Introduction
Hereditary spherocytosis (HS) is a group of
heterogeneously inherited hemolytic anemic disorders, characterized
by the presence of spherical-shaped spherocytes on peripheral blood
film, jaundice and splenomegaly (1). HS is the most frequent cause of
inherited chronic hemolysis in North America and northern Europe,
with prevalence rates of 1/5,000 and 1/2,000, respectively. HS is
usually associated with quantitative or qualitative abnormalities
in red blood cell membrane proteins, including ankyrin, band 3,
β-spectrin, spectrin-α, erythrocytic 1 and protein 4.2, encoded by
ANK1, SLC4A1, SPTB, SPTA1 and EPB42, respectively
(2). Splenectomy is widely
considered as the standard surgical treatment in moderate and
severe forms of HS (3). Newborn
infants who have HS may develop anemia, extreme hyperbilirubinemia,
bilirubin-induced neurologic dysfunction and kernicterus (4,5).
Exact diagnosis of HS is considered unreliable around birth.
However, if the early diagnosis of HS is recognized, appropriate
treatment provided at the beginning of congenital hemolysis may
prevent adverse outcomes in neonates with HS, caused by either
de novo or compound heterozygous mutations, particularly for
those with no family history of HS (6,7).
Deficiencies in ankyrin, band 3, spectrins or
protein 4.2 account for 50–60, 15–20, 20 and <5% of patients
with HS, respectively (8). Ethnic
differences may exist: For instance in Japan, ankyrin defects
account for only 5–10% of patients with HS, whereas protein 4.2
defects account for 45–50% (9).
The major structural component of the skeletal network is the
spectrin tetramer, which is formed by α-spectrin and β-spectrin
(10). Spectrins are the central
components of a complex spectrin-actin scaffold at the inner
surface of the erythrocyte membrane. The C-terminal of α-spectrin
and the N-terminal of β-spectrin associate to form heterodimers
(αβ), and two heterodimers associate at the N-terminal of
α-spectrin and the C-terminal of β-spectrin to form functional
tetramers. The tetramers constitute the filaments of this network,
and attach to the cellular membrane via interactions with various
transmembrane proteins (11).
Deficiency of α-spectrin is only clinically apparent in the
homozygous or compound heterozygous state, and these patients have
severe clinical manifestations. SPTA1 mutations cause HS in
association with recessive forms of inheritance, and the presence
of two null SPTA1 alleles is speculated to be lethal
(12,13).
In the present study, a neonate with severe
Coombs-negative hemolytic jaundice but no family history of HS was
observed. The neonate was further clinically diagnosed with HS
based on the following features: Spherocytes on the blood film,
increased osmotic fragility, normal G6PD activity, normal
hemoglobin in electrophoresis, negative thalassemia genetic
mutation screening results and negative autoimmune antibody tests.
Normal mean corpuscular hemoglobin concentration (MCHC) and mean
corpuscular volume (MCV) were observed. The molecular basis of HS
was elucidated via next-generation sequencing (NGS) of relevant
genes. Novel compound heterozygous mutations in the SPTA1
gene (c.3897-1G>C and c.5029G>A), which were inherited from
his asymptomatic mother and father, respectively, were identified
to be responsible for his clinical phenotype.
Materials and methods
Subjects
A one-month old neonate with suspected HS and his
asymptomatic parents (age, 30 years) were asked to participate in
the study in February 2017. Written informed consent to use their
blood samples and consent for publication were obtained from the
parents, and this study was formally approved by the Ethics
Committee of Tongji Hospital, Tongji Medical College, Huazhong
University of Science and Technology (Wuhan, China). All procedures
were performed in accordance with the approved guidelines.
The available clinical characteristics of the
neonate, along with hematological data, osmotic fragility,
glucose-6-phosphate dehydrogenase (G6PD) activity, thalassemia
genetic mutation screening and autoimmune antibody tests are
summarized in Table I.
 | Table I.Blood test results. |
Table I.
Blood test results.
| Hematological
parameters | Result | Reference |
|---|
| RBC |
3.77×1012/lb |
4.3–5.8×1012/l |
| MCV | 99.7 fl | 82–100 fl |
| MCH | 33.4 pg | 27–34 pg |
| MCHC | 33.5 g/dl | 31.6–35.4 g/dl |
| TBIL | 114.5
µmol/la | 3.4–20.5
µmol/l |
| IBIL | 104.7
µmol/la | ≤13.3 µmol/l |
| BRD | 9.8
µmol/la | 0–6.8 µmol/l |
| G6PD | 4,415 U/l | >1,100 U/l |
Ampliseq NGS panel design
A custom NGS panel was designed on the Ion AmpliSeq
Designer website (http://www.ampliseq.com; Ion AmpliSeq Designer; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) to sequence all exons
and adjacent introns of the ANK1, SLC4A1, SPTB, SPTA1 and
EPB42 genes, with coverage of 96.71, 96.53, 99.54, 100 and
100%, respectively. The uncovered region was amplified and
sequenced by Sanger sequencing.
NGS
Ion Torrent adapter-ligated DNA libraries were built
according to the Ampliseq™ Library Preparation kit 2.0 96Lv (Thermo
Fisher Scientific, Inc.) using 10 ng of input DNA from peripheral
blood mononuclear cells (PBMCs), quantitated by Qubit 2.0
(Invitrogen; Thermo Fisher Scientific, Inc.) in two primer-pools
with 118 and 115 amplicons, respectively. The polymerase chain
reaction (PCR) products of the two pools were mixed together, with
samples barcoded using Ion Xpress Barcodes 1–16 (Thermo Fisher
Scientific, Inc.), followed by purification using AMPure XP beads
(Beckman Coulter, Inc., Brea, CA, USA) and quantification by Ion
Library TaqMan™ Quantitation kit (Thermo Fisher Scientific, Inc.).
Prepared libraries were pooled in equal amount and diluted to 100
pM. Subsequently, 2 µl of pooled library were enriched using a Ion
One-Touch Two (OT2) system with the Ion Personal Genome Machine™
(PGM) Hi-Q™ OT2 kit (Thermo Fisher Scientific, Inc.), followed by
enrichment of Ion Sphere Particles on the Ion OneTouch Enrichment
System apparatus (Thermo Fisher Scientific, Inc.). Enriched
products were sequenced using the Ion Torrent PGM Hi-Q™ Sequencing
kit using the Ion 316™ chip v2 (Thermo Fisher Scientific,
Inc.).
Bioinformatics analysis
Raw data, following removal of adapter sequences,
were aligned to the hg19 human reference genome. Coverage analysis
and variant identification were performed on the Ion Torrent Server
software version 4.4.2 (Thermo Fisher Scientific, Inc.), with
default parameters. Variant annotation was performed with the Ion
Reporter 5.0 software (Thermo Fisher Scientific, Inc.), according
to the nomenclature recommended by the Human Genome Variation
Society (http://www.hgvs.org), to classify
variants as single nucleotide variants, multiple nucleotide
variants or indels. All identified variants and regions with
<20X coverage depth were visually verified with the Integrative
Genomics Viewer v2.3.8 (Broad Institute, Cambridge, MA, USA). The
clinical significance of annotated variants was assessed according
to The 1000 Genomes Project (1000G; http://www.internationalgenome.org/), the Human Gene
Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php), the Exome
Aggregation Consortium (ExAC; http://exac.broadinstitute.org/), the Exome Variant
Server (EVS; http://evs.gs.washington.edu/EVS/), ClinVar
(http://www.ncbi.nlm.nih.gov/clinvar/variation) and
InterVar (http://wintervar.wglab.org/)
databases (14). Functional
prediction of missense mutations was performed using multiple
software packages, including Sorting Intolerant From Tolerant
(SIFT; http://sift.jcvi.org/www/SIFT_BLink_submit.html),
MutationTaster (http://www.mutationtaster.org/) and Polymorphism
Phenotyping (PolyPhen)-2 (http://genetics.bwh.harvard.edu/pph2/) (15–17).
Sanger sequencing
Sanger sequencing was performed as previously
described (18). Briefly, genomic
DNA was extracted from PBMCs with the QIAamp DNA blood mini kit
(Qiagen GmbH, Hilden, Germany). Coding exons and splice junctions
of relevant genes were amplified for identified mutations, failed
amplicons or uncovered regions. RNA was extracted from PBMCs with
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) and
reverse transcribed into cDNA at 42°C for 1 h for sequencing.
Sanger sequencing was performed bi-directionally on an ABI 3500 Dx
Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc.). NM_003126.2 was used as the reference transcript of the
SPTA1 gene. The following primers were used in the study:
SPTA1 c.3897-1G>C: Forward, 5′-CAAGGGATTGTTATCATTAGGTG-3′ and
reverse, 5′-AAGGGGGAAGAAATCAGTGA-3′; SPTA1 c.5029G>A: Forward,
5′-CTACCTCTATCGCTCCCACAT-3′ and reverse,
5′-AACTGCTCTTCACTTCCTCCA-3′; RT PCR primers: Forward,
5′-GTTCAGGCTCTTCAGCGACG-3′ and reverse,
5′-CAAATCGTCCCGTTTCTTCAT-3′. PCR was performed using I-5™ 2X
High-Fidelity Master Mix (MCLAB, South San Francisco, CA, USA) in a
50 µl volume including 19 µl water, 25 µl master mix, 2 µl forward
primer, 2 µl reverse primer and 2 µl DNA or cDNA. The PCR was
performed as following: Initial denaturation at 98°C for 2 min,
followed by 35 cycles of 98°C for 10 sec, 62°C for 30 sec, 72°C for
3 sec, and a final extension at 72°C for 1 min.
Protein structure analysis
Protein structure analysis of the SPTA1 c.5029G>A
(p.Gly1677Arg) mutation was performed using the HOPE software
(version 1.1.1; http://www.cmbi.ru.nl/hope/) (19). The sequence relative to SPTA1 was
downloaded from UniProtKB (https://www.uniprot.org/uniprot/P02549).
Results
Clinical features of the neonate with
HS
This male neonate was delivered vaginally at 35
weeks of gestation (G2P2G35+4) following a normal pregnancy. No
family members had a history of anemia or jaundice. The neonate
weighed 5,100 g, and his Apgar scores were 9 and 10 (1 and 10 min,
respectively), with no dysmorphic features. At 30 h, jaundice was
observed, with a total serum bilirubin of 304.4 µmol/l
(transcutaneous jaundice) and hemoglobin (Hb) of 118 g/l;
phototherapy was initiated. On day 8, the neonate was transferred
to the affiliated hospital of the present study due to jaundice and
neonatal anemia, with an Hb of 85 g/l. The blood type of the
parents was A Rh(+). The clinical characteristics of the neonate on
day 8 are presented in Table I.
Spherocytes were observed in peripheral smears, Coombs test was
negative, G6PD activity was normal, no abnormal Hb was observed in
electrophoresis, α- and β-thalassemia genetic mutation screen
results were negative and autoimmune antibody tests were negative.
Osmotic fragility was also increased. Discrepancies occurred in the
patient between alterations in MCHC, MCV and clinical phenotype.
The neonate had normal MCHC, MCV, and MCHC/MCV ratio, but received
various blood transfusions in the three subsequent months due to
severe anemia. The 31-year old father and 32-year old mother were
asymptomatic.
NGS output and coverage
The sequencing of coding exons and adjacent introns
of five HS-associated genes on the PGM achieved an average output
of 425,997 mapped reads (98.9% on target). In summary, 100% of
targeted amplicons were covered at least once, 99.14% amplicons
were covered at least 20 times, and 97.42% amplicons were covered
at least 100 times. The mean uniformity of base coverage was 95.09%
in this panel. The average read depth was 1,453 fold.
Mutation detection and sanger
sequencing validation
A total of 51 variants in the neonate were found and
processed to discover pathogenic mutations. Following annotation
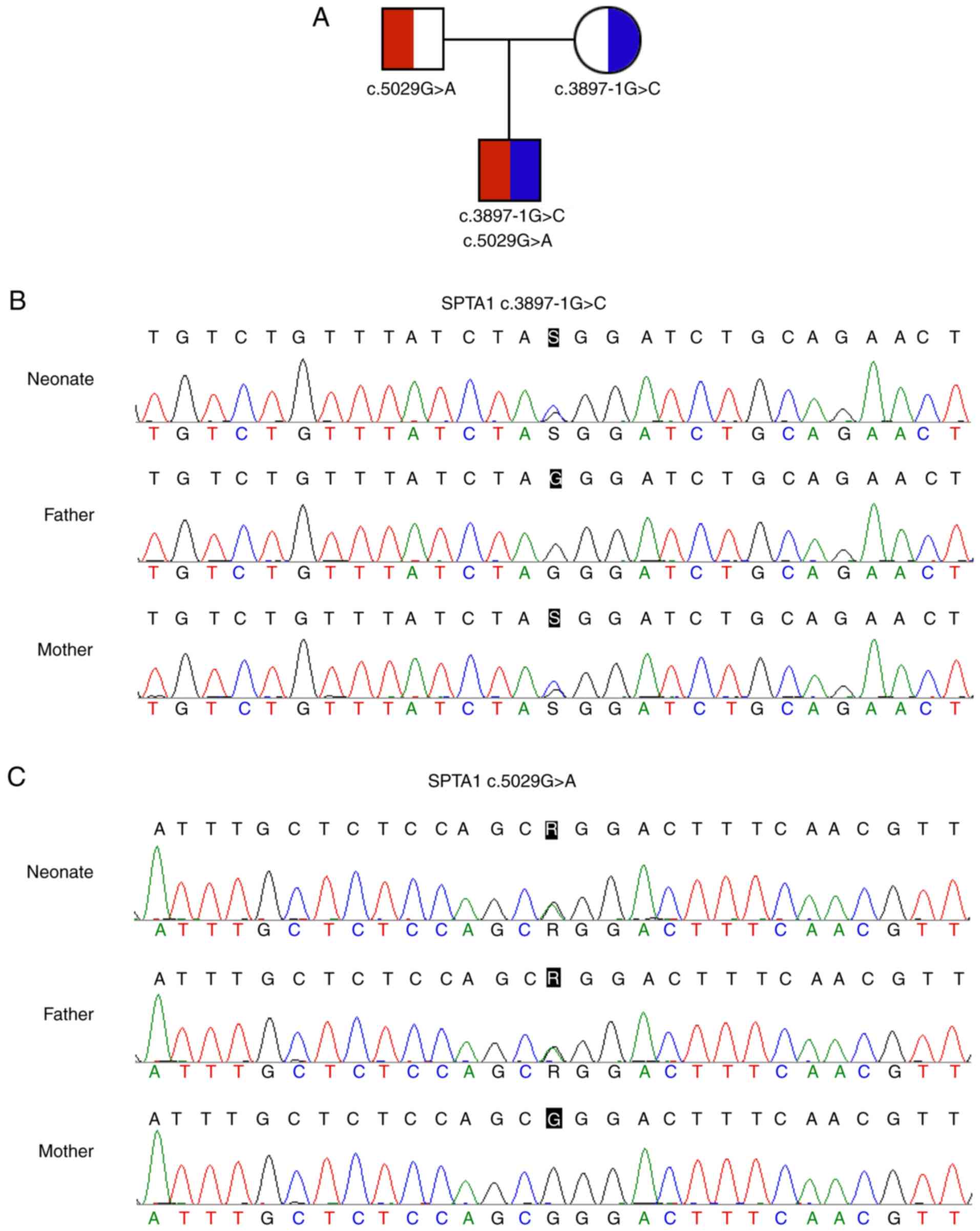
and filtration, novel compound heterozygous mutations in the
SPTA1 gene (c.3897-1G>C and c.5029G>A) were
identified. The SPTA1 c.3897-1G>C (chr1:158615385, hg19)
in intron 27-1, which may disrupt the consensus splice site, was
inherited from his asymptomatic mother. The SPTA1
c.5029G>A (p.Gly1677Arg, chr1:158607983, hg19) in trans
with the SPTA1 c.3897-1G>C mutation was inherited from
his asymptomatic father (Fig.
1).
Functional prediction of the
mutation
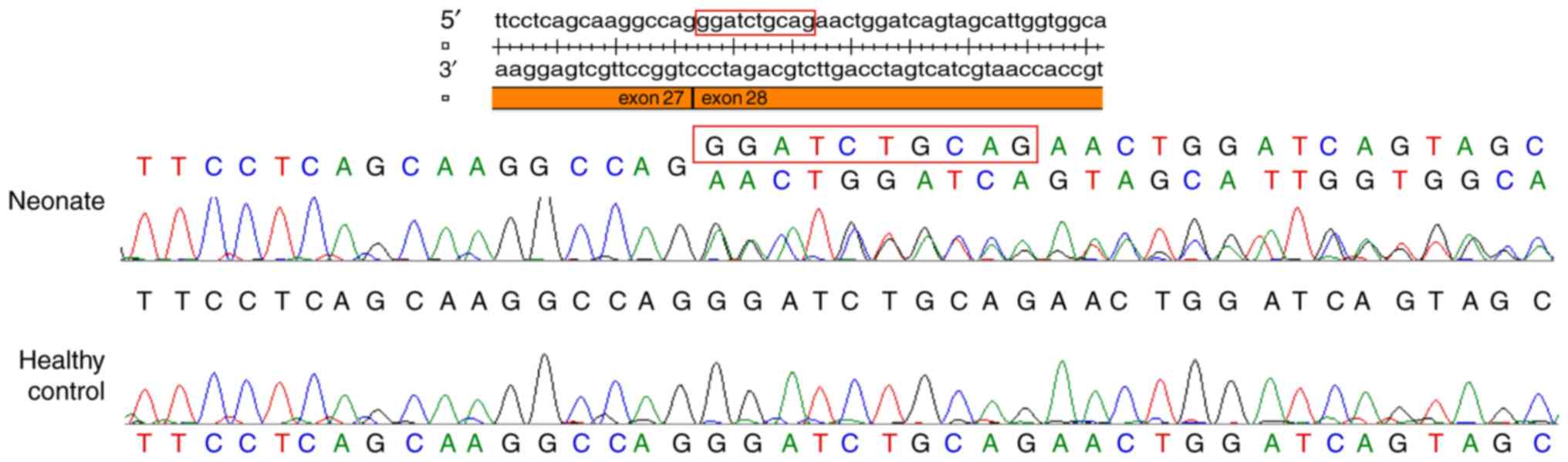
Sanger sequencing of mRNA reverse transcribed into
cDNA identified a deletion of the first 10 nucleotides of exon 28,
and confirmed that the SPTA1 c.3897-1G>C in intron 27-1
disrupted the consensus splice site (Fig. 2). This mutation was not found in
the 1000G, EVS, ExAC or HGMD databases.
The SPTA1 c.5029G>A (p.Gly1677Arg)
mutation was not found in the 1000G, EVS or HGMD databases, whereas
four carriers (one in a South Asian and three in Non-Finnish
Europeans) were found in the ExAC database, with an allele
frequency of 3.328×10−05, indicating a very low
frequency in the population. This variant was predicted to be
‘probably damaging’ in PolyPhen, ‘deleterious’ in SIFT, ‘disease
causing’ in MutationTaster, and ‘likely pathogenic’ in InterVar,
with evidence of PM2 [absent from controls in Exome Sequencing
Project, 1000 Genomes Project, or Exome Aggregation Consortium
(20–22)], PM3 (detected in trans with
a pathogenic variant), PP2 (missense variant in a gene that has a
low rate of benign missense variation and in which missense
variants are a common mechanism of disease) and PP3 (multiple lines
of computational evidence support a deleterious effect on the gene
or gene product) (23). The
SPTA1 c.5029G>A (p.Gly1677Arg) mutation may function in
trans with SPTA1 c.3897-1G>C in intron 27-1,
causing HS in the neonate.
Protein structure analysis of the
SPTA1 c.5029G>A mutation
Protein structure analysis of the SPTA1
c.5029G>A (p.Gly1677Arg) mutation was performed using the HOPE
approach (http://www.cmbi.ru.nl/hope/)
(19). The mutant residue was
larger compared with the wild-type residue. The wild-type residue
charge was neutral, and the mutant residue charge was positive. The
wild-type residue was more hydrophobic compared with the mutant
residue (Fig. 3A). The mutation
was located within a stretch of residues that was repeated in the
protein; this repeat was termed Spectrin 16. The wild-type residue
was a glycine, which is the most flexible of all residues (19). This flexibility may be necessary
for the function of the protein, and mutation of this glycine may
abolish this function (Fig. 3B).
The mutated residue was located in a domain that was important for
binding of other molecules, and was in contact with residues in a
domain that was also important for binding. The mutation may
disturb the interaction between these two domains and negatively
affect the function of the protein. Moreover, the torsion angles
for this residue were unusual. Only glycine was flexible enough to
generate these torsion angles, and mutation into another residue
may force the local backbone into an incorrect conformation and
disrupt the local structure (Fig.
3C).
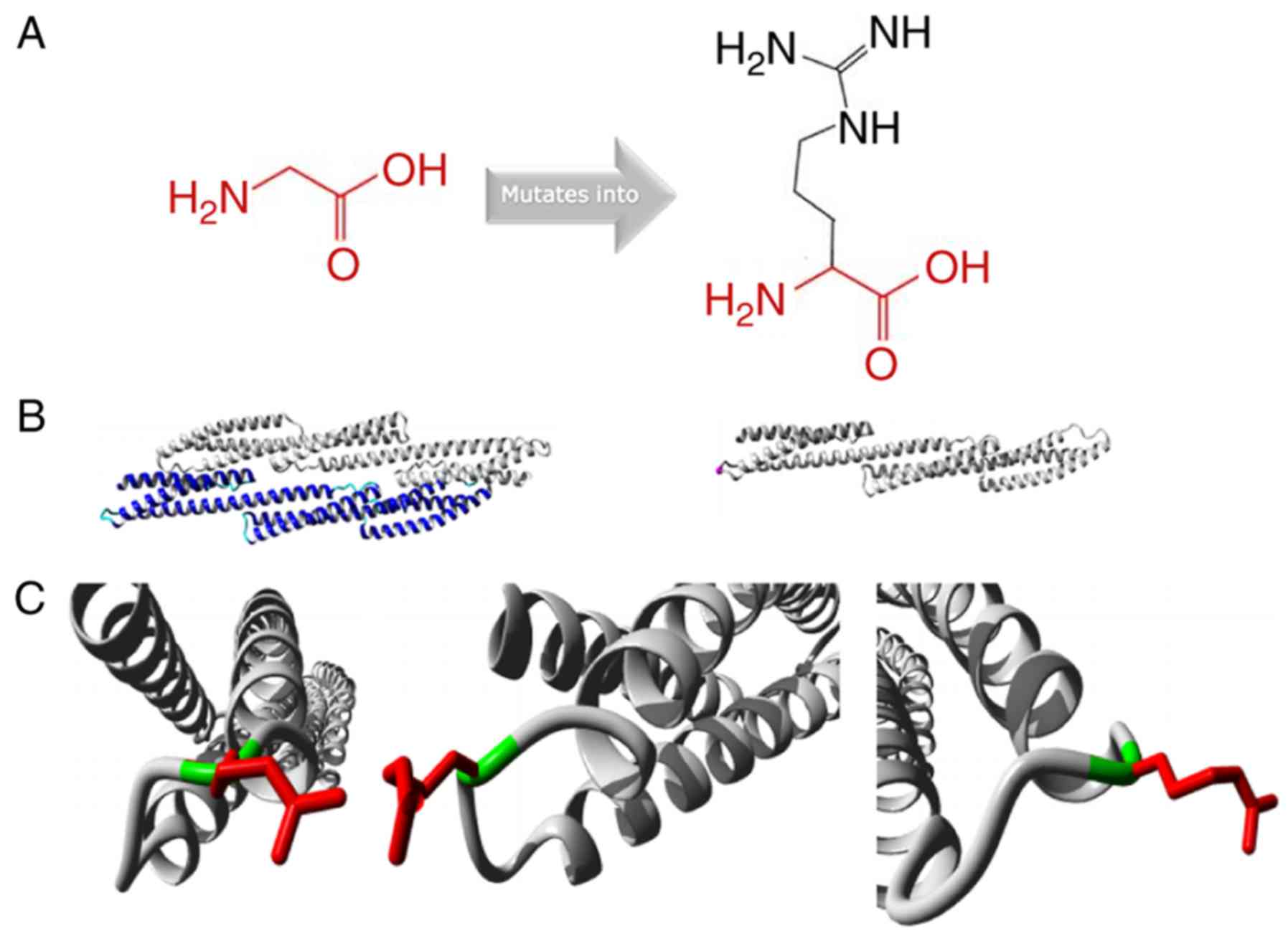 | Figure 3.Protein structure analysis of
SPTA1 c.5029G>A (p.Gly1677Arg). (A) Schematic structures
of the original (left) and the mutant (right) amino acid. The
backbone, which is the same for each amino acid, is colored red.
The side chain, unique for each amino acid, is colored black. (B)
Left is an overview of the protein in ribbon-presentation. The
protein is colored by element: α-helix, blue; β-strand, red; turn,
green; 3/10 helix, yellow; and random coil, cyan. Other molecules
in the complex are colored grey when present. Right is an overview
of the mutant protein in ribbon-presentation. The protein is
colored grey and the side chain of the mutated residue is colored
magenta (pink spheres). (C) Close-up of the mutation. The protein
is colored grey, the side chains of the wild-type and the mutant
residue are presented and colored green and red, respectively. The
structure is presented in three different angles. |
Discussion
HS is a heterogeneous disorder, and the majority of
cases are inherited in an autosomal-dominant fashion with an
unambiguous family history. The phenotype of HS in neonates ranges
from asymptomatic to hydrops fetalis (6,13).
HS is the third most common underlying hemolytic anemia, following
G6PD and ABO hemolytic disease, among neonates listed in the USA
Kernicterus Registry (6).
Moreover, HS is the leading cause of Coombs-negative hemolytic
anemia, requiring blood transfusion in the first months of life
(6). Neonates with HS may present
with extreme hyperbilirubinemia, thus a timely diagnosis may
prevent adverse outcomes in neonates with HS (24).
For neonates with Coombs-negative hemolytic jaundice
and a negative family history, the presence of spherocytes on blood
film, an increased MCHC and a decreased MCV suggest a diagnosis of
HS (25). Christensen et al
(26), and Christensen and
Sheffield (27), speculated that
an MCHC of 36.0 g/dl or more could alert caregivers to the
possibility of HS. However, the neonate described in the present
study exhibited normal MCHC, MCV and MCHC/MCV ratio. Hemolytic
jaundice and anemia were the principal signs of HS, and blood
transfusions were performed almost monthly to maintain the Hb
concentration in the blood. Neonates with normal MCHC and MCV have
also been reported previously. For example, Yaish et al
(25) described a neonate with
α-spectrin deficient HS confirmed by SDS-PAGE, and the neonate had
Coombs-negative spherocytic hemolytic jaundice in addition to
normal MCHC and MCV. These results suggested that for patients with
normal MCHC, MCV or MCHC/MCV ratio, additional tests may be
required. NGS is widely used in the diagnosis of hereditary
hemolytic anemia, including HS (28–31).
Moreover, the mutation spectrum and genetic characteristics in the
Chinese population have been identified by exome sequencing
(31). Additionally, the
eosin-5′-maleimide binding test, performed using flow cytometry,
has been also recommend for HS diagnosis (32).
The SPTA1 gene is located at 1q22-23, and
α-spectrin is an integral part of the erythrocyte cytoskeleton,
which is linked to the surface of the plasma membrane through
ankyrin and band 3. Homozygous or compound heterozygous mutations
in the SPTA1 gene may be involved in the pathogenesis of
recessive spectrin-deficient HS, and heterozygous individuals may
still produce sufficient α-spectrin to balance β-spectrin and
maintain the erythrocyte cytoskeleton (33,34).
Missense and splicing mutations account for the majority of
cases.
In this case, novel compound heterozygous mutations
in the SPTA1 gene (c.3897-1G>C and c.5029G>A) were
identified in the neonate. The SPTA1 c.3897-1G>C mutation
was not found in the 1000G, EVS, ExAC or HGMD databases. Sanger
sequencing of the mRNA reverse transcribed into cDNA identified a
deletion of the first 10 nucleotides of exon 28, and the ninth and
tenth nucleotides of exon 28 were AG. This result indicated that
the SPTA1 c.3897-1G>C mutation in the consensus splice
site led to a new AG splice acceptor site at the ninth and tenth
nucleotides of exon 28, resulting in a deletion of the first 10
nucleotides of exon 28.
The SPTA1 c.5029G>A mutation resulted in a
glycine-to-arginine substitution at amino acid position 1,677 of
α-spectrin. Various types of software predicted that this missense
mutation would have deleterious effects. Moreover, protein
structure analysis implied that this mutation may negatively affect
the function of α-spectrin as follows. First, the wild-type residue
charge was neutral, whereas the mutant residue was positive.
Additionally, the wild-type residue was more hydrophobic compared
with the mutant residue, and the mutated residue was located in a
domain important for the binding of other molecules and in contact
with residues in a domain important for binding. The mutation may
disrupt the interaction between these two domains. Only glycine is
flexible enough to generate these torsion angles, and mutation into
arginine may force the local backbone into an incorrect
conformation and disrupt the local structure. Thus, SPTA1
c.5029G>A (p.Gly1677Arg) in trans with the SPTA1
c.3897-1G>C mutation may have caused HS in this neonate.
In summary, novel compound heterozygous mutations in
the SPTA1 gene (c.3897-1G>C and c.5029G>A) may be
responsible for the clinical manifestations of the described
neonatal patient with HS. In the present study. NGS provided a
rapid and powerful approach for the genetic diagnosis of HS.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81500925).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
AL, YL and QH designed the study. XW and YL
performed the experiments and drafted the manuscript. AL and QH
cared for the patient. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Written informed consent to use blood samples from
the subjects was obtained from the parents. The present study was
formally approved by the Ethics Committee of Tongji Hospital,
Tongji Medical College, Huazhong University of Science and
Technology (Wuhan, China). All procedures were performed in
accordance with the approved guidelines.
Patient consent for publication
Written consent for publication from all subjects
was obtained.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Da Costa L, Galimand J, Fenneteau O and
Mohandas N: Hereditary spherocytosis, elliptocytosis, and other red
cell membrane disorders. Blood Rev. 27:167–178. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang X, Shen N, Huang M, Lu Y and Hu Q:
Novel hereditary spherocytosis-associated splice site mutation in
the ANK1 gene caused by parental gonosomal mosaicism.
Haematologica. 103:e219–e222. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Iolascon A, Andolfo I, Barcellini W,
Corcione F, Garçon L, De Franceschi L, Pignata C, Graziadei G,
Pospisilova D, Rees DC, et al: Recommendations regarding
splenectomy in hereditary hemolytic anemias. Haematologica.
102:1304–1313. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Johnson L, Bhutani VK, Karp K, Sivieri EM
and Shapiro SM: Clinical report from the pilot USA kernicterus
registry (1992 to 2004). J Perinatol. 29 (Suppl 1):S25–S45. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gallagher PG: Disorders of red cell volume
regulation. Curr Opin Hematol. 20:201–207. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Christensen RD, Yaish HM and Gallagher PG:
A pediatrician's practical guide to diagnosing and treating
hereditary spherocytosis in neonates. Pediatrics. 135:1107–1114.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Andres O, Eber S and Speer CP: Early
postnatal diagnosis of hereditary spherocytosis by combining light
microscopy, acidified glycerol lysis test and eosin-5′-maleimide
binding assay. Ann Hematol. 94:1959–1964. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
An X and Mohandas N: Disorders of red cell
membrane. Br J Haematol. 141:367–375. 2008.PubMed/NCBI
|
|
9
|
Yawata Y, Kanzaki A, Yawata A, Doerfler W,
Ozcan R and Eber SW: Characteristic features of the genotype and
phenotype of hereditary spherocytosis in the Japanese population.
Int J Hematol. 71:118–135. 2000.PubMed/NCBI
|
|
10
|
Speicher DW, DeSilva TM, Speicher KD,
Ursitti JA, Hembach P and Weglarz L: Location of the human red cell
spectrin tetramer binding site and detection of a related ‘closed’
hairpin loop dimer using proteolytic footprinting. J Biol Chem.
268:4227–4235. 1993.PubMed/NCBI
|
|
11
|
Lam VQ, Antoniou C, Rolius R and Fung LW:
Association studies of erythroid alpha-spectrin at the
tetramerization site. Br J Haematol. 147:392–395. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Narla J and Mohandas N: Red cell membrane
disorders. Int J Lab Hematol. 39 (Suppl 1):S47–S52. 2017.
View Article : Google Scholar
|
|
13
|
Perrotta S, Gallagher PG and Mohandas N:
Hereditary spherocytosis. Lancet. 372:1411–1426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li Q and Wang K: InterVar: Clinical
interpretation of genetic variants by the 2015 ACMG-AMP guidelines.
Am J Hum Genet. 100:267–280. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Adzhubei I, Jordan DM and Sunyaev SR:
Predicting functional effect of human missense mutations using
PolyPhen-2. Curr Protoc Hum Genet Chapter. 7:Unit7.202013.
|
|
18
|
Wang X, Zhu Y, Shen N, Peng J, Wang C, Liu
H and Lu Y: Mutation analysis of a Chinese family with
oculocutaneous albinism. Oncotarget. 7:84981–84988. 2016.PubMed/NCBI
|
|
19
|
Venselaar H, Te Beek TA, Kuipers RK,
Hekkelman ML and Vriend G: Protein structure analysis of mutations
causing inheritable diseases. An e-Science approach with life
scientist friendly interfaces. BMC Bioinformatics. 11:5482010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Auer PL, Reiner AP, Wang G, Kang HM,
Abecasis GR, Altshuler D, Bamshad MJ, Nickerson DA, Tracy RP, Rich
SS, et al: Guidelines for large-scale sequence-based complex trait
association studies: Lessons learned from the NHLBI exome
sequencing project. Am J Hum Genet. 99:791–801. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zheng-Bradley X and Flicek P: Applications
of the 1000 genomes project resources. Brief Funct Genomics.
16:163–170. 2017.PubMed/NCBI
|
|
22
|
Lek M, Karczewski KJ, Minikel EV, Samocha
KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ,
Cummings BB, et al: Analysis of protein-coding genetic variation in
60,706 humans. Nature. 536:285–291. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Christensen RD, Nussenzveig RH, Yaish HM,
Henry E, Eggert LD and Agarwal AM: Causes of hemolysis in neonates
with extreme hyperbilirubinemia. J Perinatol. 34:616–619. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yaish HM, Christensen RD and Agarwal A: A
neonate with Coombs-negative hemolytic jaundice with spherocytes
but normal erythrocyte indices: A rare case of autosomal-recessive
hereditary spherocytosis due to alpha-spectrin deficiency. J
Perinatol. 33:404–406. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Christensen RD and Henry E: Hereditary
spherocytosis in neonates with hyperbilirubinemia. Pediatrics.
125:120–125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sheffield MJ and Christensen RD:
Evaluating neonatal hyperbilirubinemia in late preterm Hispanic
twins led to the diagnosis of hereditary spherocytosis in them, and
in their sibling and in their mother. J Perinatol. 31:625–627.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Agarwal AM, Nussenzveig RH, Reading NS,
Patel JL, Sangle N, Salama ME, Prchal JT, Perkins SL, Yaish HM and
Christensen RD: Clinical utility of next-generation sequencing in
the diagnosis of hereditary haemolytic anaemias. Br J Haematol.
174:806–814. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang X, Yi B, Mu K, Shen N, Zhu Y, Hu Q
and Lu Y: Identification of a novel de novo ANK1 R1426* nonsense
mutation in a Chinese family with hereditary spherocytosis by NGS.
Oncotarget. 8:96791–96797. 2017.PubMed/NCBI
|
|
30
|
He Y, Jia S, Dewan RK and Liao N: Novel
mutations in patients with hereditary red blood cell membrane
disorders using next-generation sequencing. Gene. 627:556–562.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang R, Yang S, Xu M, Huang J, Liu H, Gu W
and Zhang X: Exome sequencing confirms molecular diagnoses in 38
Chinese families with hereditary spherocytosis. Sci China Life Sci.
61:947–953. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Farias MG: Advances in laboratory
diagnosis of hereditary spherocytosis. Clin Chem Lab Med.
55:944–948. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wichterle H, Hanspal M, Palek J and
Jarolim P: Combination of two mutant alpha spectrin alleles
underlies a severe spherocytic hemolytic anemia. J Clin Invest.
98:2300–2307. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nussenzveig RH, Christensen RD, Prchal JT,
Yaish HM and Agarwal AM: Novel α-spectrin mutation in trans
with alpha-spectrin causing severe neonatal jaundice from
hereditary spherocytosis. Neonatology. 106:355–357. 2014.
View Article : Google Scholar : PubMed/NCBI
|