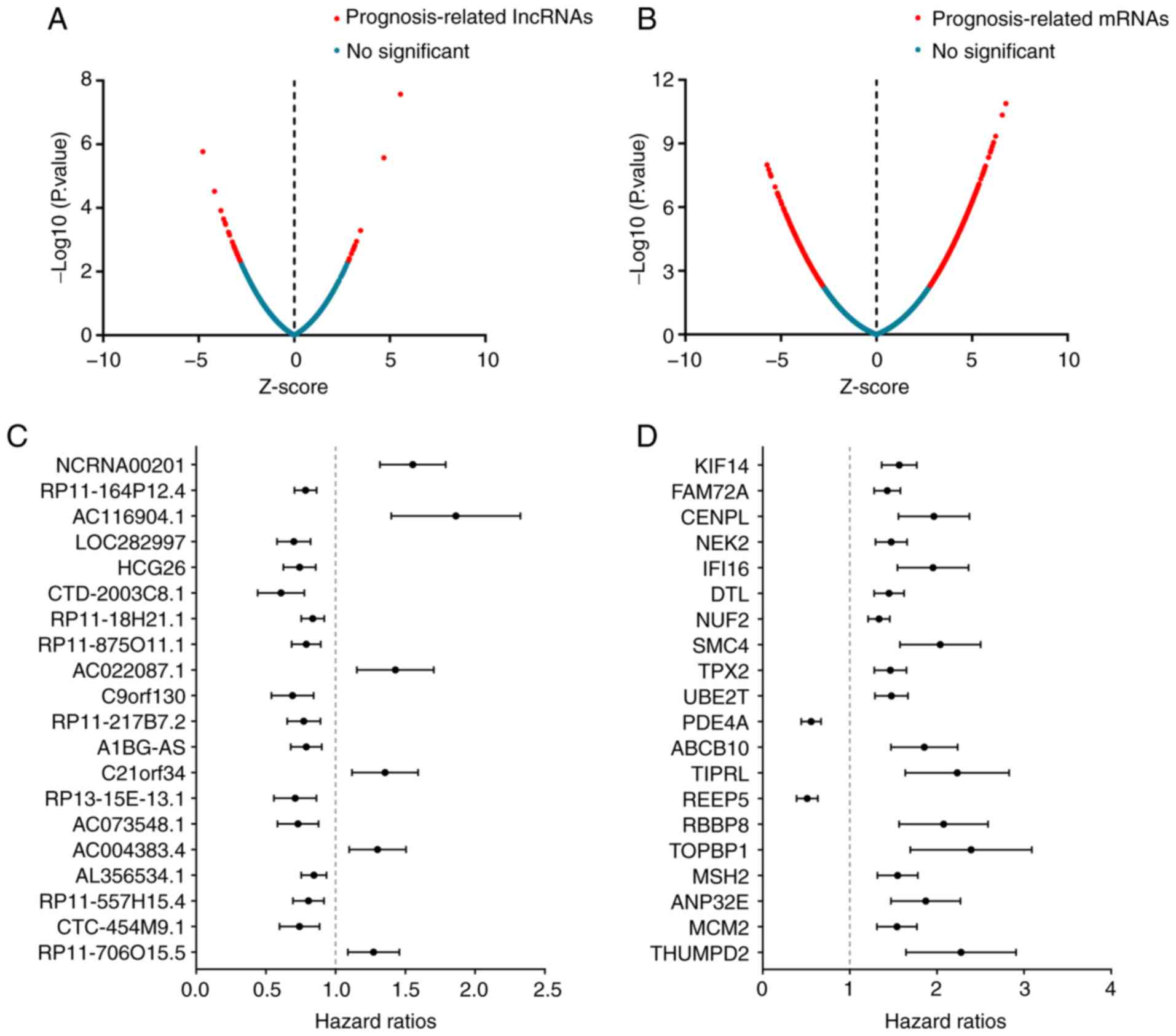
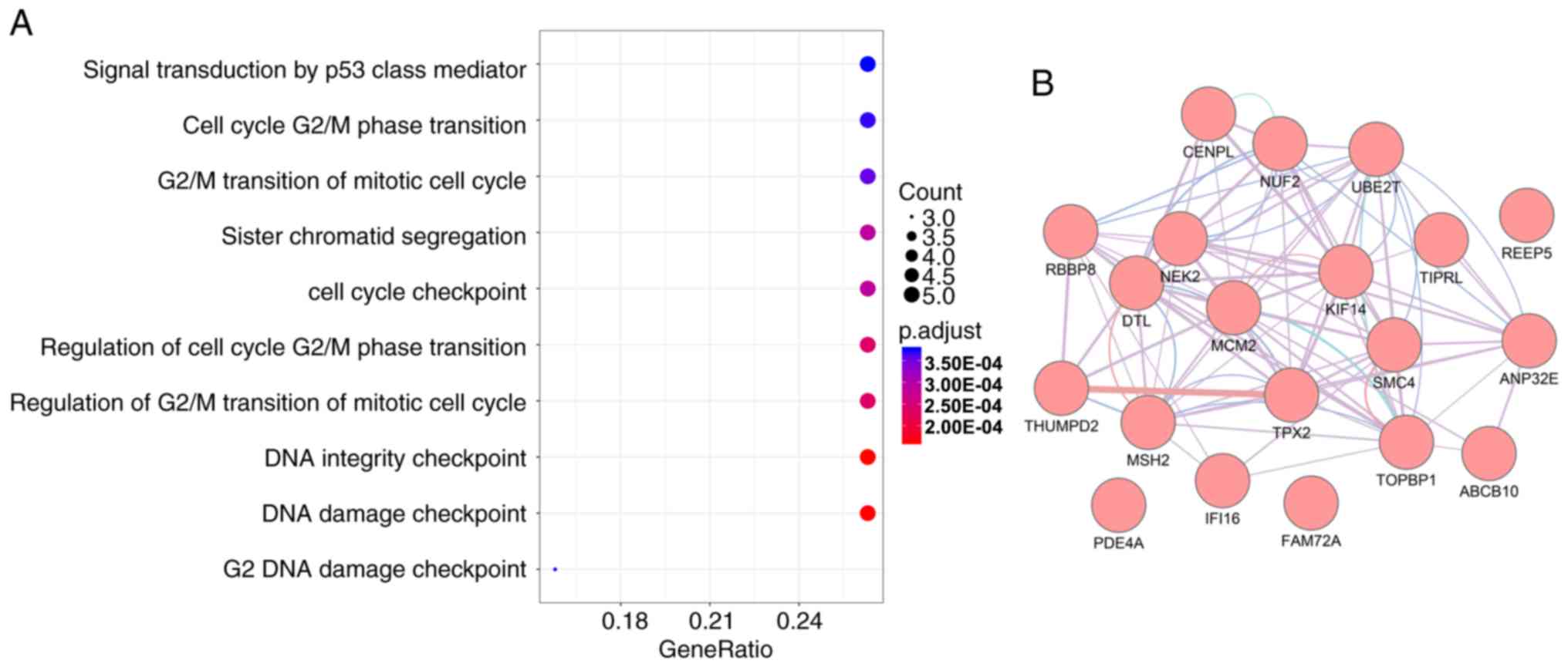
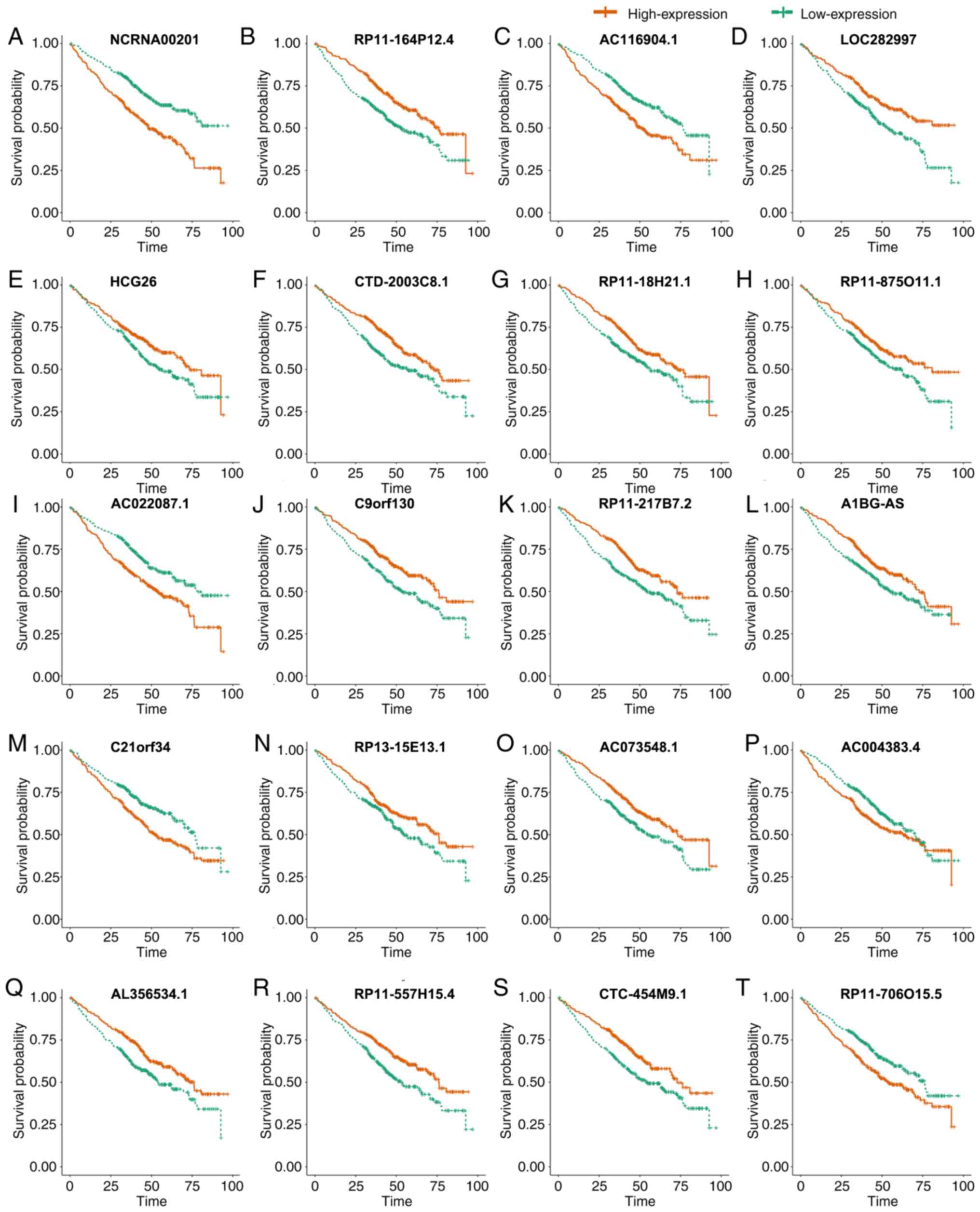
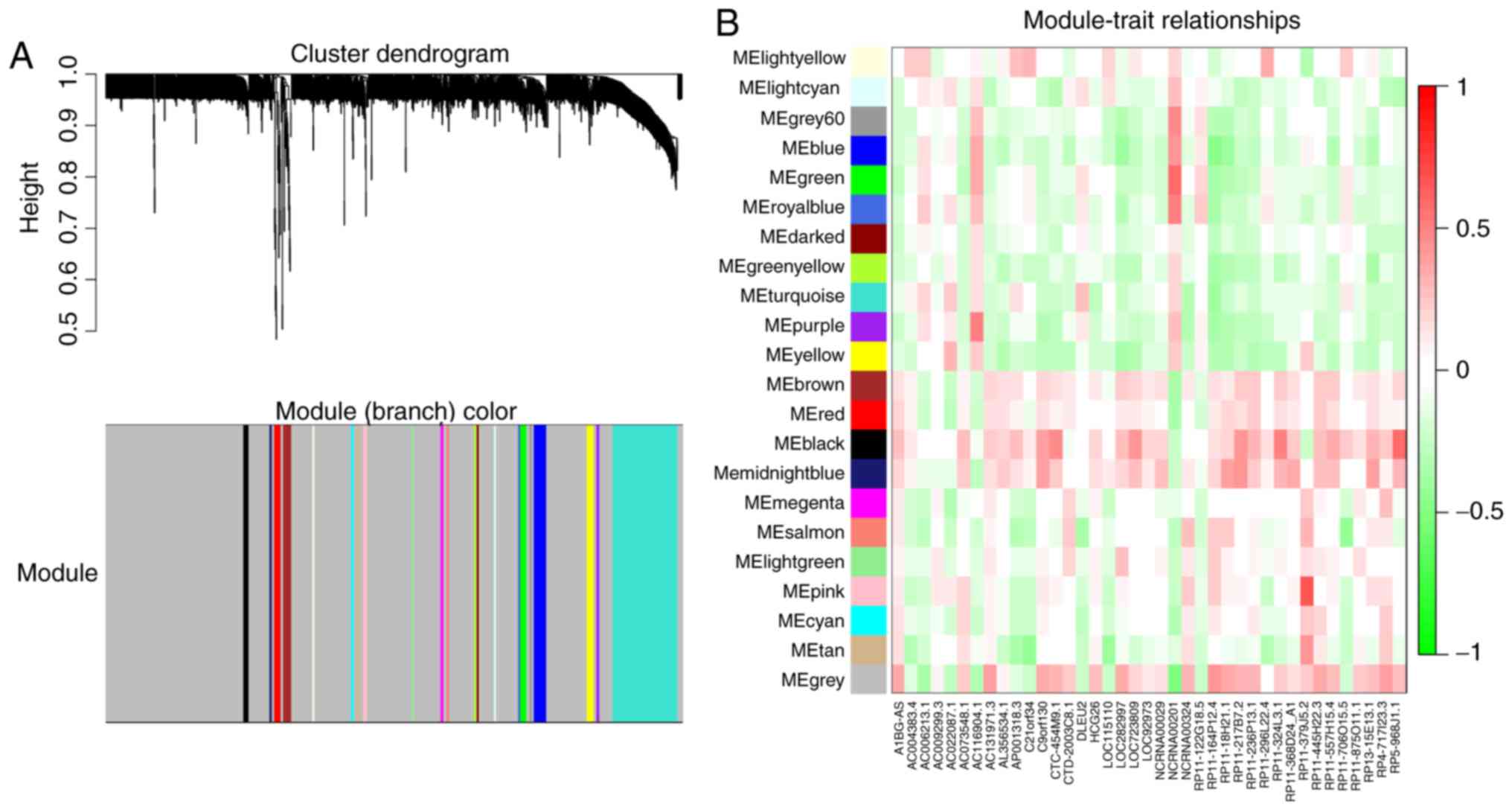
|
1
|
Xue JY, Huang C, Wang W, Li HB, Sun M and
Xie M: HOXA11-AS: A novel regulator in human cancer proliferation
and metastasis. Onco Targets Ther. 11:4387–4393. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang H, Sun J, Sun Y, Wang C, Gao S, Li W
and Hu JF: Long noncoding RNAs and their epigenetic function in
hematological diseases. Hematol Oncol. 37:15–21. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jia L, Zhang Y, Tian F, Chu Z and Xin H:
Long noncoding RNA colon cancer associated transcript-1 promotes
the proliferation, migration and invasion of cervical cancer. Mol
Med Rep. 16:5587–5591. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang R and Xia T: Long non-coding RNA
XIST regulates PDCD4 expression by interacting with miR-21-5p and
inhibits osteosarcoma cell growth and metastasis. Int J Oncol.
51:1460–1470. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li Z, Jiang X, Su Z, Li J, Kang P, Li C
and Cui Y: Current insight into a cancer-implicated long noncoding
RNA ZFAS1 and correlative functional mechanisms involved. Pathol
Res Pract. 214:1517–1523. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cai B, Zheng Y, Ma S, Xing Q, Wang X, Yang
B, Yin G and Guan F: BANCR contributes to the growth and invasion
of melanoma by functioning as a competing endogenous RNA to
upregulate Notch2 expression by sponging miR204. Int J Oncol.
51:1941–1951. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ohtsuka M, Ling H, Ivan C, Pichler M,
Matsushita D, Goblirsch M, Stiegelbauer V, Shigeyasu K, Zhang X,
Chen M, et al: H19 noncoding RNA, an independent prognostic factor,
regulates essential Rb-E2F and CDK8-β-catenin signaling in
colorectal cancer. EBioMedicine. 13:113–124. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ling ZA, Xiong DD, Meng RM, Cen JM, Zhao
N, Chen G, Li RL and Dang YW: LncRNA NEAT1 promotes deterioration
of hepatocellular carcinoma based on in vitro experiments, data
mining, and RT-qPCR analysis. Cell Physiol Biochem. 48:540–555.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li BL and Wan XP: The role of lncRNAs in
the development of endometrial carcinoma. Oncol Lett. 16:3424–3429.
2018.PubMed/NCBI
|
|
10
|
Zhu Y, Chen P, Gao Y, Ta N, Zhang Y, Cai
J, Zhao Y, Liu S and Zheng J: MEG3 activated by Vitamin D inhibits
colorectal cancer cells proliferation and migration via regulating
clusterin. EBioMedicine. 30:148–157. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu Q, Yu T, Ou X, Cao D, Xie T and Chen X:
Potential lncRNA diagnostic biomarkers for early gastric cancer.
Mol Med Rep. 16:9545–9552. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiong DD, Li ZY, Liang L, He RQ, Ma FC,
Luo DZ, Hu XH and Chen G: The LncRNA NEAT1 accelerates lung
adenocarcinoma deterioration and binds to Mir-193a-3p as a
competitive endogenous RNA. Cell Physiol Biochem. 48:905–918. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun W, Zu Y, Fu X and Deng Y: Knockdown of
lncRNA-XIST enhances the chemosensitivity of NSCLC cells via
suppression of autophagy. Oncol Rep. 38:3347–3354. 2017.PubMed/NCBI
|
|
14
|
Dong H, Jiang S, Fu Y, Luo Y, Gui R and
Liu J: Upregulation of lncRNA NR_046683 serves as a prognostic
biomarker and potential drug target for multiple myeloma. Front
Pharmacol. 10:452019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Butova R, Vychytilova-Faltejskova P,
Souckova A, Sevcikova S and Hajek R: Long non-coding RNAs in
multiple myeloma. Noncoding RNA. 5(pii): E132019.PubMed/NCBI
|
|
16
|
Yu T, Xu Z, Zhang X, Men L and Nie H: Long
intergenic non-protein coding RNA 152 promotes multiple myeloma
progression by negatively regulating microRNA-497. Oncol Rep.
40:3763–3771. 2018.PubMed/NCBI
|
|
17
|
Zhao Y, Xie Z, Lin J and Liu P: MiR-144-3p
inhibits cell proliferation and induces apoptosis in multiple
myeloma by targeting c-Met. Am J Transl Res. 9:2437–2446.
2017.PubMed/NCBI
|
|
18
|
Xia J, Xu H, Zhang X, Allamargot C,
Coleman KL, Nessler R, Frech I, Tricot G and Zhan F: Multiple
myeloma tumor cells are selectively killed by
pharmacologically-dosed ascorbic acid. EBioMedicine. 18:41–49.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao Y, Zhang E, Lv N, Ma L, Yao S, Yan M,
Zi F, Deng G, Liu X, He J, et al: Metformin and FTY720
synergistically induce apoptosis in multiple myeloma cells. Cell
Physiol Biochem. 48:785–800. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Knief J, Reddemann K, Gliemroth J, Brede
S, Bartscht T and Thorns C: ERG expression in multiple myeloma-A
potential diagnostic pitfall. Pathol Res Pract. 213:130–132. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen R, Zhang X, Gao C, Luan C, Wang Y and
Chen B: Treatment and prognostic factors for survival in newly
diagnosed multiple myeloma patients with bortezomib and
dexamethasone regimen: A single Chinese center retrospective study.
Cancer Manag Res. 9:373–380. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jin J, Wang T, Wang Y, Chen S, Li Z, Li X,
Zhang J and Wang J: SRC3 expressed in BMSCs promotes growth and
migration of multiple myeloma cells by regulating the expression of
Cx43. Int J Oncol. 51:1694–1704. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Joao C, Bergantim R, Neves M, Chacim S,
Afonso C, Barradas J, Bernardo M, Coelho H, Esteves G, Fraga C, et
al: Multiple myeloma in elderly patients-a Portuguese multicentric
real-life study. Ann Hematol. 98:1689–1701. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nooka AK, Kaufman JL, Hofmeister CC,
Joseph NS, Heffner TL, Gupta VA, Sullivan HC, Neish AS, Dhodapkar
MV and Lonial S: Daratumumab in multiple myeloma. Cancer.
125:2364–2382. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Song X, Wilson KL, Kagan J and Panjabi S:
Cost of peripheral neuropathy in patients receiving treatment for
multiple myeloma: A US administrative claims analysis. Ther Adv
Hematol. 10:20406207198390252019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen WC, Kanate AS, Craig M, Petros WP and
Hazlehurst LA: Emerging combination therapies for the management of
multiple myeloma: The role of elotuzumab. Cancer Manag Res.
9:307–314. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Willenbacher W, Seeber A, Steiner N,
Willenbacher E, Gatalica Z, Swensen J, Kimbrough J and Vranic S:
Towards molecular profiling in multiple myeloma: A literature
review and early indications of its efficacy for informing
treatment strategies. Int J Mol Sci. 19(pii): E20872018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ribatti D and Vacca A: New insights in
Anti-angiogenesis in multiple myeloma. Int J Mol Sci. 19(pii):
E20312018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zeng ZH, Chen JF, Li YX, Zhang R, Xiao LF
and Meng XY: Induction regimens for transplant-eligible patients
with newly diagnosed multiple myeloma: A network meta-analysis of
randomized controlled trials. Cancer Manag Res. 9:287–298. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Abramson HN: The multiple myeloma drug
pipeline-2018: A review of small molecules and their therapeutic
targets. Clin Lymphoma Myeloma Leuk. 18:611–627. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Handa H, Kuroda Y, Kimura K, Masuda Y,
Hattori H, Alkebsi L, Matsumoto M, Kasamatsu T, Kobayashi N, Tahara
KI, et al: Long non-coding RNA MALAT1 is an inducible stress
response gene associated with extramedullary spread and poor
prognosis of multiple myeloma. Br J Haematol. 179:449–460. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hu Y, Lin J, Fang H, Fang J, Li C, Chen W,
Liu S, Ondrejka S, Gong Z, Reu F, et al: Targeting the
MALAT1/PARP1/LIG3 complex induces DNA damage and apoptosis in
multiple myeloma. Leukemia. 32:2250–2262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ronchetti D, Agnelli L, Taiana E, Galletti
S, Manzoni M, Todoerti K, Musto P, Strozzi F and Neri A: Distinct
lncRNA transcriptional fingerprints characterize progressive stages
of multiple myeloma. Oncotarget. 7:14814–14830. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Geng W, Guo X, Zhang L, Ma Y, Wang L, Liu
Z, Ji H and Xiong Y: Resveratrol inhibits proliferation, migration
and invasion of multiple myeloma cells via NEAT1-mediated
Wnt/β-catenin signaling pathway. Biomed Pharmacother. 107:484–494.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu Y and Wang H: LncRNA NEAT1 promotes
dexamethasone resistance in multiple myeloma by targeting
miR-193a/MCL1 pathway. J Biochem Mol Toxicol. 322018.
|
|
36
|
Zhang ZS, Wang J, Zhu BQ and Ge L: Long
noncoding RNA UCA1 promotes multiple myeloma cell growth by
targeting TGF-β. Eur Rev Med Pharmacol Sci. 22:1374–1379.
2018.PubMed/NCBI
|
|
37
|
Yang X, Ye H, He M, Zhou X, Sun N, Guo W,
Lin X, Huang H, Lin Y, Yao R and Wang H: LncRNA PDIA3P interacts
with c-Myc to regulate cell proliferation via induction of pentose
phosphate pathway in multiple myeloma. Biochem Biophys Res Commun.
498:207–213. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun Y, Pan J, Zhang N, Wei W, Yu S and Ai
L: Knockdown of long non-coding RNA H19 inhibits multiple myeloma
cell growth via NF-κB pathway. Sci Rep. 7:180792017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen L, Hu N, Wang C, Zhao H and Gu Y:
Long non-coding RNA CCAT1 promotes multiple myeloma progression by
acting as a molecular sponge of miR-181a-5p to modulate HOXA1
expression. Cell Cycle. 17:319–329. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Meng YB, He X, Huang YF, Wu QN, Zhou YC
and Hao DJ: Long noncoding RNA CRNDE promotes multiple myeloma cell
growth by suppressing miR-451. Oncol Res. 25:1207–1214. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
He RQ, Zhou XG, Yi QY, Deng CW, Gao JM,
Chen G and Wang QY: Prognostic signature of alternative splicing
events in bladder urothelial carcinoma based on spliceseq data from
317 cases. Cell Physiol Biochem. 48:1355–1368. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Formicola D, Petrosino G, Lasorsa VA,
Pignataro P, Cimmino F, Vetrella S, Longo L, Tonini GP, Oberthuer
A, Iolascon A, et al: An 18 gene expression-based score classifier
predicts the clinical outcome in stage 4 neuroblastoma. J Transl
Med. 14:1422016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhou M, Zhang Z, Zhao H, Bao S and Sun J:
A novel lncRNA-focus expression signature for survival prediction
in endometrial carcinoma. BMC Cancer. 18:392018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kim HY, Lee DH, Lee JH, Cho YY, Cho EJ, Yu
SJ, Kim YJ and Yoon JH: Novel biomarker-based model for the
prediction of sorafenib response and overall survival in advanced
hepatocellular carcinoma: A prospective cohort study. BMC Cancer.
18:3072018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liang L, Zeng JH, Qin XG, Chen JQ, Luo DZ
and Chen G: Distinguishable prognostic signatures of left- and
right-sided colon cancer: A study based on sequencing data. Cell
Physiol Biochem. 48:475–490. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Edgar R, Domrachev M and Lash AE: Gene
expression omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41((Database Issue)):
D991–D995. 2013.PubMed/NCBI
|
|
48
|
Shi L, Campbell G, Jones WD, Campagne F,
Wen Z, Walker SJ, Su Z, Chu TM, Goodsaid FM, Pusztai L, et al: The
microarray quality control (MAQC)-II study of common practices for
the development and validation of microarray-based predictive
models. Nat Biotechnol. 28:827–838. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
O'Leary NA, Wright MW, Brister JR, Ciufo
S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B,
Ako-Adjei D, et al: Reference sequence (RefSeq) database at NCBI:
Current status, taxonomic expansion, and functional annotation.
Nucleic Acids Res. 44:D733–D745. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zerbino DR, Achuthan P, Akanni W, Amode
MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, et
al: Ensembl 2018. Nucleic Acids Res. 46:D754–D761. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu J, Lichtenberg T, Hoadley KA, Poisson
LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee
AV, et al: An integrated TCGA pan-cancer clinical data resource to
drive high-quality survival outcome analytics. Cell.
173:400–416.e11. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ioannidis JPA: The proposal to lower P
value thresholds to .005. JAMA. 319:1429–1430. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kanehisa M, Sato Y, Furumichi M, Morishima
K and Tanabe M: New approach for understanding genome variations in
KEGG. Nucleic Acids Res. 47:D590–D595. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kanehisa M, Furumichi M, Tanabe M, Sato Y
and Morishima K: KEGG: New perspectives on genomes, pathways,
diseases and drugs. Nucleic Acids Res. 45:D353–D361. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Warde-Farley D, Donaldson SL, Comes O,
Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT,
et al: The GeneMANIA prediction server: Biological network
integration for gene prioritization and predicting gene function.
Nucleic Acids Res. 38:W214–W220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lin P, He RQ, Ma FC, Liang L, He Y, Yang
H, Dang YW and Chen G: Systematic analysis of survival-associated
alternative splicing signatures in gastrointestinal
pan-adenocarcinomas. EBioMedicine. 34:46–60. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu LM, Xiong DD, Lin P, Yang H, Dang YW
and Chen G: DNA topoisomerase 1 and 2A function as oncogenes in
liver cancer and may be direct targets of nitidine chloride. Int J
Oncol. 53:1897–1912. 2018.PubMed/NCBI
|
|
61
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Langfelder P and Horvath S: Fast R
functions for robust correlations and hierarchical clustering. J
Stat Softw. 46(pii): i112012.PubMed/NCBI
|
|
63
|
Hu AX, Huang ZY, Zhang L and Shen J:
Potential prognostic long non-coding RNA identification and their
validation in predicting survival of patients with multiple
myeloma. Tumour Biol. 39:10104283176945632017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhou M, Zhao H, Wang Z, Cheng L, Yang L,
Shi H, Yang H and Sun J: Identification and validation of potential
prognostic lncRNA biomarkers for predicting survival in patients
with multiple myeloma. J Exp Clin Cancer Res. 34:1022015.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Thierry G, Beneteau C, Pichon O, Flori E,
Isidor B, Popelard F, Delrue MA, Duboscq-Bidot L, Thuresson AC, van
Bon BW, et al: Molecular characterization of 1q44 microdeletion in
11 patients reveals three candidate genes for intellectual
disability and seizures. Am J Med Genet A. 158A:1633–1640. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Sutaria DS, Jiang J, Azevedo-Pouly ACP,
Lee EJ, Lerner MR, Brackett DJ, Vandesompele J, Mestdagh P and
Schmittgen TD: Expression profiling identifies the noncoding
processed transcript of HNRNPU with proliferative properties in
pancreatic ductal adenocarcinoma. Noncoding RNA. 3(pii):
E242017.PubMed/NCBI
|
|
67
|
He W, Wei D, Cai, Chen S, Li S and Chen W:
Altered long non-coding RNA transcriptomic profiles in ischemic
stroke. Hum Gene Ther. 29:719–732. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liu YD, Li Y, Feng SX, Ye DS, Chen X, Zhou
XY and Chen SL: Long noncoding RNAs: Potential regulators involved
in the pathogenesis of polycystic ovary syndrome. Endocrinology.
158:3890–3899. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Low JS, Chin YM, Mushiroda T, Kubo M,
Govindasamy GK, Pua KC, Yap YY, Yap LF, Subramaniam SK, Ong CA, et
al: A genome wide study of copy number variation associated with
nasopharyngeal carcinoma in Malaysian Chinese identifies CNVs at
11q14.3 and 6p21.3 as candidate loci. PLoS One. 11:e01457742016.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Simino J, Shi G, Arnett D, Broeckel U,
Hunt SC and Rao DC: Variants on chromosome 6p22.3 associated with
blood pressure in the HyperGEN study: Follow-up of FBPP
quantitative trait loci. Am J Hypertens. 24:1227–1233. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Pei G, Lan Y, Chen D, Ji L and Hua ZC: FAK
regulates E-cadherin expression via p-SrcY416/p-ERK1/2/p-Stat3Y705
and PPARγ/miR-125b/Stat3 signaling pathway in B16F10 melanoma
cells. Oncotarget. 8:13898–13908. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yao R, Sun X, Xie Y, Sun X, Yao Y, Li H,
Li Z, Gao J and Xu K: Identification of a novel c-Myc inhibitor
with anti-tumor effects on multiple myeloma cells. Biosci Rep.
38(pii): BSR201810272018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wang H, Ding Q, Wang M, Guo M and Zhao Q:
miR-29b inhibits the progression of multiple myeloma through
downregulating FOXP1. Hematology. 24:32–38. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ronchetti D, Manzoni M, Todoerti K, Neri A
and Agnelli L: In Silico characterization of miRNA and long
non-coding RNA interplay in multiple myeloma. Genes (Basel).
7(pii): E1072016. View Article : Google Scholar : PubMed/NCBI
|