Introduction
Waardenburg syndrome (WS), a type of
auditory-pigmentary syndrome, is the most common autosomal
dominantly inherited syndrome and is characterized by bright blue
eyes, moderate to profound hearing loss (HL), pigmental
abnormalities of the hair and skin, and dystopia canthorum
(1). This syndrome has been
observed in numerous ethnic groups (1,2). WS
is responsible for 2–5% of all cases of congenital deafness in
children (3). According to the
clinical characteristics, WS is divided into four types (4,5).
Type I [WS1; Mendelian Inheritance in Man (MIM) no. 193500] and
type II (WS2; MIM no. 193510) present common features:
Sensorineural HL and pigmental abnormalities. The two types are
distinguishable by the presence or absence of dystopia canthorum
indicated by the W index (type I: W>1.957; type II: W<1.95).
Type III (WS3; Klein-Waardenburg syndrome; MIM no. 148820) is
characterized by the presence of musculoskeletal abnormalities, in
addition to the typical disorders of WS1. Type IV (WS4;
Waardenburg-Hirschprung disease; MIM no. 277580) is characterized
by the presence of an aganglionic megacolon or gastrointestinal
atresia, in addition to the typical disorders of WS2 (6). WS is phenotypically homogenous;
however, it exhibits genetic heterogeneity. Paired box gene 3,
melanocyte inducing transcription factor (MITF),
snail family transcriptional repressor 2, endothelin 3,
endothelin receptor type B (EDNRB) and SRY-box 10
(SOX10) have been previously identified to be associated
with WS and are located on chromosomes 2, 3, 8, 13, 20 and 22,
respectively (7). De novo
mutations in the SOX10 gene, which is one of the genes
responsible for WS II, are rarely observed. Between 10 and 20
mutations in the SOX10 gene have been demonstrated to be
associated with WS2 (6). The
SOX10 gene is composed of 3–5 exons, depending on the splice
variant, and it is located on the human chromosome 22q13.1
(8). SOX10 is a key transcription
factor and serves as a nucleocytoplasmic shuttle in the development
of the neural crest and peripheral nervous system (9). Additionally, SOX10 is involved in the
regulation of embryonic development and the determination of cell
fate (10).
The complete coding regions of 168 candidate genes
were amplified and sequenced to identify possible mutations in the
Chinese proband. Molecular investigation identified a novel
nonsense mutation in the second exon of SOX10.
Materials and methods
Case data
The proband, a deaf boy, received a cochlear implant
in Kunming Children's Hospital of Yunnan (Kunming, China). A
questionnaire survey was conducted to collect the clinical data of
the proband and his parents. A complete physical examination was
performed by physicians in the hospital. The inner canthus, outer
canthus, pupillary distance and W index were calculated:
{X=[2A-(0.2119C+3.909)]/C, Y=[2A-(0.249B+3.909)]/B, W=X+Y+(A/B)},
where A is the inner canthus; B is pupillary distance; C is the
outer canthus; and X and Y are indexes. Visual reinforcement
audiometry, acoustic immittance, auditory brainstem response, 40 Hz
auditory evoked relative potential and hearing aid speech tests
were performed to assess the pre-operative hearing speech level.
The hearing level of the parents was assessed by pure tone
audiometry at frequencies of 250, 500, 1,000, 2,000, 4,000 and
8,000 Hz. The diagnostic criteria were the following: Normal
hearing [<20 decibels hearing level (dBHL)], mild (20–40 dBHL),
moderate (41–70 dBHL), severe (71–95) and profound (>95 dBHL)
deafness. Additionally, temporal bone computed tomography (CT)
scans and cranial magnetic resonance imaging (MRI) were performed.
Peripheral blood samples (2 ml) from the proband and his parents
were collected and informed consent was obtained on February 7th,
2017. The study was performed in accordance with the Declaration of
Helsinki and was approved by The Ethics Committee of Kunming
Children's Hospital. Additionally, 50 individuals of normal control
aged between 7 and 30 years old were enrolled in the present study,
including 30 males and 20 females without associated hereditary
diseases. Blood samples were collected on February 7th, 2017.
Written informed consent was obtained from all participants
enrolled in the present study.
DNA library preparation
Genomic DNA was extracted from peripheral blood with
a DNA extraction kit (Tiangen Biotech Co., Ltd., Beijing, China),
according to the manufacturer's protocol. The DNA was quantified
using a Nanodrop 2000 (Thermo Fisher Scientific, Inc., Wilmington,
DE, USA). Subsequently, >3 µg DNA was used for the construction
of the indexed Illumina libraries (Illumina, Inc., San Diego, CA,
USA). A total of 3 µg genomic DNA was fragmented using Covaris-S220
(Covaris, Inc., Woburn, MA, USA). An ‘A-tail’ was ligated to the 3′
end of each DNA fragment. Illumina adapters were ligated to the
fragments. The sample size aimed for was a 350–400 base pair (bp)
product, and the size-selected product was amplified by polymerase
chain reaction (Applied Biosystems 2720 PCR; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) as follows: Initial
denaturation of 98°C for 1 min, 9 cycles of denaturation at 98°C
for 20 sec, annealing at 65°C for 30 sec, extension at 72°C for 30
sec, and a final extension of 72°C for 5 min. All samples were
checked with a Nanodrop 2000 or Qubit 4 Fluorometer (Thermo Fisher
Scientific, Inc.) to determine whether they represented a
qualifying captured library. DNA fragments between 350 and 450 bp,
and the oligonucleotides containing the adapter sequences were
selected for the DNA libraries.
Targeted gene capture and
sequencing
The exons and flanking intronic regions of 168
candidate genes associated with hearing impairment diseases were
captured and enriched with the GenCap custom enrichment kit
(Beijing Kangwei Century Biotechnology Co., Ltd., Beijing, China),
according to the manufacturer's protocol. The PCR product was
purified with solid phase reversible immobilization beads (Beckman
Coulter, Inc., Brea, CA, USA). The enriched libraries were
sequenced using the Illumina HiSeq 2500 sequencer (Illumina, Inc.)
for paired-end reads of 150 bp.
Bioinformatics analysis of candidate
variants
Data analysis and bioinformatics processing were
performed as previously described (11). Following the sorting of the raw
reads, the low-quality reads and adaptor sequences were filtered
using Cutadapt software (v1.16; http://cutadapt.readthedocs.io/en/stable).
Subsequently, the selected high-quality reads were aligned to the
reference human genome (NCBI database build 37/hg19; http://www.ncbi.nlm.nih.gov/grc) with the Burrows
Wheeler Aligner multi-vision software package (12). Single nucleotide polymorphisms
(SNPs) and insertion or deletion of bases (indels) were identified
using BWA (http://bio-bwa.sourceforge.net) and the Genome
Analysis Toolkit Indel Genotyper (http://www.broadinstitute.org/gsa/wiki/index.php),
respectively. The SNPs and the indels were filtered if their
frequency was >5% in various databases, including dbSNP138
(http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000
Genomes Project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp), ClinVar and
Exome Sequencing Project v. 6500 (https://esp.gs.washington.edu/drupal), HapMap
(ftp://ftp.ncbi.nlm.nih.gov/hapmap/), the Human Gene
Mutation Database (HGMD; http://www.ghmd.cf.ac.uk/), SIFT (http://sift.jcvi.org), PolyPhen (http://genetics.bwh.harvard.edu/pph2/)
and MutationTaster (www.mutationtaster.org) were used to predict the
pathogenic variants of genes.
Validation by Sanger sequencing
The DNA of the parents of the proband was purified
in order to perform Sanger sequencing. The polymerase chain
reaction (PCR) samples were visualized on agarose gels, purified
and sequenced using the ABI PRISM 3730 genetic analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Following the comparison among DNA sequences, genomic sites
exhibiting variations were identified with the corresponding
GenBank (www.ncbi.nlm.nih.gov) reference
sequences. A primer pair was designed to amplify exon 2 of the
SOX10 (gene ID: 6663; transcript ID: NM_006941): Exon 2
forward: 5′-CTGAGCCCACACCATGAAG-3′ and exon 2 reverse:
5′-GTTGGACTCTTTGCGAGGAC-3′. PCR amplifications were performed using
Golden Star T6 Super PCR Mix (1.1X; Qingke Xinye Biotechnology Co.,
Ltd, Beijing, China) as follows: Initial denaturation of 95°C for 5
min, 32 cycles of denaturation at 95°C for 30 sec, annealing at
60.3–59.9°C for 30 sec, extension at 72°C for 30 sec, and a final
extension of 72°C for 7 min. The purified PCR products with a size
of a 426 bp were sequenced by Sanger sequencing (Biosune, Beijing,
China; http://www.biosune.com). The sequencing
results were analysed using Mutation Surveyor 5.0.0 (SoftGenetics,
Inc., State College, PA, USA).
Results
Clinical features of the patients with
WS2
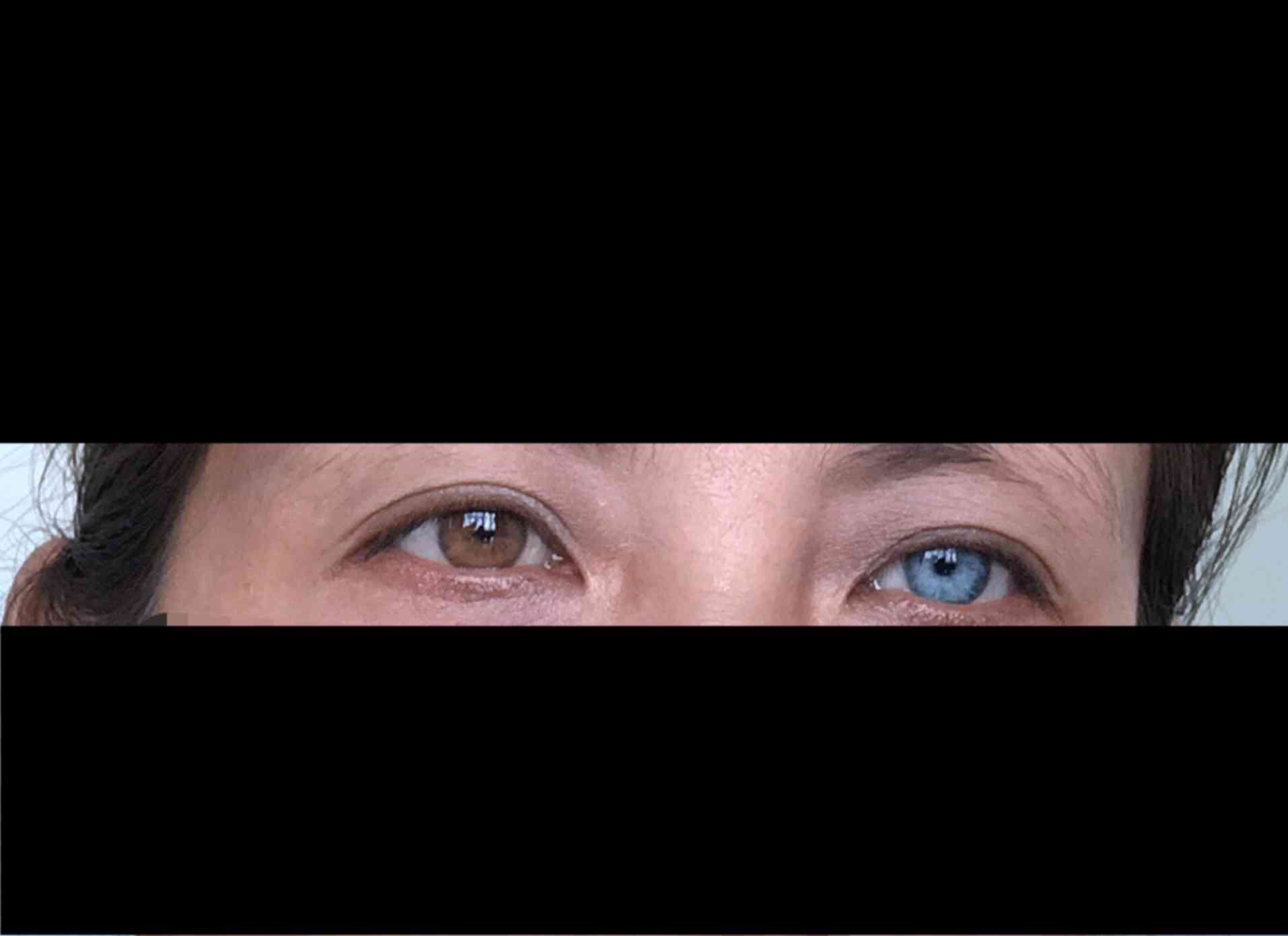
The right iris of the proband has been bright blue
since birth (Fig. 1). At 3 months
old, he did not exhibit any response to external audio stimuli and
at 1 year old he was not capable of speech. Ear injury, otitis
media and contact with ototoxic drugs were not detected. Skin
depigmentation was observed, eyesight and intelligence were normal,
no dystopia cantorum was present, and no abnormalities affecting
the digestive system or the skeletal muscles were detected. At
birth and at 42 days old, otoacoustic emissions of the two ears
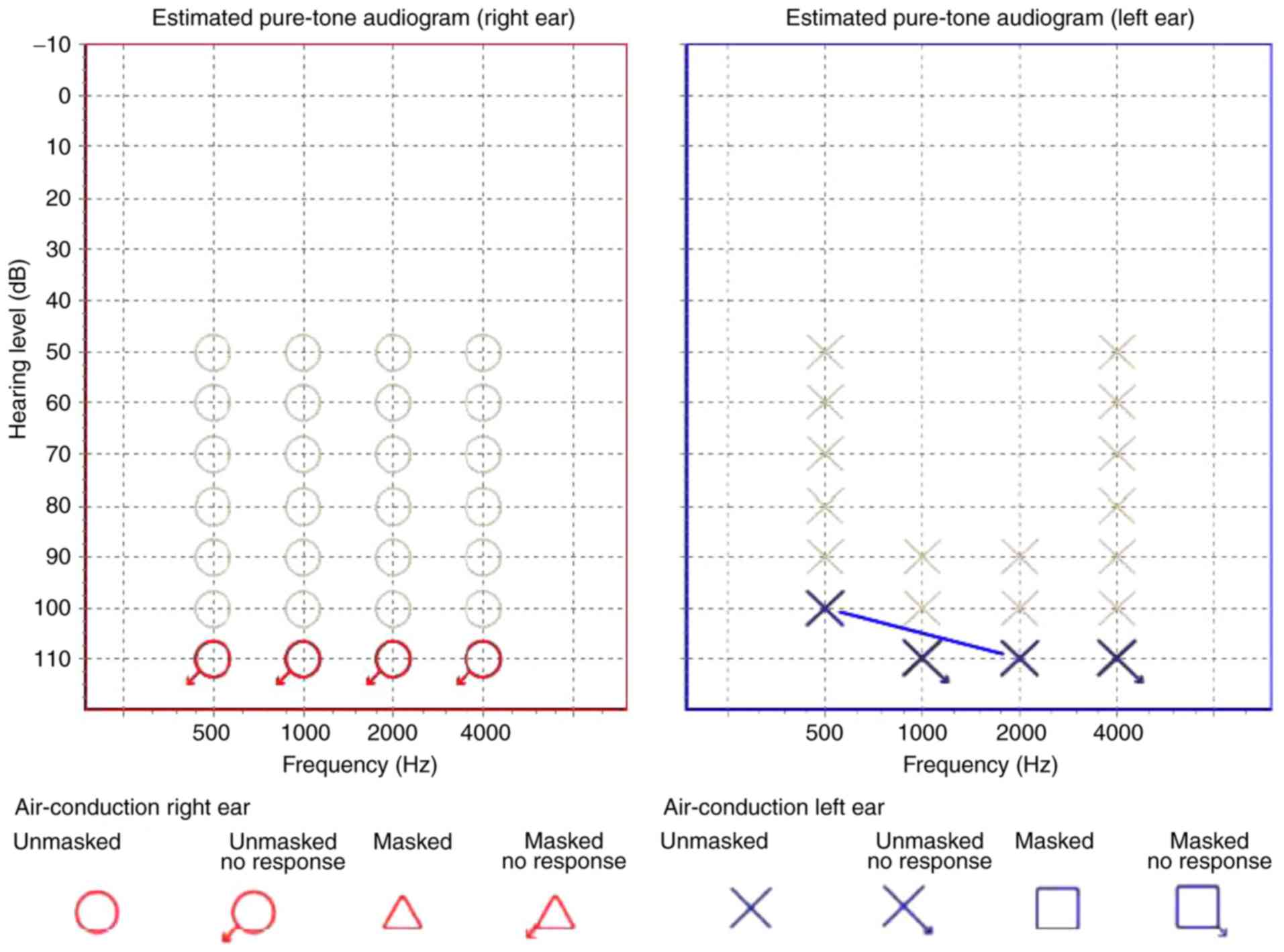
were not present. At 10 months old, bilateral congenital profound
sensorineural HL was confirmed (Fig.
2). A temporal CT scan, cranial MRI and abdomen B-scan
ultrasounds did not identify any abnormalities. The above clinical
manifestations fit the criteria of WS2. The proband was diagnosed
with WS2, according to the WS diagnostic criteria (13,14).
Left heterochromia iridis and hair hypopigmentation
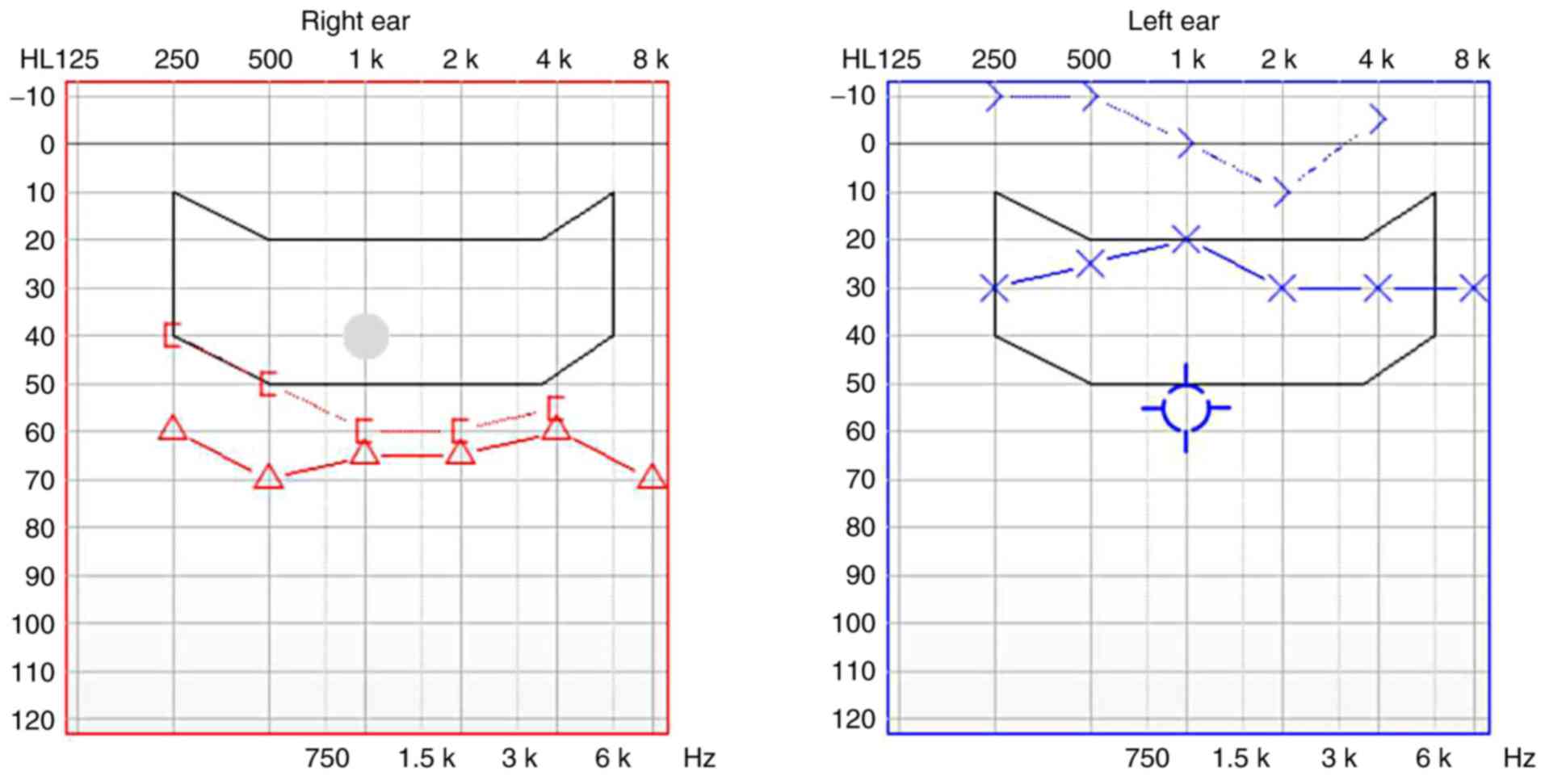
were observed in the mother of the proband (Fig. 3). However, HL was not detected by
sound field audiometry. It was identified that the average
air-conduction hearing threshold of the right ear at 500, 1,000,
2,000 and 3,000 Hz on the audiogram was 60 dBHL (moderate HL) and
the hearing threshold of the left ear was 30 dBHL (mild HL;
Fig. 4). The mother of the proband
exhibited normal speech ability. Transmission of hearing
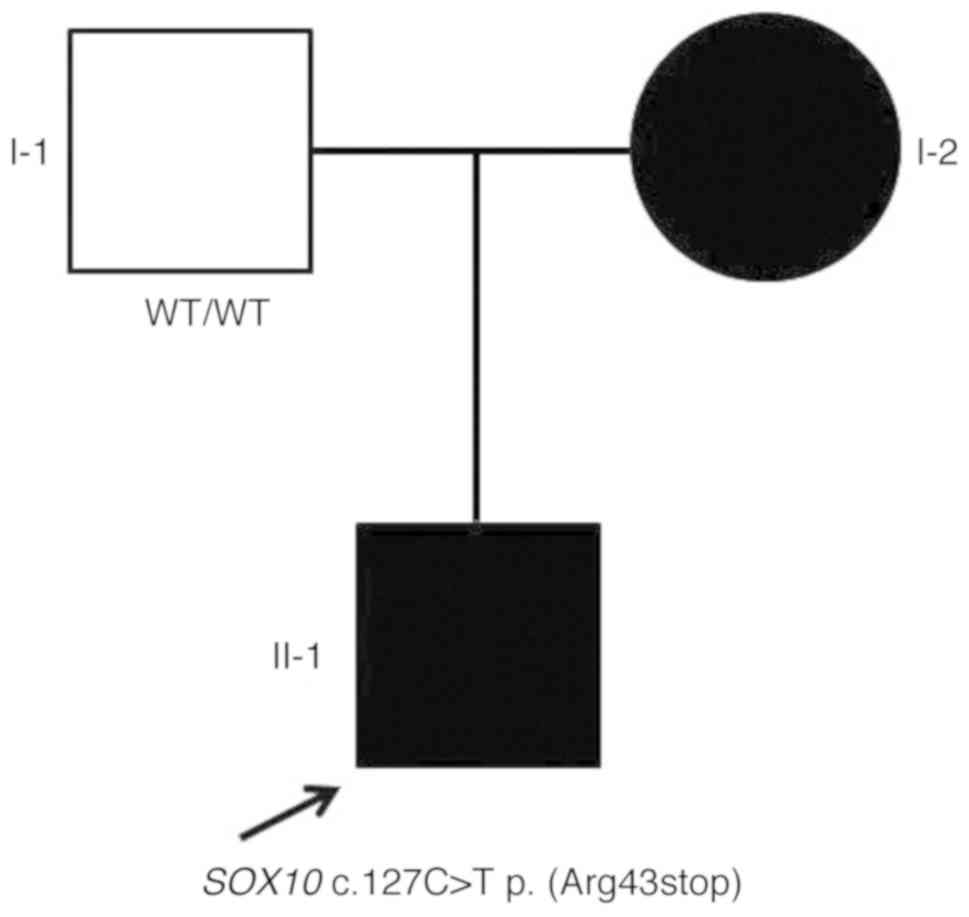
impairment, as highlighted by the pedigree (Fig. 5), suggested a possible autosomal
dominant mode of inheritance with incomplete penetrance. The father
of the proband did not exhibit heterochromia iridis, HL or
pigmentation abnormality. The parents of the proband state in the
interviews that no obvious abnormalities were present in other
family members.
Identification of a novel SOX10
termination mutation
The genomic DNA of the proband was extracted, and
the coding exons plus ~100 bp of the flanking intronic sequences of
168 deafness-associated genes were sequenced. The raw data
comprised of ~805 Mb. The coverage of the target regions was 98.6%.
The average sequencing depth of the target coverage area was 346; a
total of 539 nucleotide variations were detected, including 48
indels.
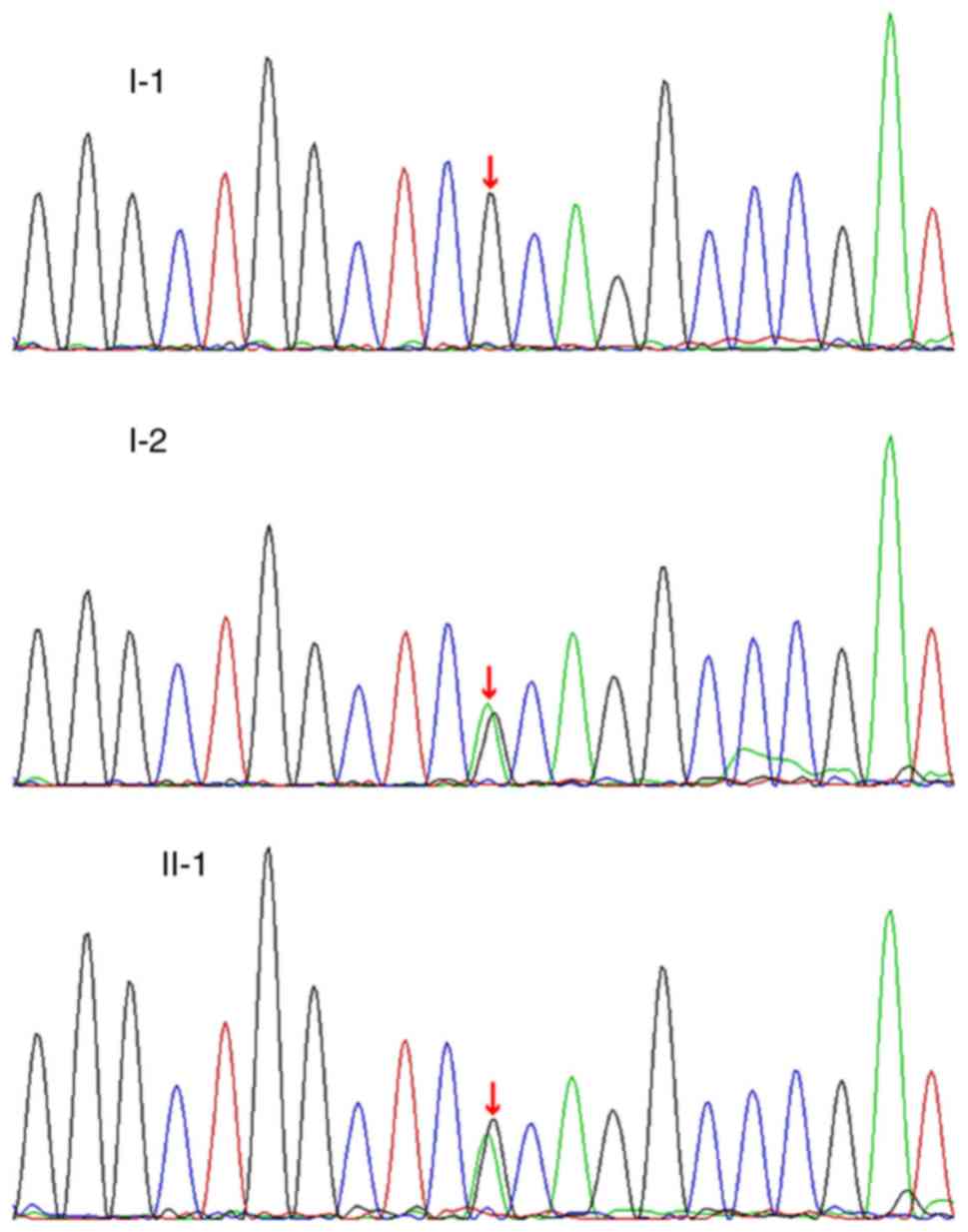
A heterozygous mutation cytosine (C) to thymine (T)
in position 127 (c.127C>T) was located in the second exon of
SOX10, leading to a substitution of the 43rd codon: The
codon arginine was replaced by a stop codon, causing premature
termination of protein translation. According to the standards and
the guidelines of The American College of Medical Genetics and
Genomics (15), this type of
variant is considered to be likely pathogenic and represents a
novel variant that was not identified in the following databases:
dbSNP, HapMap, 1000 Genomes Project and the HGMD. Additionally,
this variant was absent in 50 normal control individuals. The
mother exhibited the same variant (SOX10, exon 2,
c.127C>T). The father exhibited a wild-type genotype (Fig. 6). Furthermore, no additional
mutations associated with WS were identified in the proband.
Discussion
Among the congenital genetic diseases, the WS is the
most common syndrome involving HL, and this syndrome exhibits a
single-gene pathogenic autosomal dominant inheritance with
incomplete penetrance (16). WS is
divided in four types according to clinical characteristics and
various accompanying phenotypes. Children with WS2 exhibit
significant phenotypic characteristics, including sensorineural
prelingual palsy, unilateral bright blue irises and hypopigmented
hair, and a W index of 1.85 (<1.95). Therefore, the clinical
diagnosis of the present proband was WS2. SOXl0 serves an
important role in the pathogenesis of WS and melanocyte
development, and mutations in this gene may lead to WS2 and WS4
(17). It is estimated that ~15%
of the WS2 pathogenesis is associated with mutations in the
SOX10 gene (17), and the
genetic data derived from patients with WS2 is increasing in
numerous countries; however, the molecular mechanism underlying a
large proportion of WS2 cases remains unclear (17). SOX10 belongs to the SOX gene
superfamily (18), that is
characterized by a highly conserved and active domain, the high
mobility group (HMG) (19), whose
principal function is to recognize and bind to the promoters of the
target genes.
SOX10 is a key transcription factor involved in the
migration and differentiation of neural crest cells and it is able
to function alone or in combination with other transcription
factors by binding to the promoters or enhancers of target genes
(20,21). The downstream target genes of SOX10
are the following: MITF, tyrosinase, tyrosinase related protein
1, dopachrome tautomerase, myelin protein zero, gap junction
protein β 1, ret proto-oncogene and EDNRB (20,21).
These target genes are directly or indirectly involved in melanin
synthesis, and their expression is regulated by SOX10. The majority
of mutations affecting the SOX10 gene result in a
termination codon that may cause WS4 and WS2. In addition to WS,
mutations in the SOX10 gene may lead to other neural
crest-associated diseases (22,23),
including peripheral demyelinating neuropathy, central
dysmyelinating leukodystrophy and Hirschsprung's disease.
WS is a genetic disorder characterized by numerous
and complex genetic mutations that exhibit various clinical
manifestations. Individuals with the same type of WS, even in the
same family, may exhibit unique phenotypic traits. The present
study identified that the proband and his mother carried a
c.127C>T heterozygous mutation in exon 2 of the SOX10
gene. However, the hearing defects between the proband and his
mother were phenotypically distinct. The proband exhibited severe
bilateral HL, and his mother exhibited moderate HL in the left ear
and mild HL in the right ear. Chen et al (24) demonstrated that the mutation
c.760C>T causing WS2 was located in the seventh exon of
MITF, leading to a premature termination codon, as Wilcox
(25) observed. However, families
carrying the same mutation exhibited distinct phenotypes,
suggesting that additional genetic variations may be responsible
for the partial penetrance and the variability of the clinical
manifestations of WS. Notably, this phenomenon was observed among
individuals of the same family, carrying the same mutation. In the
present study, the proband and his mother exhibited the same
pathogenic variation; however, the clinical phenotypes were
heterogeneous. The present results suggested that the genetic
background serves an important role in the clinical phenotype. The
proband presented the typical symptoms of WS, including
sensorineural deafness and iris discoloration, in addition to
hypochromic hair alterations in the body, which demonstrated the
multiple functions of the SOX10 protein. The c.127C>T mutation
is a heterozygous mutation in the second exon of SOX10, that
causes an alteration in the translation product; the arginine in
the 43rd position is replaced by a stop codon. The early stop codon
leads to a premature termination of the protein translation,
causing the loss of the majority of the HMG domain. In 50 healthy
controls selected randomly, this mutation was not identified.
Therefore, the mutation SOX10 (c.127C> T, Arg43stop) was
considered a causative mutation.
In the present study, clinical analyses and
diagnoses based on detailed clinical data of patients were
performed. In addition, a genetic mutation associated with WS was
identified, and the patient received a genetic diagnosis. The novel
mutation identified in the SOX10 gene provides additional
insight for the mechanism underlying the disease and refines the
human gene mutation database. The experimental data presented may
help in examining the associations between various types of WS and
in reassessing the current clinical classification of WS.
Additionally, the present results provided novel insight to improve
the understanding of SOX10 function. The present study may
represent an important basis for prenatal genetic diagnosis and
provides novel insight for the treatment of patients with WS.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from The
National Natural Science Fund of China (grant no. 81660175; China),
The Basic Applied Study Planning Projects of Yunnan Province (grant
no. 2015FB090), The High-level Health and Family Planning Technical
Personnel Training Projects of Yunnan Province (grant no. D201637),
and Yunnan Children's Hearing Impairment and Speech Disease
Comprehensive Prevention Innovation Team.
Availability of data and materials
The analyzed data sets generated during the present
study are available in the Figshare repository, https://figshare.com/account/home#/data.
Authors' contributions
JM, ZZ, TSZ and BR were responsible for the
conceptualization of the study. ZZ, HCJ, HS and LPZ were
responsible for the data curation. JM and ZZ acquired the funding.
JM, ZZ, YQG, ZCL, YX and GLW performed the investigation. JM, ZZ,
HCJ and CM were responsible for the methodology. TSZ and BR were
responsible for project administration. CM, YQG and MHS were
responsible for software. TSZ and BR were responsible for
supervision. JM, ZZ and HCJ wrote the original draft. JM, TSZ and
BR reviewed and edited the manuscript.
Ethics approval and consent to
participate
The study was performed in accordance with the
Declaration of Helsinki and was approved by The Ethics Committee of
Kunming Children's Hospital (Kunming, China). Written informed
consent was obtained from all participants enrolled in the present
study.
Patient consent for publication
The mother of the proband provided consent for the
publication of images of her and the proband.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Otman SG and Abdelhamid NI: Waardenburg
syndrome type 2 in an African patient. Indian J Dermatol Venereol
Leprol. 71:426–427. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Morín M, Viñuela A, Rivera T, Villamar M,
Moreno Pelayo MA, Moreno F and Del Castillo I: A de novo missense
mutation in the gene encoding the SOX10 transcription factor in a
Spanish sporadic case of Waardenburg syndrome type IV. Am J Med
Genet A. 146A:1032–1037. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cullen RD, Zdanski C, Roush P, Brown C,
Teagle H, Pillsbury HR III and Buchman C: Cochlear implants in
Waardenburg syndrome. Laryngoscope. 116:1273–1275. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bondurand N, Dastot-Le MF, Stanchina L,
Collot N, Baral V, Marlin S, Attie-Bitach T, Giurgea I, Skopinski
L, Reardon W, et al: Deletions at the SOX10 gene locus cause
Waardenburg syndrome types 2 and 4. Am J Hum Genet. 81:1169–1185.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pingault V, Ente D, Dastot-Le MF, Goossens
M, Marlin S and Bondurand N: Review and update of mutations causing
Waardenburg syndrome. Hum Mutat. 31:391–406. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ma J, Zhang T, Lin K, Sun H, Jiang HC,
Yang YL, Low F, Gao YQ and Ruan B: Waardenburg syndrome type II in
a Chinese patient caused by a novel nonsense mutation in the SOX10
gene. Int J Pediatr Otorhinolaryngol. 85:56–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Unzicker A, Pingault V, Meyer T, Rauthe S,
Schütz A and Kunzmann S: A novel SOX10 mutation in a patient with
PCWH who developed hypoxic-ischemic encephalopathy after E. coli
sepsis. Eur J Pediatr. 170:1475–1480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuhlbrodt K, Herbarth B, Sock E,
Hermans-Borgmeyer I and Wegner M: Sox10, a novel transcriptional
modulator in glial cells. J Neurosci. 18:237–250. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen K, Zong L, Liu M, Zhan Y, Wu X, Zou W
and Jiang H: De novo dominant mutation of SOX10 gene in a Chinese
family with Waardenburg syndrome type II. Int J Pediatr
Otorhinolaryngol. 78:926–929. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang XF, Xiang P, Chen J, Xing DJ, Huang
N, Min Q, Gu F, Tong Y, Pang CP, Qu J and Jin ZB: Targeted exome
sequencing identified novel USH2A mutations in Usher syndrome
families. PLoS One. 8:e638322013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Milunsky JM: Waardenburg syndrome type I.
GeneReviews. In. 2017.
|
|
14
|
Tabor D: Waardenburg syndrome. DermNet NZ.
In. 2015.
|
|
15
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kochhar A, Hildebrand MS and Smith RJ:
Clinical aspects of hereditary hearing loss. Genet Med. 9:393–408.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bondurand N, Dastot-Le MF, Stanchina L,
Collot N, Baral V, Marlin S, Attie-Bitach T, Giurgea I, Skopinski
L, Reardon W, et al: Deletions at the SOX10 gene locus cause
Waardenburg syndrome types 2 and 4. Am J Hum Genet. 81:1169–1185.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harris ML, Baxter LL, Loftus SK and Pavan
WJ: Sox proteins in melanocyte development and melanoma. Pigment
Cell Melanoma Res. 23:496–513. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schreiner S, Cossais F, Fischer K, Scholz
S, Bösl MR, Holtmann B, Sendtner M and Wegner M: Hypomorphic Sox10
alleles reveal novel protein functions and unravel developmental
differences in glial lineages. Development. 134:3271–3281. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Murisier F, Guichard S and Beermann F: The
tyrosinase enhancer is activated by Sox10 and Mitf in mouse
melanocytes. Pigment Cell Res. 20:173–184. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hou L, Arnheiter H and Pavan WJ:
Interspecies difference in the regulation of melanocyte development
by SOX10 and MITF. Proc Natl Acad Sci USA. 103:9081–9085. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Inoue K, Khajavi M, Ohyama T, Hirabayashi
S, Wilson J, Reggin JD, Mancias P, Butler IJ, Wilkinson MF, Wegner
M and Lupski JR: Molecular mechanism for distinct neurological
phenotypes conveyed by allelic truncating mutations. Nat Genet.
36:361–369. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pingault V, Guiochon-Mantel A, Bondurand
N, Faure C, Lacroix C, Lyonnet S, Goossens M and Landrieu P:
Peripheral neuropathy with hypomyelination, chronic intestinal
pseudo-obstruction and deafness: A developmental ‘neural crest
syndrome’ related to a SOX10 mutation. Ann Neurol. 48:671–676.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen J, Yang SZ, Liu J, Han B, Wang GJ,
Zhang X, Kang DY, Dai P, Young WY and Yuan HJ: Mutation screening
of MITF gene in patients with Waardenburg syndrome type 2. Yi
Chuan. 30:433–438. 2008.(In Chinese). View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lalwani AK, Attaie A, Randolph FT,
Deshmukh D, Wang C, Mhatre A and Wilcox E: Point mutation in the
MITF gene causing Waardenburg syndrome type II in a
three-generation Indian family. Am J Med Genet. 80:406–409. 1998.
View Article : Google Scholar : PubMed/NCBI
|