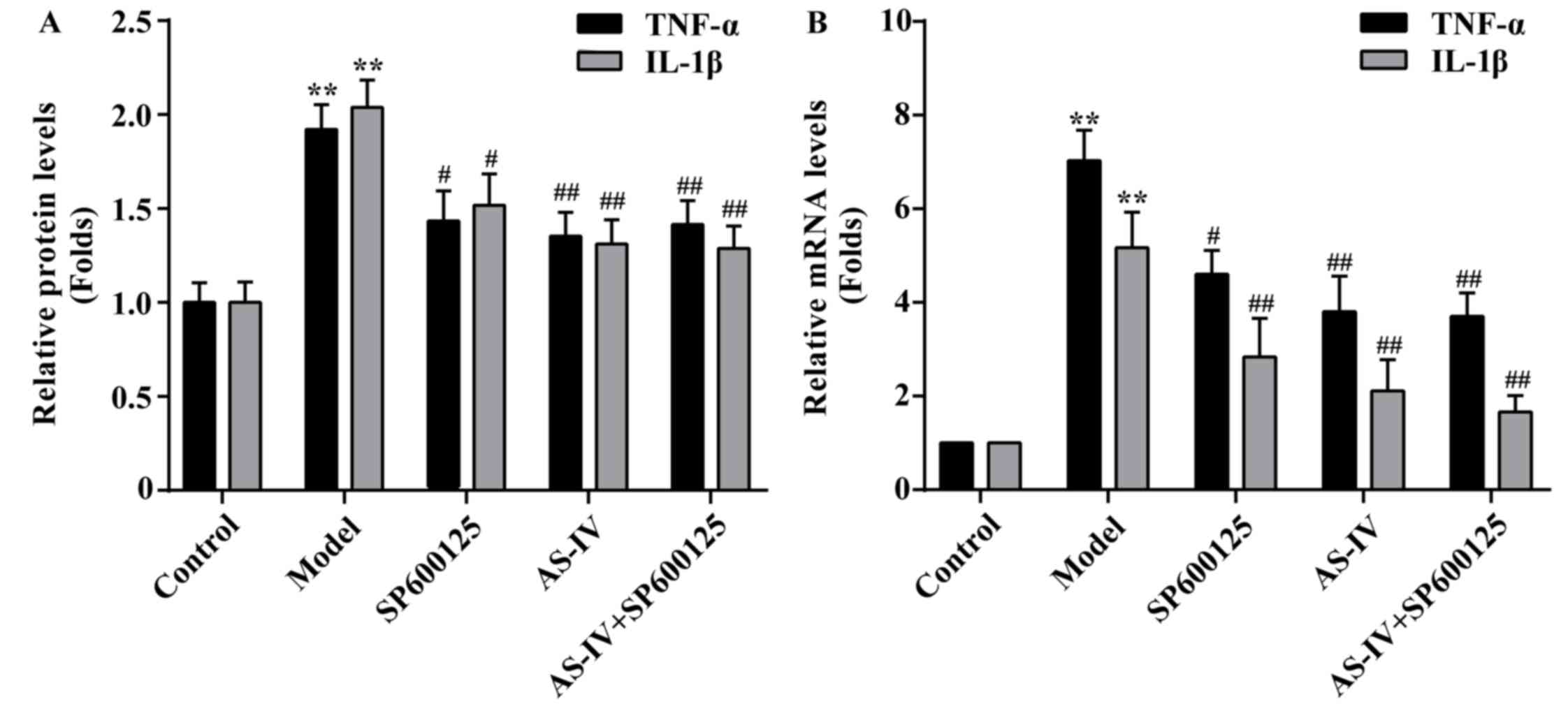
The cytokine levels of IL-1β and TNF-α in HUVECs
were measured using an ELISA assay. As indicated in Fig. 5A, treatment of HUVECs with high
glucose resulted in the significantly increased IL-1β and TNF-α
levels compared with the control (P<0.01). However, IL-1β and
TNF-α levels were significantly reduced following pre-treatment
with AS-IV (P<0.01).
Effect of AS-IV on the mRNA expression
levels of IL-1β and TNF-α
To further investigate the expression of
inflammatory factors, mRNA expression levels of IL-1β and TNF-α
were measured by RT-qPCR. As indicated in Fig. 5B, stimulation with high glucose
increased IL-1β and TNF-α mRNA expression levels in HUVECs.
However, pretreatment with AS-IV with could significantly decrease
the mRNA expression levels of IL-1β and TNF-α in HUVECs
(P<0.01).
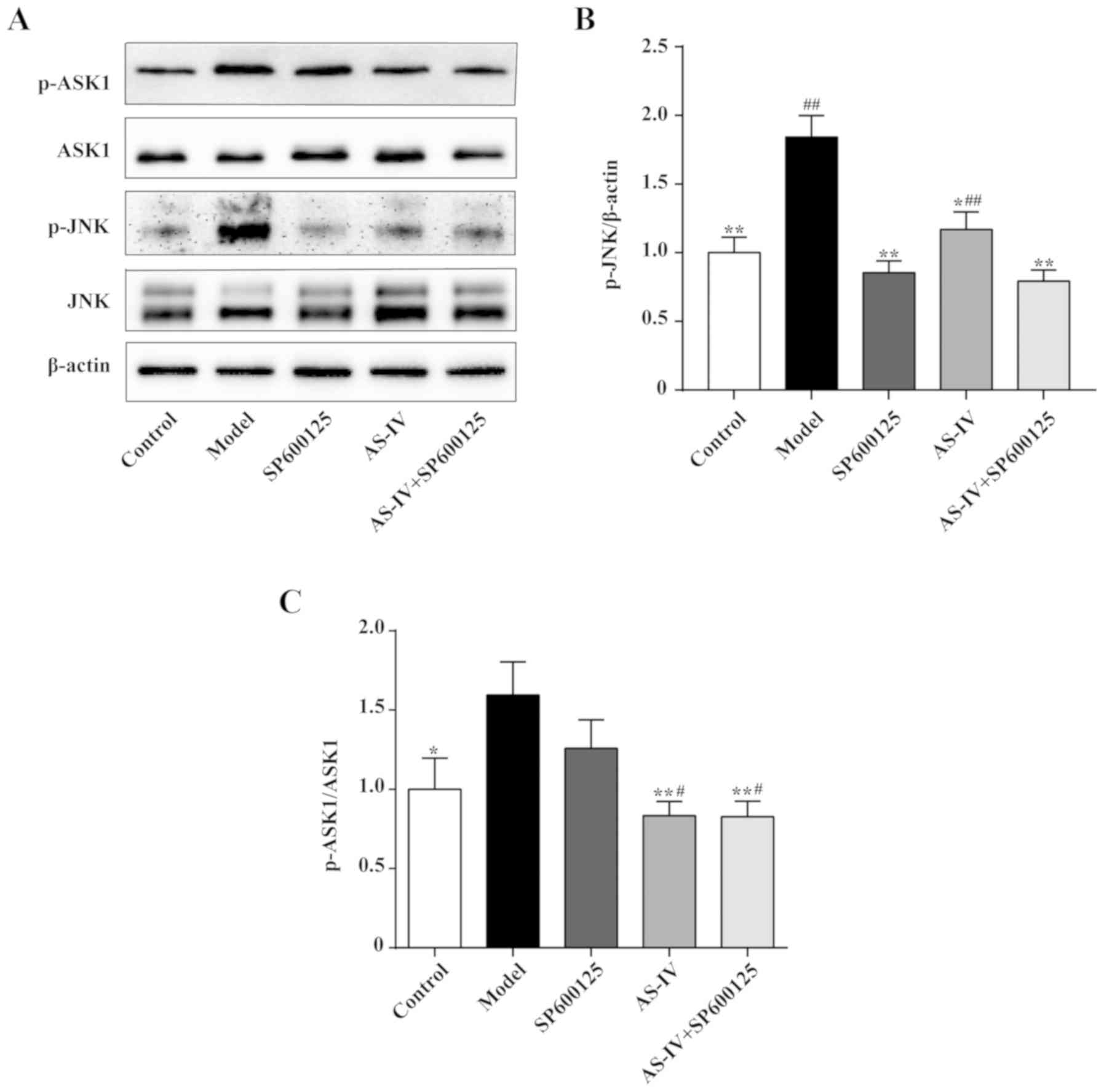
Effect of AS-IV on the expression of
the JNK signaling pathway
The JNK signaling pathway helps regulate the
inflammatory response. Western blot analysis was used to detect the
protein expression levels of JNK, p-JNK, ASK1 and p-ASK1 in HUVECs
in order to investigate the mechanism of action of AS-IV. As
indicated in Fig. 6, stimulation
with high glucose increased JNK and ASK1 phosphorylation in HUVECs.
AS-IV and the combination of AS-IV and JNK inhibitor could decrease
JNK and ASK1 phosphorylation (P<0.01 or P<0.05). Notably, the
JNK inhibitor could significantly decrease JNK phosphorylation
(P<0.01); however, the JNK inhibitor was unable to significantly
decrease ASK1 phosphorylation.
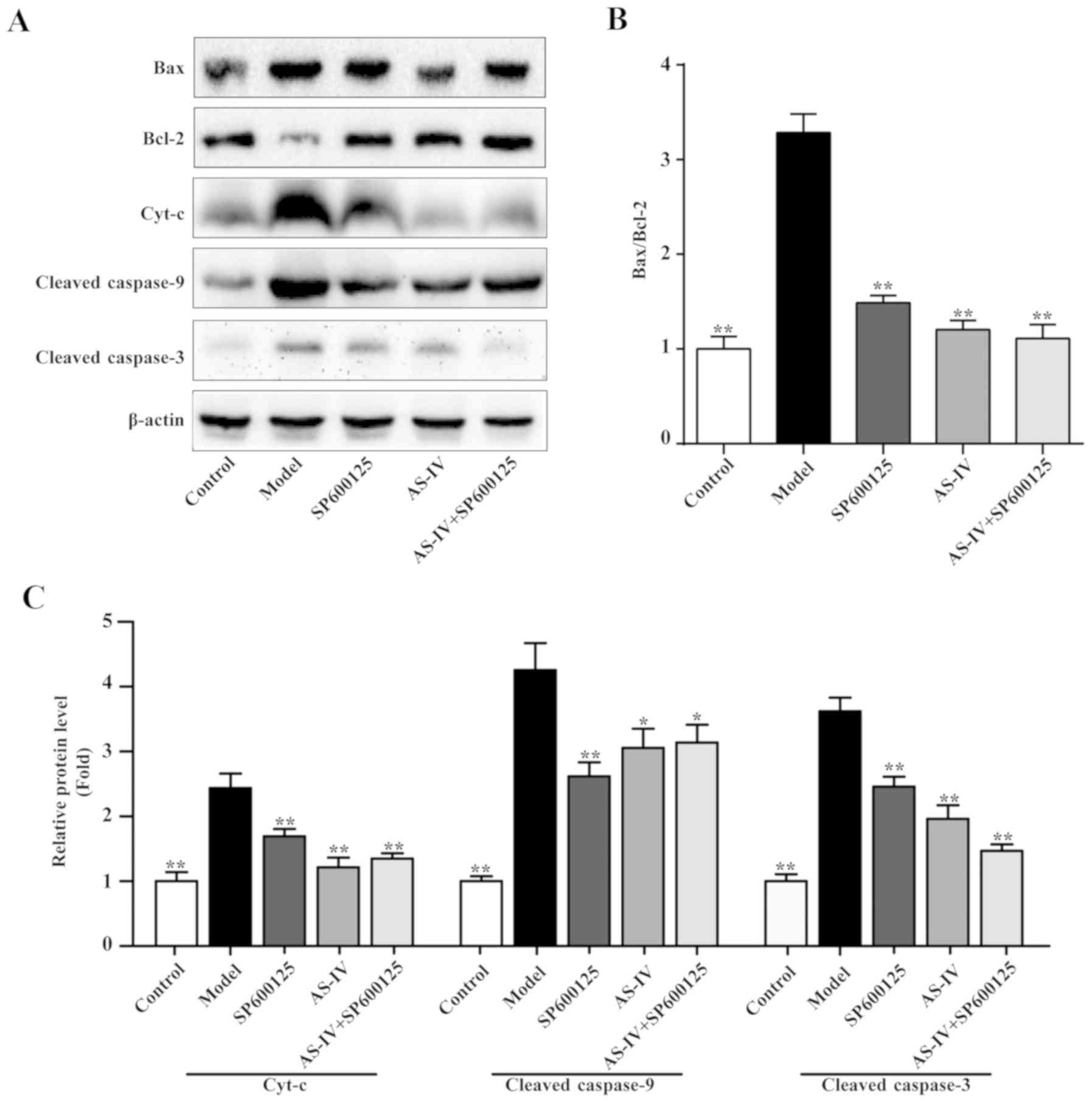
Effect of AS-IV on the expression
levels of Bax and Bcl-2
In order to determine the effect of AS-IV on the
protein expression levels of components associated with apoptosis
and the JNK signaling pathway in HUVECs, the Bax and Bcl-2 protein
expression levels were assessed. As indicated in Fig. 7, stimulation with high glucose
increased the protein expression levels of Bax and decreased the
expression levels Bcl-2 protein in HUVECs. AS-IV, the JNK inhibitor
and the combination of AS-IV and JNK inhibitor could decrease the
protein expression levels of Bax, increased the protein expression
levels of Bcl-2 and significantly decreased the ratio of Bax/Bcl-2
(P<0.01; Fig. 7).
Effects of AS-IV on the expression
levels of Cyt-c, cleaved-caspase-9 and cleaved-caspase-3
Apoptosis regulatory proteins Bax and Bcl-2 are
associated with the cell mitochondrial apoptotic pathways.
Therefore, western blot analysis was performed to detect the
expression of a series of key proteins involved in the
mitochondrial apoptosis pathway, including Cyt-c, cleaved-caspase-9
and cleaved-caspase-3. As indicated in Fig. 7, stimulation with high glucose
increased the protein expression levels of Cyt-c, cleaved-caspase-9
and cleaved-caspase-3; however, AS-IV, JNK inhibitor and the
combination of AS-IV with JNK inhibitor could significantly
decrease the expression levels of these proteins (P<0.05).
Discussion
The glucotoxicity of diabetes induces sustained
hyperglycemia, but also participates in the development of chronic
diabetic complications (31,32).
A large amount of medical evidence indicates that glucotoxicity is
associated with endothelial dysfunction, which is an important
factor involved in the development of macrovascular and
microvascular diabetic complications (32,33).
Notably, a number of diabetic incidents are associated with
inflammatory reactions (34–36).
The inflammatory response is the important mechanism of endothelial
dysfunction that can be promoted by glucotoxicity. Upon exposure to
high glucose conditions, HUVECs produce high levels of
proinflammatory cytokines, including TNF-α and IL-1β (37,38).
In the present study, the results indicated that high glucose
conditions decreased the HUVEC survival rate, increased cell
apoptosis and promoted the protein and mRNA expression levels of
TNF-α and 1L-1β. Previous studies have demonstrated that AS-IV
inhibits inflammation and protects endothelial function (26,39–41).
AS-IV has also been revealed to decrease the serum levels of TNF-a
in diabetic rats (39) and can
inhibit the expression of inflammatory factors in TNF-α-induced
HUVECs (40). Furthermore, AS-IV
can improve the structure of the aortic endothelium wall, inhibit
over-activation of monocyte chemoattractant protein-1 and TNF-α in
diabetic rats (26) and protect
endothelial cells from high glucose-induced barrier impairment
(41). In the present study, no
direct cytotoxic effect of ASIV was identified in HUVECs.
Additionally, the present findings suggested that ASIV enhanced the
HUVEC viability under high glucose conditions but also exerted an
inhibitory effect on high-glucose-induced HUVEC apoptosis and
inflammation. Notably, SP600125, an inhibitor of JNK, inhibited
high-glucose-induced TNF-α and IL-1β expression levels and cell
apoptosis. Similar effects were also observed with AS-IV and
SP600125 treatment. These results suggest that the effects of AS-IV
are associated with the JNK signaling pathway.
The JNK signaling pathway is sensitive to the
inflammatory response in diabetes complications. It has previously
been suggested that, under the stimulation of inflammatory
cytokines, which are involved in the endothelial dysfunction
process, the JNK signaling pathway is activated and raises the
number of endothelial cells damaged and cell apoptosis (18–20).
Inflammatory cytokines are the primary activators of the JNK
signaling pathway, which is a key regulator of pro-inflammatory
gene expression (16). It has been
reported that TNF-α induces increased phosphorylation of JNK in
HUVECs (42). ASK1 is a member of
the MAPK kinase kinase family, which activates JNK in response to a
variety of stress stimuli (43). A
previous study revealed that high glucose conditions induced the
increase of ASK1 expression and its activity in HUVECs (44). In the present study, high glucose
conditions in HUVECs resulted in increased ASK1 phosphorylation and
JNK expression. However, a significant decrease in JNK and ASKI
phosphorylation was indicated following treatment with AS-IV. In
addition, an inhibitor of JNK, SP600125 (45), was used as a reference to compare
the effectiveness of ASIV in the JNK signaling pathway. The results
indicated that AS-IV could significantly inhibit the
phosphorylation of high glucoseinduced JNK and ASK1, as well as the
expression of TNF-α and IL-1β. Furthermore, SP600125 significantly
suppressed the phosphorylation of JNK but not ASK1. Compared with
SP600125, the inhibitory effect of AS-IV on JNK phosphorylation was
decreased compared with SP600125; however, there was no notable
difference between SP600125 or AS-IV in combination with SP600125.
Furthermore, SP600125 and the combination of AS-IV with SP600125
had the ability to inhibit ASK1 phosphorylation, whereas SP600125
did not.
The increase of endothelial cell apoptosis is one of
the primary characteristics of endothelial dysfunction (4) and one of the most important effects
on HUVECs in high glucose conditions (46). The present data indicated that high
glucose conditions induced cell apoptosis. The JNK signaling
pathway activates apoptotic signaling, either by promoting the
expression of apoptotic genes in the nucleus or through modulating
the activities of associated mitochondrial apoptotic proteins by
phosphorylation events in the cytoplasm (23). In higher organisms,
mitochondria-mediated apoptosis is the predominant pathway of
cellular apoptosis (32). The
pathways associated with mitochondrial JNK-mediated apoptosis
include intrinsic apoptosis and extrinsic apoptosis pathways
(23). In the intrinsic apoptosis
pathway, JNK activates the internal cell mitochondrial apoptotic
pathway and changes the mitochondrial structure and function,
causing the release of Cyt-c into the cytoplasm (47). Following this, Cyt-c and an adaptor
protein are combined with active caspase-9, forming the apoptotic
bodies Cyt-c, caspase-9 and apoptotic protease activating factor-1,
which further activates caspase-3 and causes cell apoptosis
(48). Furthermore, JNK can adjust
the activity of Bcl-2 members by promoting the activation of
apoptosis proteins Bax, which are dimers in the cytoplasm. This
subsequently inhibits the protein expression of Bcl-2 and regulates
cell apoptosis (48,49). In the present study, the expression
levels of Bax, Cyt-c, cleaved-caspase-9 and cleaved-caspase-3 were
increased and Bcl-2 expression was decreased in high
glucose-stimulated HUVECs. However, the expression levels of Bax,
Cyt-c, cleaved-caspase-9 and cleaved-caspase-3 were reduced and
Bcl-2 expression was increased by treatment with ASIV. Compared
with ASIV, the effects of SP600125 and the combination of AS-IV
with SP600125 were not significantly different. This suggests that
the ability of AS-IV and JNK inhibitors in regulating apoptotic
proteins is similar. These data strongly support the
mitochondria-mediated apoptosis pathway being involved in the
pathogenesis of injury induced by high glucose conditions and ASIV
could reduce cell apoptosis associated with the
mitochondria-mediated apoptosis pathway. However, the effect of
ASIV on mitochondrial morphology and mitochondrial membrane
potential requires further observation in future studies to
identify the mechanism of apoptosis inhibition.
In conclusion, the present study indicates that high
glucosestimulation in HUVECs causes decreased cell activity,
increased the expression of apoptosis and inflammatory factors and
activated the JNK signaling pathway, and mitochondrial apoptosis
pathway. The findings suggested that ASIV may be valuable for the
prevention and treatment of diabetic vascular complications by
reducing cell apoptosis and inflammatory reactions through the
inhibition of the JNK signaling pathway and mitochondria-mediated
apoptosis pathway. However, other relevant mechanisms underlying
the effects of ASIV require further investigation.
Acknowledgements
The authors would like to thank the Key Laboratory
of Xinan Medicine of Chinese Ministry of Education (Hefei, China)
for their technical assistance.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81774286,
81573944 and 81273646), the Scientific Research Base of Traditional
Chinese Medicine Clinical of State Administration of Traditional
Chinese Medicine (grant no. JDZX2015001) and the National Basic
Public Health Services Project of State Administration of
Traditional Chinese Medicine (grant no. 20131012).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author on reasonable request.
Authors' contributions
ZF, GS and LY conceived the present study and edited
the manuscript. LY, QW, YH, SY, LW, MH and YL performed the
experiments and acquisition of data. ML and AJ were involved in
analysis and interpretation of data. All authors discussed the
results and implications and commented on the manuscript at all
stages, as well as in the final approval of the version to be
published.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Huang D, Refaat M, Mohammedi K, Jayyousi
A, Al Suwaidi J and Abi Khalil C: Macrovascular complications in
patients with diabetes and prediabetes. Biomed Res Int 2017.
78391012017.
|
|
2
|
Kaur R, Kaur M and Singh J: Endothelial
dysfunction and platelet hyperactivity in type 2 diabetes mellitus:
Molecular insights and therapeutic strategies. Cardiovasc Diabetol.
17:1212018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Galley HF and Webster NR: Physiology of
the endothelium. Br J Anaesth. 93:105–113. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Q, Zhang M, Ding Y, Wang Q, Zhang W,
Song P and Zou MH: Activation of NAD(P)H oxidase by
tryptophan-derived 3-hydroxykynurenine accelerates endothelial
apoptosis and dysfunction in vivo. Circ Res. 114:480–492. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hotamisligil GS: Inflammation and
metabolic disorders. Nature. 444:860–867. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Donath MY: Targeting inflammation in the
treatment of type 2 diabetes: Time to start. Nat Rev Drug Discov.
13:465–476. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yan SF, Ramasamy R, Naka Y and Schmidt AM:
Glycation, inflammation, and RAGE: A scaffold for the macrovascular
complications of diabetes and beyond. Circ Res. 93:1159–1169. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haidari M, Zhang W, Willerson JT and Dixon
RA: Disruption of endothelial adherens junctions by high glucose is
mediated by protein kinase C-beta-dependent vascular endothelial
cadherin tyrosine phosphorylation. Cardiovasc Diabetol. 13:1052014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sajja RK, Prasad S and Cucullo L: Impact
of altered glycaemia on blood-brain barrier endothelium: An in
vitro study using the hCMEC/D3 cell line. Fluids Barriers CNS.
11:82014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao XY, Wang XF, Li L, Zhang L, Shen DL,
Li DH, Jin QS and Zhang JY: Effects of high glucose on human
umbilical vein endothelial cell permeability and myosin light chain
phosphorylation. Diabetol Metab Syndr. 7:982015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen G, Chen Y, Chen H, Li L, Yao J, Jiang
Q, Lin X, Wen J and Lin L: The effect of NF-kappaB pathway on
proliferation and apoptosis of human umbilical vein endothelial
cells induced by intermittent high glucose. Mol Cell Biochem.
347:127–133. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kageyama S, Yokoo H, Tomita K,
Kageyama-Yahara N, Uchimido R, Matsuda N, Yamamoto S and Hattori Y:
High glucose-induced apoptosis in human coronary artery endothelial
cells involves up-regulation of death receptors. Cardiovas
Diabetol. 10:732011. View Article : Google Scholar
|
|
13
|
Ning RB, Zhu J, Chai DJ, Xu CS, Xie H, Lin
XY, Zeng JZ and Lin JX: RXR agonists inhibit high glucose-induced
upregulation of inflammation by suppressing activation of the NADPH
oxidase-nuclear factor-kappaB pathway in human endothelial cells.
Genet Mol Res. 12:6692–6707. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Owen GR, Achilonu I and Dirr HW: High
yield purification of JNK1beta1 and activation by in vitro
reconstitution of the MEKK1-->MKK4-->JNK MAPK phosphorylation
cascade. Protein Expr Purif. 87:87–99. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ip YT and Davis RJ: Signal transduction by
the c-Jun N-terminal kinase (JNK)-from inflammation to development.
Curr Opin Cell Biol. 10:205–219. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Newton K and Dixit VM: Signaling in innate
immunity and inflammation. Cold Spring Harb Perspect Biol. 4(pii):
a0060492012.PubMed/NCBI
|
|
17
|
Foletta VC, Segal DH and Cohen DR:
Transcriptional regulation in the immune system: All roads lead to
AP-1. J Leukoc Biol. 63:139–152. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kralisch S, Sommer G, Stangl V, Köhler U,
Kratzsch J, Stepan H, Faber R, Schubert A, Lössner U, Vietzke A, et
al: Secretory products from human adipocytes impair endothelial
function via nuclear factor kappaB. Atherosclerosis. 196:523–531.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shen C, Li Q, Zhang YC, Ma G, Feng Y, Zhu
Q, Dai Q, Chen Z, Yao Y, Chen L, et al: Advanced glycation
endproducts increase EPC apoptosis and decrease nitric oxide
release via MAPK pathways. Biomed Pharmacother. 64:35–43. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamawaki H, Saito K, Okada M and Hara Y:
Methylglyoxal mediates vascular inflammation via JNK and p38 in
human endothelial cells. Am J Physiol Cell Physiol.
295:C1510–C1517. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Breton-Romero R, Feng B, Holbrook M, Farb
MG, Fetterman JL, Linder EA, Berk BD, Masaki N, Weisbrod RM,
Inagaki E, et al: Endothelial dysfunction in human diabetes is
mediated by Wnt5a-JNK signaling. Arterioscler Thromb Vasc Biol.
36:561–569. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sabio G and Davis RJ: TNF and MAP kinase
signalling pathways. Semin Immunol. 26:237–245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dhanasekaran DN and Reddy EP:
JNK-signaling: A multiplexing hub in programmed cell death. Genes
Cancer. 8:682–694. 2017.PubMed/NCBI
|
|
24
|
Li L, Hou X, Xu R, Liu C and Tu M:
Research review on the pharmacological effects of astragaloside IV.
Fundam Clin Pharmacol. 31:17–36. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
You LZ, Lin YX, Fang ZH, Shen GM, Zhao JD
and Wang TT: Research advances on astragaloside-IV in treatment of
diabetes mellitus and its complications pharmacological effects.
Zhongguo Zhong Yao Za Zhi. 42:4700–4706. 2017.(In Chinese).
PubMed/NCBI
|
|
26
|
Yin Y, Qi F, Song Z, Zhang B and Teng J:
Ferulic acid combined with astragaloside IV protects against
vascular endothelial dysfunction in diabetic rats. Biosci Trends.
8:217–226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang WJ and Frei B: Astragaloside IV
inhibits NF-κB activation and inflammatory gene expression in
LPS-treated mice. Mediators Inflamm 2015. 2743142015.
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhong W, Zou G, Gu J and Zhang J:
L-arginine attenuates high glucose-accelerated senescence in human
umbilical vein endothelial cells. Diabetes Res Clin Pract.
89:38–45. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sheu ML, Chiang CK, Tsai KS, Ho FM, Weng
TI, Wu HY and Liu SH: Inhibition of NADPH oxidase-related oxidative
stress-triggered signaling by honokiol suppresses high
glucose-induced human endothelial cell apoptosis. Free Radic Biol
Med. 44:2043–2050. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Campos C: Chronic hyperglycemia and
glucose toxicity: Pathology and clinical sequelae. Postgrad Med.
124:90–97. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ceriello A: Point: Postprandial glucose
levels are a clinically important treatment target. Diabetes Care.
33:1905–1907. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Grassi D, Desideri G, Necozione S,
Ruggieri F, Blumberg JB, Stornello M and Ferri C: Protective
effects of flavanol-rich dark chocolate on endothelial function and
wave reflection during acute hyperglycemia. Hypertension.
60:827–832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jourdan T, Godlewski G, Cinar R, Bertola
A, Szanda G, Liu J, Tam J, Han T, Mukhopadhyay B, Skarulis MC, et
al: Activation of the Nlrp3 inflammasome in infiltrating
macrophages by endocannabinoids mediates beta cell loss in type 2
diabetes. Nat Med. 19:1132–1140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Masters SL, Dunne A, Subramanian SL, Hull
RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen
Z, et al: Activation of the NLRP3 inflammasome by islet amyloid
polypeptide provides a mechanism for enhanced IL-1β in type 2
diabetes. Nat Immunol. 11:897–904. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wei X, Song H, Yin L, Rizzo MG, Sidhu R,
Covey DF, Ory DS and Semenkovich CF: Fatty acid synthesis
configures the plasma membrane for inflammation in diabetes.
Nature. 539:294–298. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang W, Wang WH, Azadzoi KM, Dai P, Wang
Q, Sun JB, Zhang WT, Shu Y, Yang JH and Yan Z: Alu RNA accumulation
in hyperglycemia augments oxidative stress and impairs eNOS and
SOD2 expression in endothelial cells. Mol Cell Endocrinol.
426:91–100. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang X, Wu Z and He Y, Zhang H, Tian L,
Zheng C, Shang T, Zhu Q, Li D and He Y: Humanin prevents high
glucose-induced monocyte adhesion to endothelial cells by targeting
KLF2. Mol Immunol. 101:245–250. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang WJ, Hufnagl P, Binder BR and Wojta
J: Antiinflammatory activity of astragaloside IV is mediated by
inhibition of NF-kappaB activation and adhesion molecule
expression. Thromb Haemost. 90:904–914. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gui D, Huang J, Guo Y, Chen J, Chen Y,
Xiao W, Liu X and Wang N: Astragaloside IV ameliorates renal injury
in streptozotocin-induced diabetic rats through inhibiting
NF-κB-mediated inflammatory genes expression. Cytokine. 61:970–977.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li HB, Ge YK, Zhang L and Zheng XX:
Astragaloside IV improved barrier dysfunction induced by acute high
glucose in human umbilical vein endothelial cells. Life Sci.
79:1186–1193. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lo HM, Lai TH, Li CH and Wu WB: TNF-α
induces CXCL1 chemokine expression and release in human vascular
endothelial cells in vitro via two distinct signaling pathways.
Acta Pharmacol Sin. 35:339–350. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hattori K, Naguro I, Runchel C and Ichijo
H: The roles of ASK family proteins in stress responses and
diseases. Cell Commun Signal. 7:92009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yokoi T, Fukuo K, Yasuda O, Hotta M,
Miyazaki J, Takemura Y, Kawamoto H, Ichijo H and Ogihara T:
Apoptosis signal-regulating kinase 1 mediates cellular senescence
induced by high glucose in endothelial cells. Diabetes.
55:1660–1665. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shin M, Yan C and Boyd D: An inhibitor of
c-jun aminoterminal kinase (SP600125) represses c-Jun activation,
DNA-binding and PMA-inducible 92-kDa type IV collagenase
expression. Biochim Biophys Acta 1589. 311–316. 2002.
|
|
46
|
Zhu W, Yuan Y, Liao G, Li L, Liu J, Chen
Y, Zhang J, Cheng J and Lu Y: Mesenchymal stem cells ameliorate
hyperglycemia-induced endothelial injury through modulation of
mitophagy. Cell Death Dis. 9:8372018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tournier C: Requirement of JNK for
stress-induced activation of the cytochrome c-mediated death
pathway. Science. 288:870–874. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chauhan D, Li G, Hideshima T, Podar K,
Mitsiades C, Mitsiades N, Munshi N, Kharbanda S and Anderson KC:
JNK-dependent release of mitochondrial protein, Smac, during
apoptosis in multiple myeloma (MM) cells. J Biol Chem.
278:17593–17596. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kim BJ, Ryu SW and Song BJ: JNK- and p38
kinase-mediated phosphorylation of Bax leads to its activation and
mitochondrial translocation and to apoptosis of human hepatoma
HepG2 cells. J Biol Chem. 281:21256–21265. 2006. View Article : Google Scholar : PubMed/NCBI
|