Introduction
Hereditary nephropathy is a progressive fatal renal
disease caused by genetic changes in the somatic or germ cells
(1). The main types, as previously
described, include: Glomerular diseases, such as focal segmental
glomerular sclerosis (FSGS); renal cystic lesions, such as
autosomal dominant polycystic kidney disease; and renal tubular
diseases, such as nephronophthisis (NPH) (2). The proportion of nephropathy cases in
China through 2003–2014 that were primary glomerulonephritis,
secondary glomerulonephritis, tubulointerstitial disease or
hereditary renal diseases was 67.1, 26.4, 2.9 and 2.5%,
respectively (3). FSGS represents
20% of all nephrotic syndrome cases in children and is one of the
five most common pathological changes in China, especially in
Southern China, with a detection rate of 5.6% (4,5).
However, kidney tubular disease such as NPH is also commonly
observed in juvenile and adolescent subjects (6). Due to the non-specific pathological
changes mentioned above, focal glomerular segmental sclerosis alone
cannot be used to confirm the true pathogenic mechanism of FSGS.
Therefore, genetic screening is adopted for diagnostic testing,
especially for diagnosing patients with a history of genetic
disease.
Genetic analysis has become a more accurate
diagnostic method due to advances in medical technology and
updating of the human genetic variation database (7,8). The
single nucleotide polymorphism (SNP) technique for detecting single
base pair changes plays a vital role in the diagnosis of a number
of diseases, including kidney damage (9). One genetic mutation can lead to
different phenotypic changes, while one phenotypic change could be
induced by multiple gene mutations. High-throughput mutation
analyses have found mutations in Wilms tumor 1 (WT1),
actinin α 4 (ACTN4), nephrocystin 1 (NPHP1),
nephrosis 2 (NPHS2), phospholipase C ε 1 (PLCE1),
angiotensin I converting enzyme (ACE) and multidrug
resistance mutation 1 genes in patients with pathological changes
in FSGS (2,10–12).
Mutations in 15 genes, including nephrocystin 1–13, uromodulin
(UMOD), Abelson helper integration site 1, and coiled-coil
and C2 domain containing 2A have been found in patients with NPH
worldwide (13–15). New mutation sites in the NPH
population have been found with familial aggregation
characteristics (13,16–19).
In the present study, a Chinese family with
hereditary renal injury was screened, and SNPs of NPHS2, WT1,
PLCE1, ACTN4, ACE, NPHP1 and UMOD were sequenced to
explore the main causes of kidney damage and determine the genetic
mutations in this Chinese family with hereditary nephropathy. This
study also aimed to confirm the importance of genetic screening in
the diagnosis of complex hereditary diseases.
Materials and methods
Familial data and sample
collection
A total of 10 subjects (7 male and 3 female) were
enrolled at the Guangzhou Red Cross Hospital between October and
December 2012, the mean age of all patients was 27.90±19.92. The
families of the affected patients were enrolled, and the families
of non-affected siblings were also recruited for bias reduction.
Full medical and family histories were collected for pedigree
analysis. Blood and urine samples were collected from each patient.
Routine and biochemical tests were performed. Levels of albumin,
parathyroid hormone and inflammatory factors were determined. Color
Doppler ultrasonography and magnetic resonance imaging (MRI) of the
kidney were also performed. Percutaneous renal samples were
collected guided by B-ultrasound after obtaining informed consent
from all the participants or their guardians. Consent forms were
signed by the patients or their guardians. This study was approved
by the ethics committee of the Guangzhou Red Cross Hospital (permit
no. 20121228) and adhered to the tenets of the Declaration of
Helsinki and the Guidance on Sample Collection of Human Genetic
Diseases given by the Ministry of Public Health of China [Health
Science and Education Planning Memo (2003) no. 80].
Hematoxylin-eosin staining
Kidney biopsy samples were routinely fixed in 2%
glutaraldehyde buffer for 2–4 h at room temperature, embedded in
optimal cutting temperature compound (OCT) for 15 min and cut into
~5 µm thick sections. The sections were soak in hematoxylin for 7
min for nuclear dying, then washed 3 times, differentiated with
0.5% hydrochloric acid alcohol and washed once before being placed
into 1% ammonia solution for several seconds, and finally stained
with 1% eosin for about 3 min, all at room temperature.
Hematoxylin-eosin (HE) sections were observed under light
microscopy at 200× magnification.
Transmission Electron microscopy
A sample (1 mm3) was cut from the
cortical end of kidney tissue and placed in 2.5% glutaraldehyde
buffer for 2–4 h at 4 °C, rinsed with PBS, and then fixed in a l%
citrate fixative solution for 1–2 h. Following dehydration for 15
min with a graded ethanol series (50, 70, 80 and 90%), the sections
were finally dehydrated with 100% ethanol for 30 min. The tissue
was embedded with fresh EPON812 resin at gradient temperature (35,
45 and 60°C), each for 12 h. The tissue was cut into 50 nm sections
and double stained with 2% uranyl acetate and pH 12 lead citrate at
room temperature. Nanoparticle morphological properties of these
kidney samples were confirmed using a transmission electron
microscope at 13,500× magnification (Thermo Fisher Scientific,
Inc.).
Immunohistochemistry of UMOD
Kidney tissue was embedded with 50% OCT for 10 h at
room temperature and cut into ~5 µm thick sections. The sections
were fixed in 4% paraformaldehyde solution for 15 min at room
temperature and washed 3 times with PBS before being incubated in
0.4% pepsin for 30 min at 37°C for antigen retrieval and then
blocked in 3% BSA for 30 min at room temperature.
Immunohistochemical analysis of UMOD was performed using a
mouse monoclonal antibody against human UMOD (1:300, cat.
no. ab207170, Abcam) at 4°C overnight, goat anti-mouse IgG was used
as secondary antibody (1:500, cat. no. Ab150113, Abcam) at 37°C for
0.5 h. I–VIEW DAB Universal Kit (Ventana Medical Systems, Inc.) was
used for color reaction, after the termination of color
development, hematoxylin was used for nuclear counterstain and
observations were made under light microscopy at 400×
magnification.
Immunofluorescence detection
Kidney samples were sliced into ~5 µm sections and
fixed with 4% acetone for 15 min and washed 3 times with PBS at
room temperature. Fluorescein-labeled antibodies IgG (1:150, cat.
no. A0423, Dako, Agilent Technologies, Inc.), IgA (1:150, cat. no.
A0262, Dako, Agilent Technologies, Inc.), IgM (1:200, cat. no.
A0425, Dako, Agilent Technologies, Inc.), C3 (1:100, cat. no.
F0201, Dako, Agilent Technologies, Inc.), C1q (1:100, cat. no.
F0254, Dako, Agilent) (Dako, Agilent Technologies, Inc.),
podocalyxin (1:300, cat. no. 14-8883-80, Invitrogen, Thermo Fisher
Scientific, Inc.), fabrillarin (Fib) (1:200, cat. no. PA5-29801,
Invitrogen, Thermo Fisher Scientific, Inc.) were added, and
incubation was performed for 30 min at 37°C. The samples were
finally sealed with glycerin and observed under a fluorescence
microscope at 400× magnification.
Targeted sequencing
Genomic DNA of the patients was extracted from the
peripheral blood samples as per the instruction manual of the
TIANamp Genomic DNA kit (Tiangen Biotech Co., Ltd.). Targeted
first-generation sequencing of NPHS2, WT1, PLCE1, ACTN4, ACE
and UMOD was performed in all family members except P7 who
had died. Targeted second-generation sequencing of NPHP1 was
performed in P4, P5, P17, P18, P19, P20 and P21. Primers (presented
in Tables SI–SVII) were designed and synthesized by
Primer Premier 5 (Premier Biosoft International). These genes were
sequenced on the Applied Biosystems 3730 DNA Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.).
Total RNA was extracted from the peripheral blood
samples using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Total RNAs were reverse
transcribed to cDNA using a FastKing RT kit (Tiangen Biotech Co.,
Ltd.) in a 10 µl system with 2.5 µl RNA sample, 10X King RT Buffer,
1 µl FastKing RT Enzyme Mix, 2 µl FQ-RT Primer Mix and 2.5 µl
RNase-Free ddH2O at 42°C for 15 min, then transferred to
95°C for 3 min. The expression of NPHP1 and mutated genes
was verified using TaqMan™ quantitative (q)PCR analysis (Thermo
Fisher Scientific, Inc.). PCR reaction conditions: DNA was
pre-denatured at 95°C for 3 min, cooled to 55–60°C for 35 sec, the
primers were added, and then it was rapidly warmed to 72°C for 35
cycles. After Taq DNA polymerase was added, the primer strand was
extended for 40–50 sec and put through repair extension at 72°C for
5–8 min. The quantification method of qPCR experiments was
2−∆∆Cq (20). The
relative expression levels of NPHP1 were normalized to those
of GAPDH (F: CAAGGTCATCCATGACAACTTTG; R:
GTCCACCACCCTGTTGCTGTAG).
Diagnostic criteria for familial
adolescent NPH
Patients with a family history of NPH were
presented. The main clinical characteristics of NPH include:
Polyuria and polydipsia on account of renal concentration
defection; growth retardation; anemia; and chronic renal failure.
The renal pathological features of adolescent NPH include: Small to
normal-sized kidneys; increased echogenicity and reduced
corticomedullary differentiation; renal cysts on the
corticomedullary border; and a dilated bladder.
Statistical analysis
The quantitative data were expressed as the mean ±
SD and the frequencies of qualitative data were described.
Bioinformatics processing was performed following the sequencing
procedure. The relationship between genes and diseases
(polymorphism or causative) was evaluated with in combination with
the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) and Online
Mendelian Inheritance in Man database (https://omim.org/) according to the ACMG guidelines
(21). Mutated genes were
evaluated using GeneMANIA v3.4.1 in Cytoscape v3.6.1 (https://cytoscape.org/) for gene co-location analysis
(22). The GeneMANIA Cytoscape
plugin was an efficient way of fast gene function prediction,
including 6 categories: Co-expression, co-localization, genetic
interaction, physical interaction, predicted and shared protein
domain. Co-occurrence sequences are an important factor to related
genome function, which can be determined by co-localization
analysis. Colocalization analysis determines genomic
co-localization characteristics and is used to assess nucleotide or
spatial proximity of overlapping sequences between genes. The
amount of overlaps or spatial proximity are two important
evaluation indicators for gene co-localization analysis.
Results
Clinical and biochemical
detection
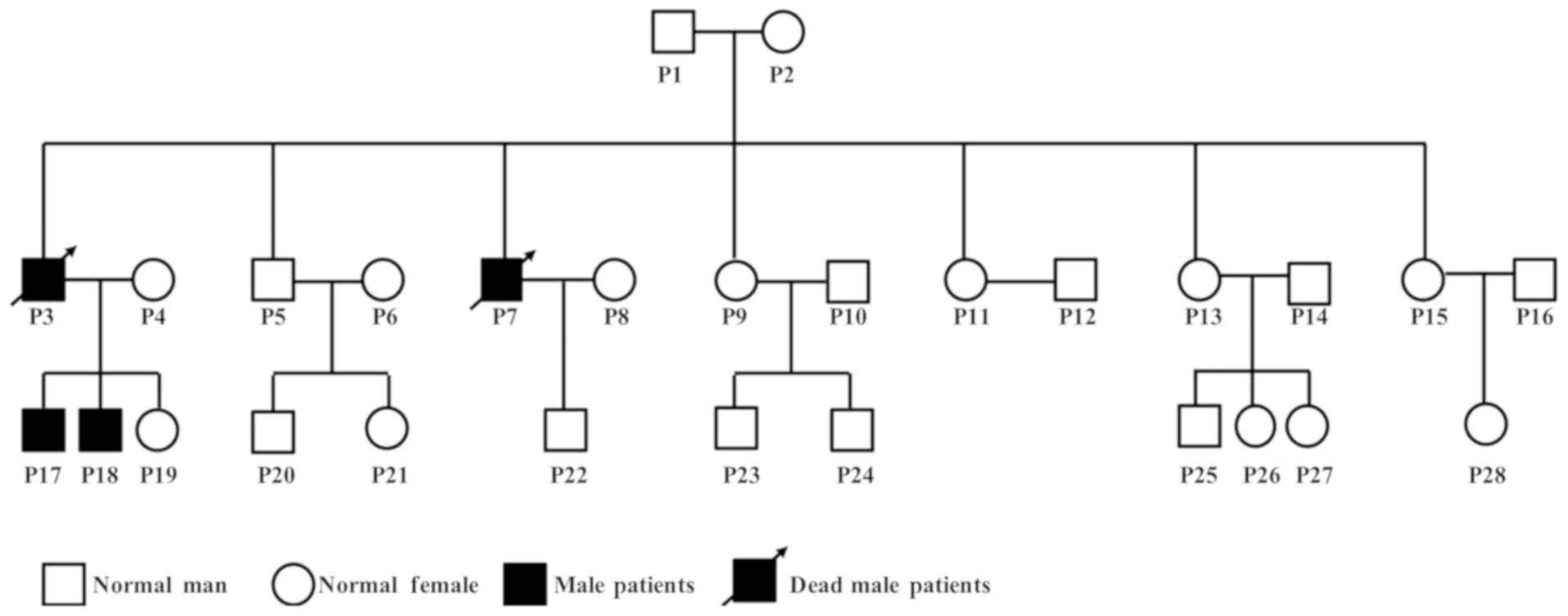
P17 and P18 were the index patients; they were
regarded as the starting point of this study. Disease history was
recorded, and laboratory tests were performed for 10 family members
across 3 generations (P1, P4, P5, P7, P17, P18, P19, P20, P21 and
P22). Pedigree of the family showed that father P3 and uncle P7 of
the two index patients were diagnosed with uremia and died at the
age of 33 and 34 years, respectively. They were considered to be
strong evidence of hereditary kidney disease, although specimens
could not be obtained (Fig.
1).
The 10 enrolled patients included three females and
seven males with a mean age of 27.8±19.97 years and an average BMI
of 17.71±3.0 kg/m2. Three members had hyperuricemia
without gout, and four members had hematuria. Connective
tissue-associated nephropathy, HIV-related nephropathy, purpuric
nephritis, hepatitis virus-associated nephritis, diabetic
nephropathy, hypertensive renal damages and other related renal
damages were excluded based on their clinical tests.
P17, a 20-year-old male had growth retardation, with
a height of 162 cm, weight of 38.5 kg, and a low BMI of 14.7
kg/m2. This patient was diagnosed with hereditary
nephrosis at Guangzhou Red Cross Hospital. Serum testing showed an
impaired glomerular filtration rate (urea, 26.5 mmol/l; creatinine,
980 µmol/l) and disturbed metabolism (parathyroid hormone, 165
pmol/l).
P18, a 16-year-old male and the brother of P17,
experienced growth retardation with a low BMI of 17.7
kg/m2 (height of 160 cm and weight of 45.4 kg) and was
diagnosed with hereditary nephropathy at Guangzhou Red Cross
Hospital. Laboratory testing showed slight gastrointestinal
bleeding with weak positive hematuria, microalbuminuria, impaired
glomerular filtration rate (urea, 28.9 mmol/l; creatinine, 743
µmol/l; calcium, 2.68 mmol/l; phosphorus, 1.79 mmol/l), and
disordered metabolism (parathyroid hormone, 185 pmol/l; Table I).
 | Table I.Clinical characteristics of the
Chinese hereditary nephrotic family. |
Table I.
Clinical characteristics of the
Chinese hereditary nephrotic family.
|
|
Patient |
|
|---|
|
|
|
|
|---|
| Characteristic | P17 | P18 | P1 | P4 | P5 | P7 | P19 | P20 | P21 | P22 | Reference
ranges |
|---|
| Demographic
data |
|
|
|
|
|
|
|
|
|
|
|
|
Sex | M | M | M | F | M | M | F | M | F | M | –– |
| Age
(years) | 20 | 16 | 73 | 44 | 33 | 40 | 18 | 14 | 12 | 9 | –– |
| BMI
(kg/m2) | 14.7 | 17.7 | 18 | 16 | – | 19 | 19 | 15 | 16 | 15 | 18.5–24.9 |
| eGFR
(ml/min/1.73m2) | 13.5 | 17.7 | 81.56 | 89.48 | 5.7 | 101.78 | 128.9 | 247.8 | 179.2 | 146.7 |
|
| Urine routine |
|
|
|
|
|
|
|
|
|
|
|
|
Hematuria | Anuria | T | Mo | Mo | T | T | N | N | N | T | Negative |
| White
cell count (/µl) | 0 | 0 | 0 | 0 | – | 0 | 0 | 0 | 0 | 0 | 0.00~28.00 |
| Albumin
(g/l) | T | 0.3 | N | N | – | N | N | N | N | N | Negative |
| pH | 6 | 6 | 5.5 | 6.5 | 6.0 | 7 | 6.5 | 6.5 | 6 | 6 | 5.4~8.4 |
|
Proportion | 1.01 | 1.02 | ≤1.005 | 1.015 | 1.01 | 1.02 | ≤1.005 | ≤1.005 | 1.025 | 1.01 | 1.003~1.030 |
| Blood biochemical
test |
|
|
|
|
|
|
|
|
|
|
|
| Urea
(mol/l) | 13.8 | 17.6 | 6.8 | 5.4 | 26.5 | 6.5 | 4 | 5.2 | 4.8 | 4.7 | 1.9~6.8 |
|
Creatinine (µmol/l) | 706 | 562 | 77 | 61 | 980 | 71 | 61 | 57 | 49 | 43 | F: 53~9; M:
62~105 |
| Uric
acid (µmol/l) | 272.5 | 607.7 | 289 | 243.1 | 537 | 374.7 | 251.5 | 392 | 428.9 | 421.8 | F: 155~357; M:
208~428 |
| Albumin
(g/l) | 52.9 | 51.2 | 40.7 | 50.6 | 45 | 50.4 | 49.6 | 47.5 | 49.9 | 45.3 | 34~53 |
| Calcium
(mmol/l) | 2.68 | 2.26 | 2.30 | 2.51 | – | 2.52 | 2.54 | 2.42 | 2.56 | 2.41 | 2.1~2.6 |
|
Phosphorus (mmol/l) | 1.79 | 1.39 | 1.29 | 1.39 | – | 1.25 | 1.28 | 1.3 | 1.54 | 1.40 | 1.00~1.60 |
|
25-hydroxyvitamin D3
(ng/ml) | 59.6 | 47.2 | 33.1 | 37.4 | – | 38.3 | 28.4 | 29.9 | 25.3 | 15.1 | 30.1~100.0 |
|
Parathyroid hormone
(pmol/l) | 17.65 | 16.66 | 3.41 | 4.02 | – | 2.23 | 3.1 | 3.09 | 3.34 | 5.06 | 1.6–6.9 |
Imaging test
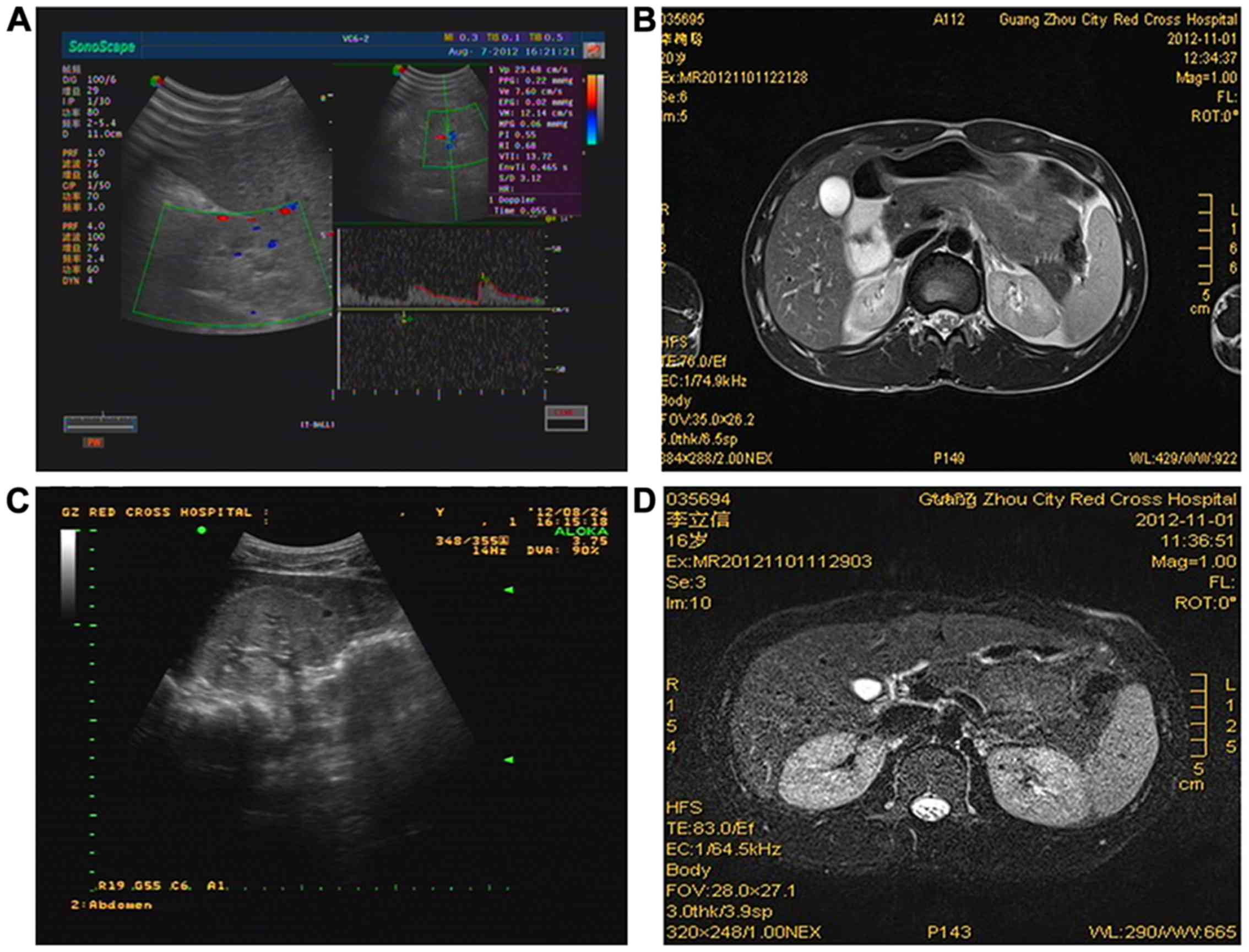
In P17, Color Doppler ultrasonography showed
slightly narrowed, hyperechogenic kidneys (62×28 and 54×26 mm, left
and right kidneys, respectively) with decreased blood flow
distribution and a blurred medullary boundary (Fig. 2A). Further analysis with MRI found
a 0.8×0.8 cm cyst near the medullary boundary (Fig. 2B). In P18, Color Doppler
ultrasonography showed enlarged, hyperechogenic kidneys (100×49 and
100×49 mm, left and right kidneys, respectively) with an unclear
medulla boundary; 5×4 mm-sized cysts were also observed (Fig. 2C). Further analysis with MRI found
multiple small cysts in both of the kidneys (Fig. 2D).
Pathological examination
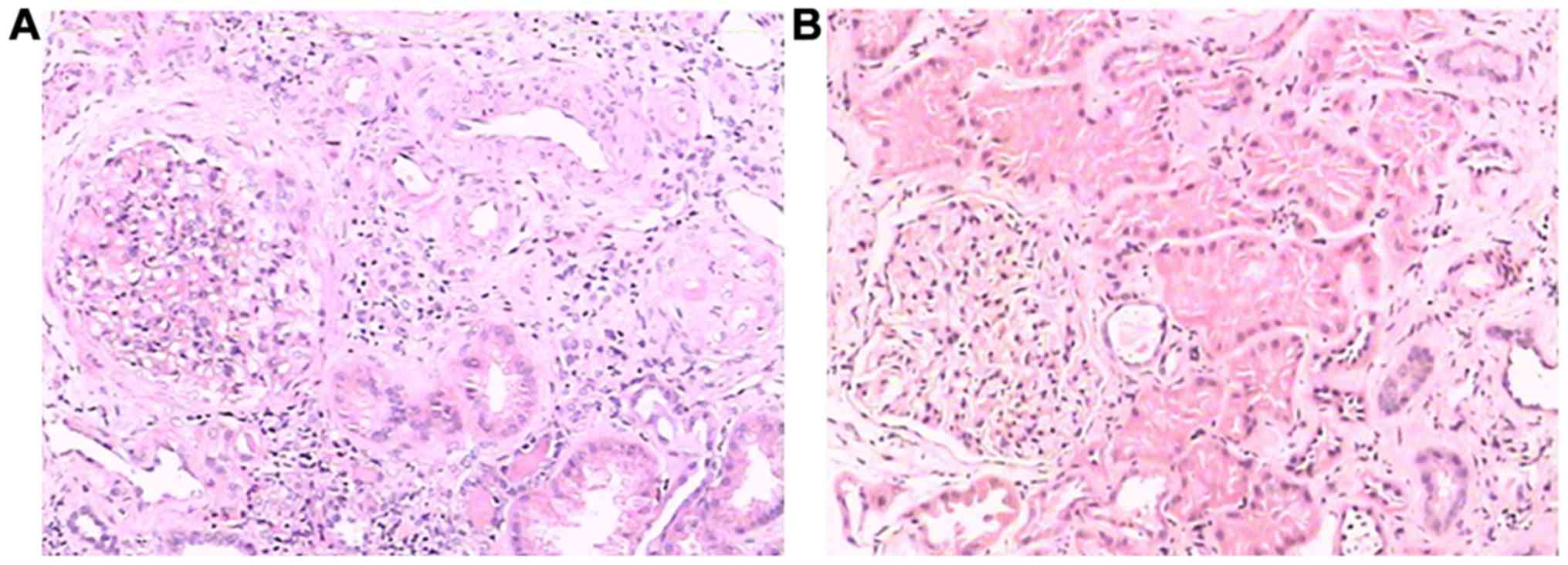
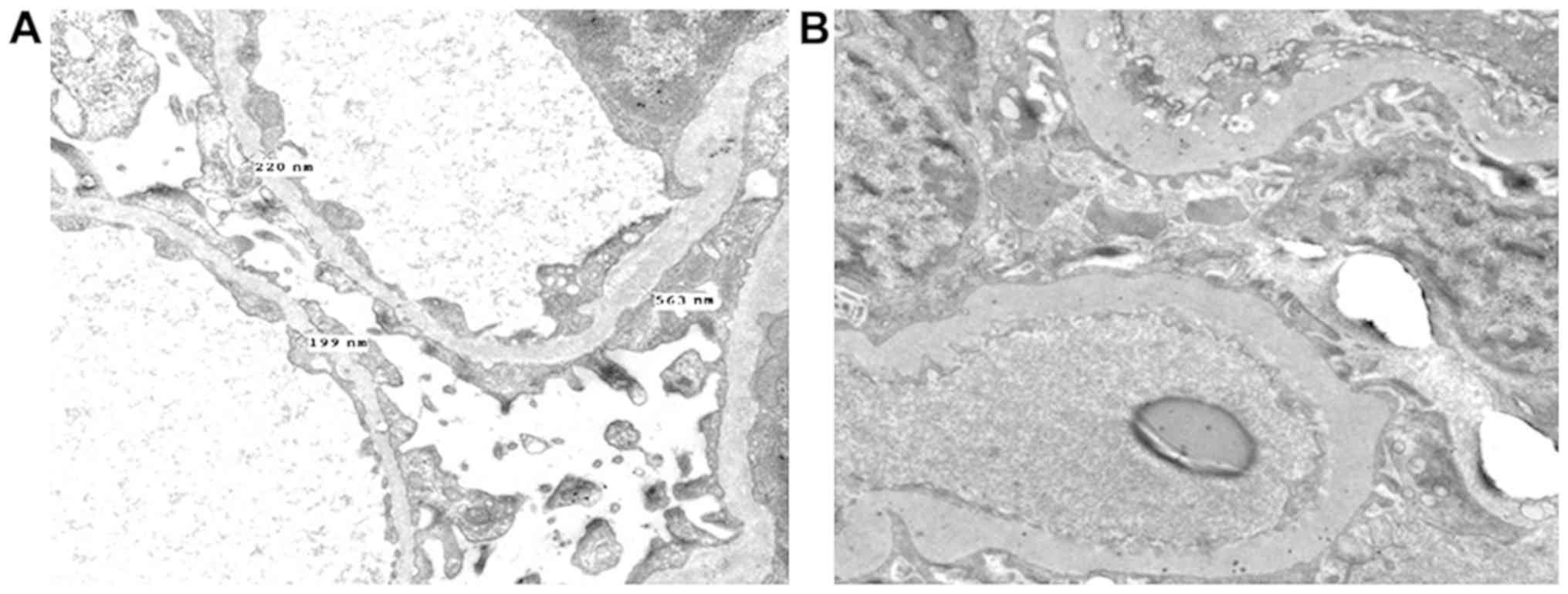
In P17, HE staining and electron microscopy revealed
sclerosing glomerulonephritis, and increased lymphocyte and
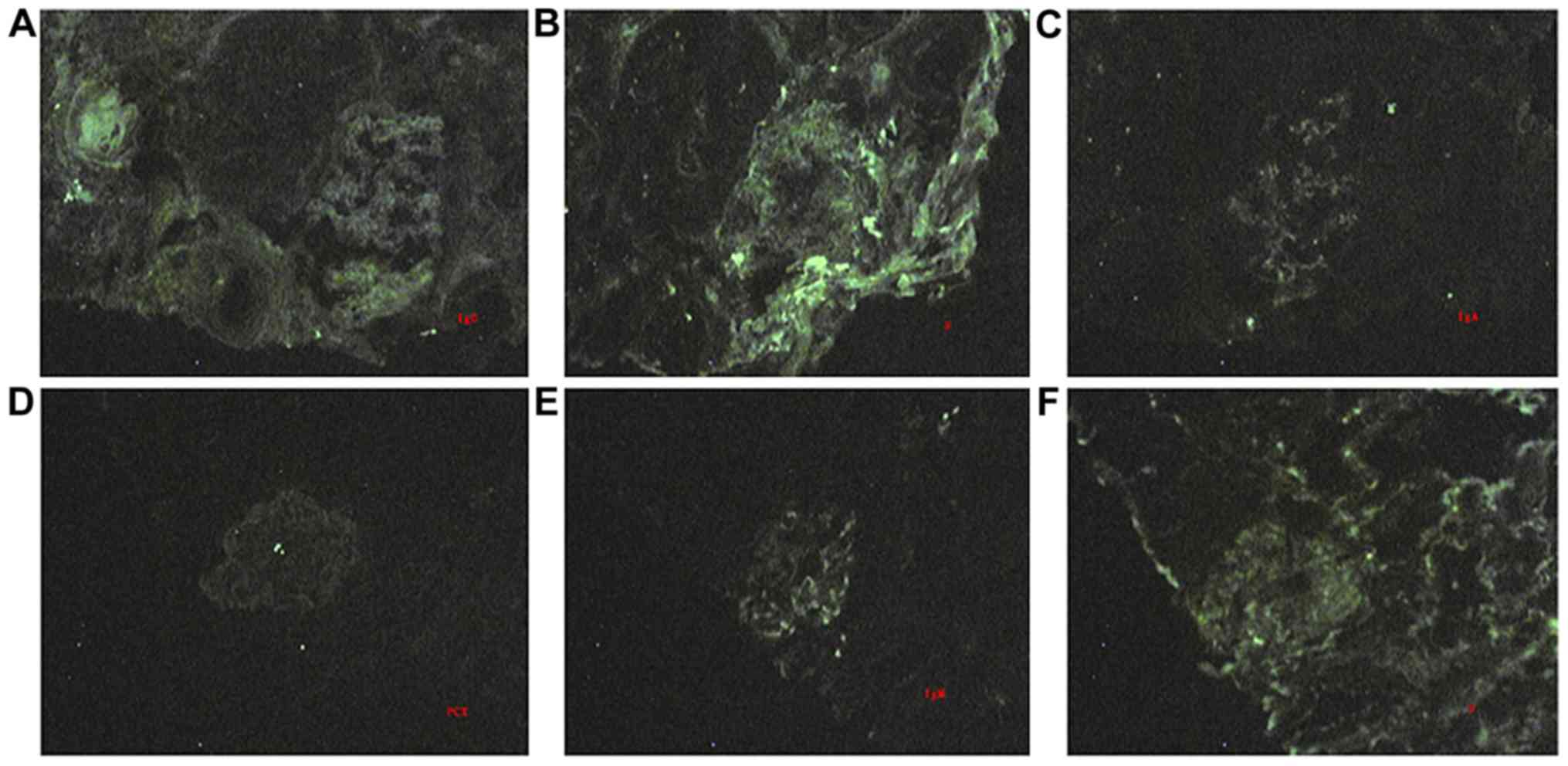
monocyte infiltration in the renal interstitium (Figs. 3A and 4A). Immunofluorescence staining revealed
IgG, IgA and IgM depositions in the mesangium matrix (Fig. 5A-C). In P18, HE staining and
electron microscopy showed glomerular sclerosis, glomerular
fibrosis crescents, tubular epithelial cell histiocytosis, and
increased infiltration of lymphocytes and monocytes in the
interstitium (Figs. 3B and
4B). Collagen fibers and monocytes
infiltrations were also observed via electron microscopy (Fig. 4B). IgM and Fib depositions were
observed in the mesangium matrix during immunofluorescence staining
(Fig. 5D-F). Significantly dilated
capillary loops, glomerular capsules and renal tubules were
found.
Genetic investigations
NPHS2 analysis
Sequencing of NPHS2 revealed two heterozygous
variants. 102A>G was detected in exon 1 of all nine members,
with no differences in the 34-glycine podocin protein. Another
variant, 954C>T, was discovered in exon 8 of the two index
patients and four other family members, including P4, P5, P19 and
P20, suggesting that this variant was inherited from the mother P4
and passed on to her three children. P5 and his son P20 also
carried this variation (Fig.
6A).
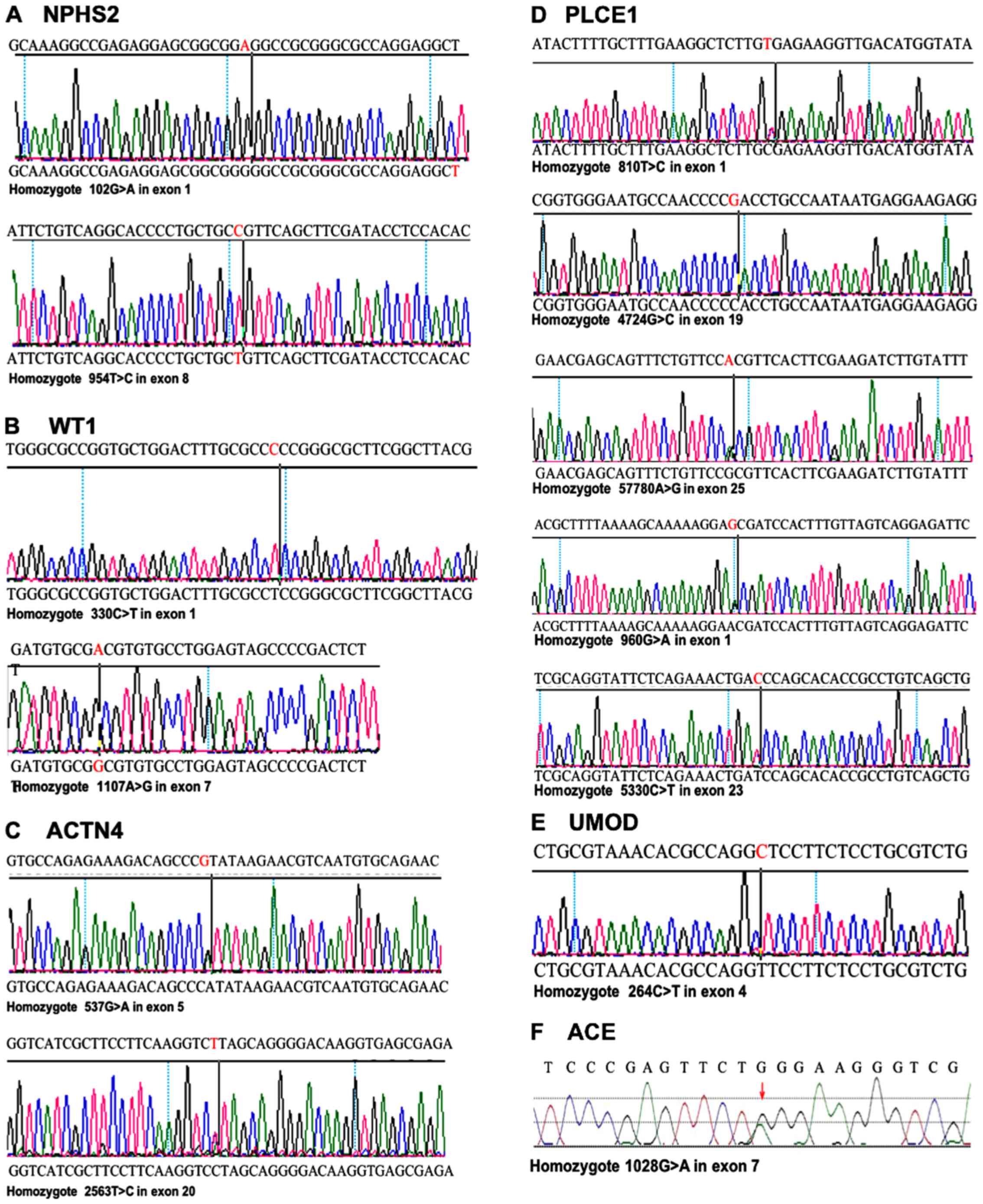 | Figure 6.Variants of NPHS2, WT1, ACTN4,
PLCE1, UMOD and ACE identified by gene sequencing.
Variants of 6 high prevalent genes in 10 family members were
identified. (A) Two nonsense mutation of NPHS2, including
102A>G and 954C>T. (B) Two novel variants of WT1,
including 330C>T and 1107A>G. (C) Two heterozygous variants
of 537G>A and 2563T>C in ACTN4. (D) Five heterozygous
variants of PLCE1 including 810T>C, 960G>A,
5330C>T, 5780A>G, and 4724G>C. (E) Nonsense mutation
264C>T in UMOD. (F) Causative nonsense mutation
1028G>A in ACE. WT1, wilms tumor 1; ACTN4,
actinin α 4; NPHS2, nephrosis 2; PLCE1, phospholipase C ε 1;
ACE, angiotensin I converting enzyme; UMOD, uromodulin. |
WT1 analysis
Two novel variations were found through WT1
sequencing, including 330C>T and 1107A>G. 330C>T was
detected in exon 1 in P17, P18, P1, P4, P5, P19 and P22. This
variation was inherited from their grandfather P1 and was passed on
to his son P5; P4 and P22, as mother and sister of P17 and P18,
also carried the variation. 1107A>G was present in exon 7 in
P17, P18, P1, P4 and P9, suggesting that P9 inherited this
variation from his father, P1. P4 also carried the variation and
passed it on to her three children P17, P18 and P20 (Fig. 6B).
ACTN4 analysis
In ACTN4 sequencing, two variations
considered as gene polymorphisms were identified. 537G>A and
2563T>C variants were only detected in exon 5 and exon 20 in two
patients, P17 and P18. No mutations of this gene were found in
other family members (Fig.
6C).
PLCE1 analysis
Five heterozygous variants in PLCE1 were
found. 810T>C and 960G>A in exon 1 were detected in P17, P4
and P22. In exon 23 and 25 of P17, P4, P21 and P22, variants
including 5330C>T and 5780A>G were found. Furthermore,
4724G>C in exon 19 was identified in P17, P18, P4 and P19
(Fig. 6D). Of these affected
members, P17, P18 and P19 were siblings and the children of P3 and
P4, and P22 is the son of P7 and P8.
UMOD analysis
Variants of 264C>T in exon 4 were confirmed in
index patient P17. Variants in UMOD also occurred in four
healthy family members, including first generation P1, his daughter
P9 who belonged to the second generation, as well as P21 and P22
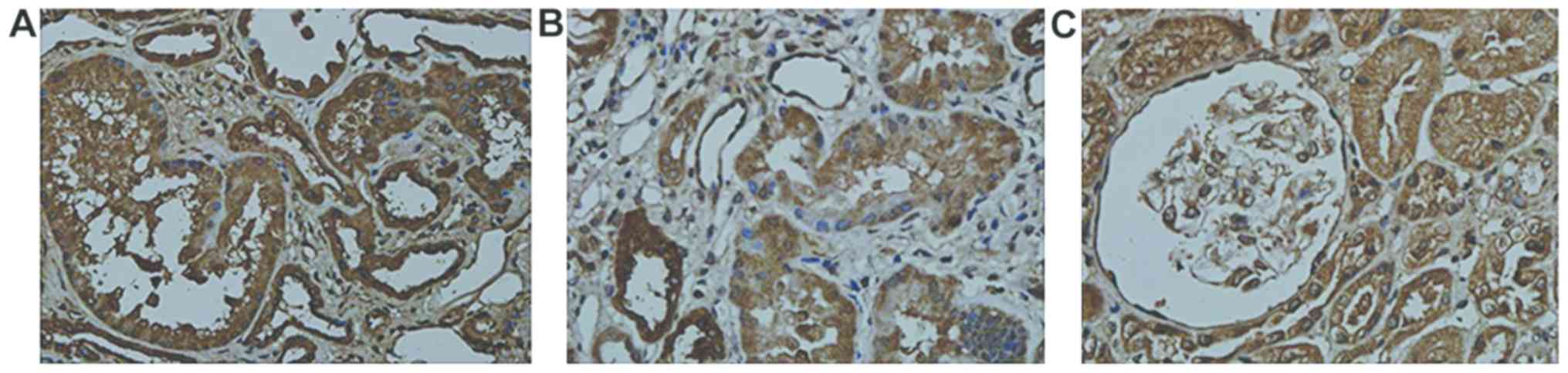
who belonged to the third generation (Fig. 6E). The deposition of UMOD
was further confirmed with immunohistochemistry detection. Uneven
UMOD depositions were found in the renal tubular epithelial
cells of the two index patients (Fig.
7).
ACE analysis
A causative variant 1028G>A was identified in
ACE in the index patients P17 and P18 and presented with no
variants in the other family members (Fig. 6F).
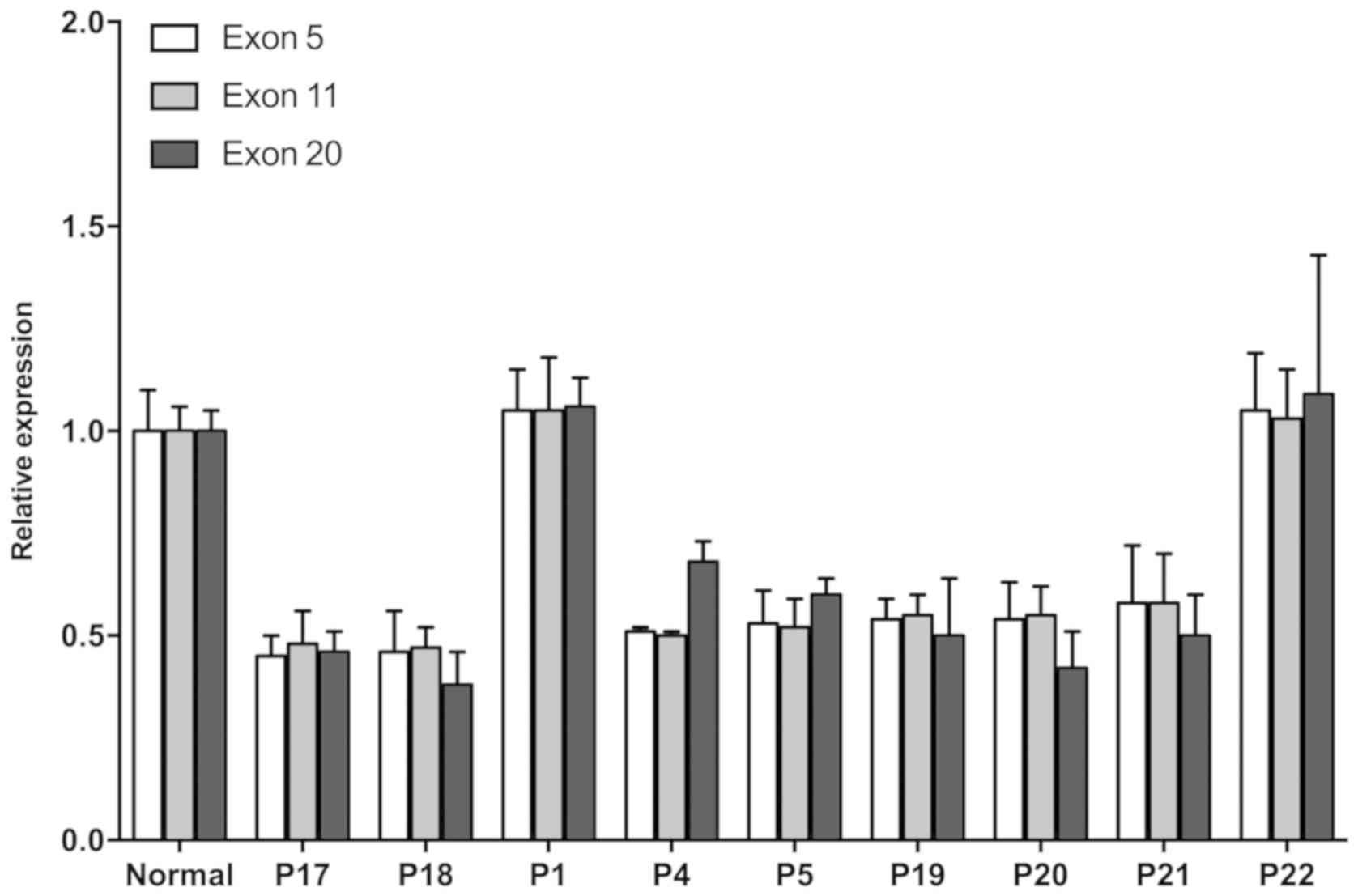
NPHP1 analysis
Homozygous deletion mutation of NPHP1 was
found in exon 5, 11 and 20 in both P17 and P18. Their sister P19,
as well as cousins P20 and P21 who belonged to the third
generation, also experienced loss of heterozygosity (LOH) in
NPHP1. LOH was also confirmed in two of the
second-generation members: P4, the mother of the two index
patients, and P5, the father of P20 and P21 (Fig. 8).
In silico analysis
Homozygous deletions in NPHP1 and
heterozygous deletion in ACE were found in the following
mutation identification analysis. Homozygous deletion mutation of
NPHP1 in exons 5, 11 and 20 could be passed down to the next
generation via X-linked recessive inheritance. In contrast, a
variant in ACE was shown to be a disease-causing mutation
that was predicted to alter 343-bit amino acid from Trp to a stop
codon. Mutations in both NPHP1 and ACE can lead to a
frame shift and a truncated protein, thus stopping the production
of the target protein and leading to a loss of function, resulting
in disease. In addition, no pathogenic variants were found in other
genes of the panel with sequencing, indicating that these missense
variations are gene polymorphisms that don't cause disease.
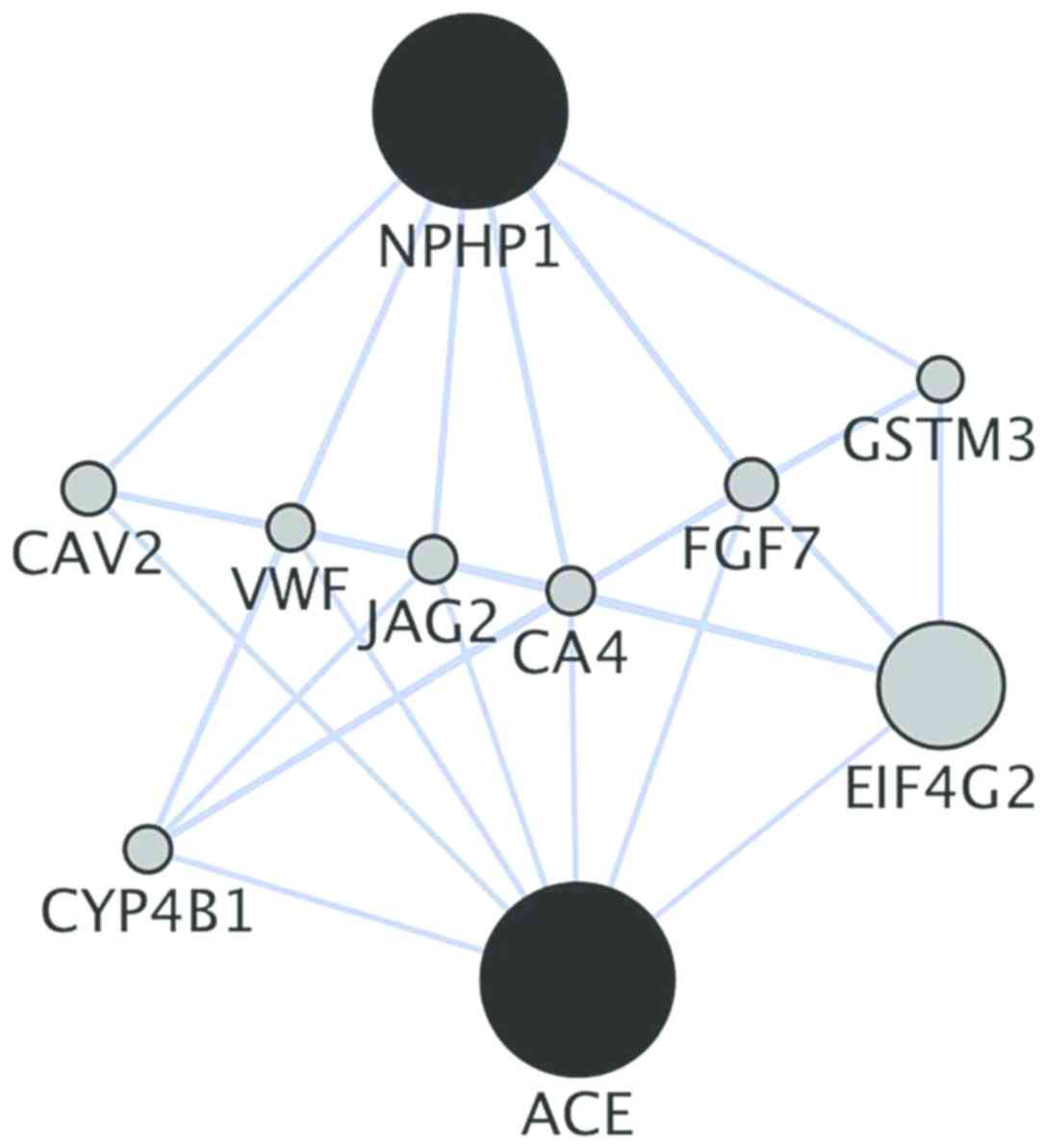
Additionally, co-location analysis of the genes,
using GeneMANIA, showed direct and indirect co-locations for
NPHP1 and ACE. Caveolin 2, von Willebrand factor,
jagged canonical notch ligand 2, carbonic anhydrase 4 and
fibroblast growth factor 7 are direct co-location genes, while the
other genes can influence these six direct genes and further
activate NPHP1 or ACE (Fig. 9).
Discussion
In the present study, cases in a family with
heredity nephropathy with heterozygous genetic variants in
NPHS2, WT1, ACTN4, PLCE1, ACE, UMOD and NPHP1
deletion were presented. A renal biopsy of two index patients
showed sclerosing glomerulonephritis, dilated tubules and
lymphocyte/monocyte infiltration in the interstitium. Combined with
the results of routine serum/urine tests and family history
investigation, hereditary tubular disease was suggested.
Disease-causing mutations included homozygous deletions in
NPHP1; therefore, familial adolescent NPH was diagnosed for
the two index patients. Network analysis discovered direct and
indirect co-location genes of NPHP1 and ACE.
FSGS is characterized as a
morphological/histological injury rather than a specific glomerular
disease (23). Even though
sclerosing nephritis and focal segmental glomerulosclerosis were
found in the index patients, significant dilated renal tubules and
high infiltration in the interstitium also showed a high
possibility of hereditary tubular disease. Considering the three
main pathological changes of NPH, including renal tubular basement
membrane destruction, tubular atrophy and cystic changes, it was
hypothesized that glomerular injury, secondary to the tubular and
interstitial damages, may have occurred in the patients (6).
Further SNP screening analysis indicated no
causative mutations in common FSGS-related genes, including
NPHS2, WT1, ACTN4 and PLCE1; this further lowered the
probability of FSGS. Homozygous deletion of NPHP1 is
reported to be the most common mutation in NPH (24,25).
NPHP1 deficiency occurs in Iranian, Turkish, Japanese and
French families with NPH, indicating a strong possibility of NPH
diagnosis in this Chinese nephrotic family (26–28).
This study showed that NPHP1 was vertically transmitted
between the second and third generation, indicating the possibility
of familial adolescent NPH diagnosis. Notably, the mother of the
index patients carried a LOH of NPHP1, her two sons showed
homozygous deletion in NPHP1 and her daughter had LOH of
NPHP1, indicating that mutations or mutant isomers carried
by their mother were very likely to be inherited by her children
(29).
NPH is an inherited renal disease associated with
tubule-interstitial damage that causes end-stage renal disease
(30). Depending on the age at
onset, it is categorized as infantile, juvenile and adolescent NPH;
adolescent NPH is the most common form (31). Adolescent-onset NPH patients
present with polyuria and polydipsia symptoms at ~4–6 years of age,
and end-stage renal disease (ESRD) developed at an average age of
19 years (32). The index patients
progressed to ESRD at the age of 20 and 16 years, consistent with
adolescent NPH. In addition, the patients showed an extremely high
degree of hyperuricemia, when compared to the reference ranges.
Unlike primary hyperuricemia, hyperuricemia in NPH-medullary cystic
kidney disease (MCKD) is caused by decreased tubular uric acid
excretion (33). High uric acid
retention that was detected in the two index patients was further
evidence for the diagnosis of adolescent NPH.
A heterozygous pathological mutation in ACE
was also found in this study. Research has suggested that
ACE-deficient mice can develop symptoms characteristic of
NPHP pathology, including decreased urine concentration,
hypotension and progressive renal failure (34,35).
However, the possibility of these two genes as pathogenic genes was
excluded from the genetic screening of three Italian families with
confirmed nephropathy (36).
Patients with NPH can show a variety of different phenotypes, such
as Oculomotor apraxia type Cogan, liver fibrosis, MCKD and
Joubert/Meckel-Gruber syndromes, and other multi-system injuries
(15). Considering the co-location
genes between NPHP1 and ACE, it was hypothesized that
ACE could be involved, however further validation is needed
(32,37,38).
The present study has certain limitations. Firstly,
this was single-center research, therefore the generalizability of
these results for all adolescent patients with NPH with hereditary
nephropathy is limited. Multi-center research is needed to confirm
our results. Secondly, samples were limited; only a single large
family with nephropathy history was studied. Although a total of
seven family branches were included, patients from different
families should be enrolled and investigated in the future.
Thirdly, the lack of in vitro experiments to validate the
present results was also a limitation of this study. Additional
in vitro experiments are needed to verify the possible
pathogenic factors. In spite of these limitations, these results
provide information regarding the prevalence of familial adolescent
NPH in China and suggest that NPH should be considered as a common
cause of hereditary nephropathy.
In conclusion, familial adolescent NPH was diagnosed
in two index patients in this study. Therefore, it is recommended
that comprehensive gene mutation screening combined with kidney
biopsy detection is used for the diagnosis of hereditary nephrotic
disease, and concern should be raised about gene variants related
to multiple organ system comorbidities.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by Guangdong
Provincial Science and Technology Project Fund for Key Scientific
Research Base for Metabolic Disease Related Clinical Nutrition
(grant no. 2014B030303002) and Guangdong Provincial Academician
workstation Fund (grant no. 2017B09090 4027).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
CT and DZ contributed to analyzing data and drafting
the manuscript; RT and XZ contributed to the interpretation of data
and manuscript revision; XX, DQ, YuL and JH collected and analyzed
the data; YaL contributed to the conception and design of the
present study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by The Ethics
Committee of the Guangzhou Red Cross Hospital (permit no. 20121228)
and adhered to the tenets of the Declaration of Helsinki and the
Guidance on Sample Collection of Human Genetic Diseases given by
the Ministry of Public Health of China. Consent forms were signed
by patients or their guardians.
Patient consent for publication
This study has followed the principles of anonymity;
no direct or indirect identifiers of our participants were used for
publication.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
FSGS
|
focal segmental glomerular
sclerosis
|
|
NPH
|
nephronophthisis
|
|
SNP
|
single nucleotide polymorphism
|
|
MRI
|
magnetic resonance imaging
|
|
LOH
|
loss of heterozygosity
|
|
MCKD
|
medullary cystic kidney disease
|
References
|
1
|
Tianjun G, Zhihong L, Zhaohong C, Wweixin
H, XIiaodan Y and Zheng T: Association studies of gene polymorphism
in lupus nephritis. Chin J Nephrol Dial Transplant. 5–10.
100:1997.
|
|
2
|
Sharif B and Barua M: Advances in
molecular diagnosis and therapeutics in nephrotic syndrome and
focal and segmental glomerulosclerosis. Curr Opin Nephrol
Hypertens. 27:194–200. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hou JH, Zhu HX, Zhou ML, Le WB, Zeng CH,
Liang SS, Xu F, Liang DD, Shao SJ, Liu Y and Liu ZH: Changes in the
spectrum of kidney diseases: An analysis of 40,759 biopsy-proven
cases from 2003 to 2014 in China. Kidney Dis (Basel). 4:10–19.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sperry ZJ, Na K, Parizi SS, Chiel HJ,
Seymour J, Yoon E and Bruns TM: Flexible microelectrode array for
interfacing with the surface of neural ganglia. J Neural Eng.
15:0360272018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rosenberg AZ and Kopp JB: Focal segmental
glomerulosclerosis. Clin J Am Soc Nephrol. 12:502–517. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wolf MTF and Hildebrandt F:
Nephronophthisis. Pediatr Nephrol. 26:181–194. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi M, Scholl UI, Ji W, Liu T, Tikhonova
IR, Zumbo P, Nayir A, Bakkaloğlu A, Ozen S, Sanjad S, et al:
Genetic diagnosis by whole exome capture and massively parallel DNA
sequencing. Proc Natl Acad Sci USA. 106:19096–19101. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
McInerney-Leo AM, Marshall MS, Gardiner B,
Benn DE, McFarlane J, Robinson BG, Brown MA, Leo PJ, Clifton-Bligh
RJ and Duncan EL: Whole exome sequencing is an efficient and
sensitive method for detection of germline mutations in patients
with phaeochromcytomas and paragangliomas. Clin Endocrinol (Oxf).
80:25–33. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Clifford RJ, Edmonson MN, Nguyen C,
Scherpbier T, Hu Y and Buetow KH: Bioinformatics tools for single
nucleotide polymorphism discovery and analysis. Ann N Y Acad Sci.
1020:101–109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Benetti E, Caridi G, Malaventura C,
Dagnino M, Leonardi E, Artifoni L, Ghiggeri GM, Tosatto SC and
Murer L: A novel WT1 gene mutation in a three-generation family
with progressive isolated focal segmental glomerulosclerosis. Clin
J Am Soc Nephrol. 5:698–702. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dhandapani MC, Venkatesan V, Rengaswamy
NB, Gowrishankar K, Nageswaran P and Perumal V: Association of ACE
and MDR1 gene polymorphisms with steroid resistance in children
with idiopathic nephrotic syndrome. Genet Test Mol Biomarkers.
19:454–456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sako M, Nakanishi K, Obana M, Yata N,
Hoshii S, Takahashi S, Wada N, Takahashi Y, Kaku Y, Satomura K, et
al: Analysis of NPHS1, NPHS2, ACTN4, and WT1 in Japanese patients
with congenital nephrotic syndrome. Kidney Int. 67:1248–1255. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Halbritter J, Porath JD, Diaz KA, Braun
DA, Kohl S, Chaki M, Allen SJ, Soliman NA, Hildebrandt F and Otto
EA; GPN Study Group, : Identification of 99 novel mutations in a
worldwide cohort of 1,056 patients with a nephronophthisis-related
ciliopathy. Hum Genet. 132:865–884. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kang HG, Lee HK, Ahn YH, Joung JG, Nam J,
Kim NK, Ko JM, Cho MH, Shin JI, Kim J, et al: Targeted exome
sequencing resolves allelic and the genetic heterogeneity in the
genetic diagnosis of nephronophthisis-related ciliopathy. Exp Mol
Med. 48:e2512016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wolf MTF, Lee J, Panther F, Otto EA, Guan
KL and Hildebrandt F: Expression and phenotype analysis of the
nephrocystin-1 and nephrocystin-4 homologs in Caenorhabditis
elegans. J Am Soc Nephrol. 16:676–687. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Waldherr R, Lennert T, Weber HP, Födisch
HJ and Schärer K: The nephronophthisis complex-A clinicopathologic
study in children. Virchows Arch A Pathol Anat Histol. 394:235–254.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chaki M, Hoefele J, Allen SJ, Ramaswami G,
Janssen S, Bergmann C, Heckenlively JR, Otto EA and Hildebrandt F:
Genotype-phenotype correlation in 440 patients with NPHP-related
ciliopathies. Kidney Int. 80:1239–1245. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wolf MTF, Mucha BE, Attanasio M, Zalewski
I, Karle SM, Neumann HP, Rahman N, Bader B, Baldamus CA, Otto E, et
al: Mutations of the Uromodulin gene in MCKD type 2 patients
cluster in exon 4, which encodes three EGF-like domains. Kidney
Int. 64:1580–1587. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rinschen MM, Schermer B and Benzing T:
Vasopressin-2 receptor signaling and autosomal dominant polycystic
kidney disease: From bench to bedside and back again. J Am Soc
Nephrol. 25:1140–1147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Montojo J, Zuberi K, Rodriguez H, Kazi F,
Wright G, Donaldson SL, Morris Q and Bader GD: GeneMANIA cytoscape
plugin: Fast gene function predictions on the desktop.
Bioinformatics. 26:2927–2928. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Angioi A and Pani A: FSGS: From
pathogenesis to the histological lesion. J Nephrol. 29:517–523.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Soliman NA, Hildebrandt F, Allen SJ, Otto
EA, Nabhan MM and Badr AM: Homozygous NPHP1 deletions in Egyptian
children with nephronophthisis including an infantile onset
patient. Pediatr Nephrol. 25:2193–2194. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang X, Kanegane H, Nishida N, Imamura T,
Hamamoto K, Miyashita R, Imai K, Nonoyama S, Sanayama K, Yamaide A,
et al: Clinical and genetic characteristics of XIAP deficiency in
Japan. J Clin Immunol. 32:411–420. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gheissari A, Harandavar M, Hildebrandt F,
Braun DA, Sedghi M, Parsi N, Merrikhi A, Madihi Y and Aghamohammadi
F: Gene mutation analysis in Iranian children with
nephronophthisis: A two-center study. Iran J Kidney Dis. 9:119–125.
2015.PubMed/NCBI
|
|
27
|
Hoefele J, Nayir A, Chaki M, Imm A, Allen
SJ, Otto EA and Hildebrandt F: Pseudodominant inheritance of
nephronophthisis caused by a homozygous NPHP1 deletion. Pediatr
Nephrol. 26:967–971. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Saunier S, Calado J, Benessy F, Silbermann
F, Heilig R, Weissenbach J and Antignac C: Characterization of the
NPHP1 locus: Mutational mechanism involved in deletions in familial
juvenile nephronophthisis. Am J Hum Genet. 66:778–789. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Caridi G, Dagnino M, Rossi A, Valente EM,
Bertini E, Fazzi E, Emma F, Murer L, Verrina E and Ghiggeri GM:
Nephronophthisis type 1 deletion syndrome with neurological
symptoms: Prevalence and significance of the association. Kidney
Int. 70:1342–1347. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sugimoto K, Takemura Y, Yanagida H, Fujita
S, Miyazawa T, Sakata N, Okada M and Takemura T: Renal tubular
dysgenesis and tubulointerstitial nephritis antigen in juvenile
nephronophthisis. Nephrology (Carlton). 16:495–501. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Salomon R, Saunier S and Niaudet P:
Nephronophthisis. Pediatr Nephrol. 24:2333–2344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hildebrandt F, Strahm B, Nothwang HG,
Gretz N, Schnieders B, Singh-Sawhney I, Kutt R, Vollmer M and
Brandis M; Members of the APN Study Group, : Molecular genetic
identification of families with juvenile nephronophthisis type 1:
Rate of progression to renal failure. Kidney Int. 51:261–269. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bleyer AJ, Woodard AS, Shihabi Z, Sandhu
J, Zhu H, Satko SG, Weller N, Deterding E, McBride D, Gorry MC, et
al: Clinical characterization of a family with a mutation in the
uromodulin (Tamm-Horsfall glycoprotein) gene. Kidney Int. 64:36–42.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Krege JH, John SWM, Langenbach LL, Hodgin
JB, Hagaman JR, Bachman ES, Jennette JC, O'Brien DA and Smithies O:
Male-female differences in fertility and blood pressure in
ACE-deficient mice. Nature. 375:146–148. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Esther CR Jr, Howard TE, Marino EM,
Goddard JM, Capecchi MR and Bernstein KE: Mice lacking
angiotensin-converting enzyme have low blood pressure, renal
pathology, and reduced male fertility. Lab Investig. 74:953–965.
1996.PubMed/NCBI
|
|
36
|
Omran H, Häffner K, Vollmer M, Pigulla J,
Wagner G, Caridi G and Hildebrandt F: Exclusion of the candidate
genes ACE and Bcl-2 for six families with nephronophthisis not
linked to the NPH1 locus. Nephrol Dial Transplant. 14:2328–2331.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sang L, Miller JJ, Corbit KC, Giles RH,
Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, et al:
Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy
disease genes and pathways. Cell. 145:513–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tory K, Lacoste T, Burglen L, Morinière V,
Boddaert N, Macher MA, Llanas B, Nivet H, Bensman A, Niaudet P, et
al: High NPHP1 and NPHP6 mutation rate in patients with Joubert
syndrome and nephronophthisis: Potential epistatic effect of NPHP6
and AHI1 mutations in patients with NPHP1 mutations. J Am Soc
Nephrol. 18:1566–1575. 2007. View Article : Google Scholar : PubMed/NCBI
|