Introduction
Sepsis is the most common cause of acute kidney
injury (AKI) development. Clinical data have demonstrated that, in
developed countries, sepsis accounts for 26–50% of all cases of AKI
(1,2). In addition, sepsis-induced AKI is
associated with a 6–8-fold risk of pneumonia and an increase in
risk of progression to chronic kidney disease (3). Despite this, the mechanisms by which
sepsis induces kidney injury are yet to be elucidated.
Autophagy is a highly regulated lysosomal
intracellular degradation pathway involved in removing aggregated
protein and maintaining intracellular homeostasis (4,5).
Autophagy is associated with several diseases, including kidney
disease (6). Autophagy is
considered to be a degradation system that occurs under conditions
of stress in order to meet energy and nutrient requirements.
Autophagy is very important for a number of fundamental biological
activities (4,5). Dysregulation of autophagy has been
emphasized in the occurrence of a variety of diseases, as the
targets of selective autophagy, including key organelles, such as
mitochondria and lysosomes, are involved in diseases (7,8).
An increasing number of studies have revealed that
autophagy plays a potential role in kidney disease and aging, which
is associated with the genetic modification of autophagy-associated
genes (9–13). A growing amount of evidence
indicates that dysregulation of the autophagic pathway is involved
in the pathogeneses of kidney diseases, such as AKI, polycystic
kidney disease, and focal and segmental glomerulosclerosis (FSGS),
and other kidney diseases (14–17).
With the identification of autophagy-associated genes (ATGs), using
autophagy-defective animals have fascinated researchers to study
the characteristics of the molecular regulators that participate in
autophagy (18). The production of
autophagosomes starts from the phagophore, which is a
double-membrane structure formed by expansion and closure of a
vesicle. Phagophores are formed at the phagophore assembly site or
in the pre-autophagosommal structure. The formation of the
phagophore is catalyzed by 16 ATG proteins, which constitute the
conserved core molecular mechanism of ATG, and expands into
autophagosomes (19). Several
genes are involved in the generation and maturation of
autophagosomes, including ATGs, beclin I gene (BECN1) and
microtubule-associated protein 1 light chain 3 (LC3A/B) (20–23).
In a recent study, the suppression of autophagy through
knocking-out ATG7 significantly ameliorated Van-induced kidney
injury (24). Notably,
Atg5-deficient mice exhibited deteriorated kidney function in an
acute hyperuricemic kidney injury mouse model and demonstrated a
mild kidney injury phenotype (9).
A recent study demonstrated that the deficiency of LC3-associated
phagocytosis (LAP) leads to the formation of autoantibody
deposition and systemic inflammation in the mouse kidney,
indicating that LC3-associated phagocytosis and LAP-associated
genes (Atg5, Atg7, Becn1, Cybb/Nox2 and
Rubcn/Rubicon) are likely to play a potential role in kidney
injury.
MicroRNAs (miRNA) are single-stranded, small
(22–24) nucleotides, non-coding RNAs that can
inhibit endogenous gene expression by specifically binding to the
3′-untranslated region (3′-UTR) of the target gene mRNAs to induce
translational repression or mRNA cleavage. miRNAs play significant
roles in several biological processes, including cell proliferation
and death, hematopoiesis, neuronal patterning and organ development
(25–31). A number of studies have identified
miRNAs as biomarkers for various diseases, not just in the
progression of disease, but also in the prognosis, due to its
important role and conservatism (32,33).
In addition, it has been reported that miRNA dysregulation played
an important role in inflammation and the development of a number
of diseases, such as cancer (32,34,35).
A recent study demonstrated that miR-155-5p is significantly
increased in the kidney tubules of patients with diabetic kidney
disease (36) and downregulated
expression of sirt1, and autophagy-associated proteins LC3II, ATG5
and ATG7 (37). Thus, the present
study speculated whether autophagy is involved in sepsis-induced
kidney injury, and whether miRNA plays an important role in this
process.
The present study demonstrated that excessive
autophagy is evident in sepsis-induced kidney injury. It was also
revealed that miR-526b is significantly downregulated in
sepsis-induced kidney injury. Furthermore, the present study
identified ATG7 as a directed target gene of miR-526b.
Materials and methods
Cell culture and transfection
HK2 cells were cultured in 75 cm2 and 25
cm2 cell culture flasks and 6 or 12 well plates
(Corning-Star) in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), antibiotics (Penicillin-Streptomycin solution;
Sigma Aldrich; Merck KGaA) at 37°C in a 5% CO2
atmosphere. The miR-526b mimic, ATG7 overexpression plasmid,
miR-526b inhibitor, ATG7 siRNA were designed and synthesized by
Synthgene (Nanjing, China). Transient transfection was performed
using X-tremeGENE HP DNA Transfection reagent (Roche) according to
the manufacturer's protocol. Vector-NC (empty pcDNA3.1 vector),
siRNA-NC, mimics-NC, or inhibitors-NC were used as the
corresponding negative controls. ATG7 was used at a concentration
of 4 µg per well while ATG7 overexpression, siAtg7 and miR-526b
mimic were used at a final concentration of 50 nM and miR-526b
inhibitors were used at a final concentration of 100 nM. The
sequence of miR-526b mimic is 5′-GAAAGUGCUUCCUUUUAGAGGC-3′ (guide
strand) and 5′-CUCUUGAGGGAAGCACUUUCUGU-3′ (passenger strand). The
sequence of miR-526b inhibitor is 5′-CUCUUGAGGGAAGCACUUUCUGU-3′.
The ATG7-pcDNA3.1 overexpression plasmid was purchased from
Synthgene (Nanjing, China). The sequence of siAtg7 is
5′-GGUUCUUGAUCAAUAUGAACG-3′ (sense strand) and
5′-UUCAUAUUGAUCAAGAACCUG-3′ (antisense strand). The sequence of
siRNA-NC is 5′-UUCUCCGAACGUGUCACGUUUdTdT-3′ (guide strand) and
5′-AAACGUGACACGUUCGGAGAAdTdT-3′ (passenger strand). The sequence of
mimics-NC is 5′-UUCUCCGAACGUGUCACGU-3′ (sense strand) and
5′-ACGUGACACGUUCGGAGAA-3′ (passenger strand). The sequence of
inhibitor NC is 5′-ACGUGACACGUUCGGAGAA-3′.
Cell viability assay
Cells were plated in 96-well plates at a density of
5,000 cells per well. After overnight incubation, cells were
stimulated with LPS (Sigma Aldrich; Merck KGaA) at a concentration
of 100 ng/ml for 12, 24 and 48 h. Cell viability was then assayed
with MTT according to the manufacturer's protocol.
Immunofluorescence confocal laser
scanning microscopy
Following transfection of LC3-GFP plasmids for 12 h,
the HK2 cells were treated with LPS (100 ng/ml) for 12, 24 and 48
h. HK2 cells were then fixed with 4% paraformaldehyde for 15 min at
room temperature. Slides were counterstained with DAPI (0.1 µg/ml)
and examined using the Nikon A1 confocal laser microscope
system.
Mouse inflammation models
BALB/c mice (male, 6 weeks old, 20 g) were purchased
from the Model Animal Research Center of Nanjing University, Ltd.
Animal care and euthanasia were performed with the approval of the
Institutional Animal Care and Use Committee (IACUC) of Nanjing
Medical University. Mice were challenged with CLP but anesthetized
with sodium pentobarbital (30 mg/kg). The sham surgery, which
included the same procedure except for ligation and perforation of
the cecum, was performed on control mice. After 4 days, mice were
intravenously injected with sodium pentobarbital (90 mg/kg). Death
was confirmed by observing respiratory arrest, no nerve reflex and
muscle relaxation. Kidney tissues were collected after mice were
sacrificed and assessed for mRNA expression.
Protein extraction and western
blotting
Total proteins were extracted from cells using RIPA
lysis and extraction buffer (Beyotime Institute of Biotechnology)
containing protease inhibitor and phosphatase inhibitor (Thermo
Fisher Scientific, Inc.). Equivalent amounts of protein were
separated via SDS-PAGE (12% gel), and transferred onto Immobilon-NC
Membranes (EMD Millipore). After blocking with 5% non-fat milk
solution for 1 h at room temperature, the membranes were washed
with TBS-Tween-20 (0.1%, v/v) and then incubated with primary
antibody at 4°C overnight followed by incubating with horseradish
peroxidase-conjugated secondary antibody for 1 h at room
temperature. ATG7, Beclin, LC3 and β-actin antibodies were
purchased from Abcam. β-actin served as a loading control and
protein bands were quantified using Image J Software. The
antibody-antigen complexes were visualized via chemiluminescence
with the enhanced ECL immunoblotting system (Tanon).
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from tissues or cultured
cells using TRIzol® reagent (Thermo Fisher Scientific,
Inc.) and RNA was reverse transcribed to cDNA from 1 µg of total
RNA using AMV reverse transcriptase (Takara) and a RT primer
according to the manufacturer's protocol. The reaction conditions
were: 16°C for 30 min, 42°C for 30 min and 85°C for 5 min. RT-PCR
was performed using a Taqman PCR kit on an Applied Biosystems 7300
sequence detection system (Applied Biosystems). GAPDH levels were
used to normalize the relative abundance of ATG7 and mRNAs. U6
levels were used to normalize the relative abundance of miR-526b.
The reactions were performed in a 96-well plate at 95°C for 10 min,
followed by 40 cycles of 95°C for 15 sec, 56°C for 15 sec and 72°C
for 30 sec. Polymerase chain reaction primers: ATG7 FP:
5′-CAGCAGTGACGATCGGATGA-3′; ATG7 RP: 5′-TCAAGAACCTGGTGAGGCAC-3′;
GAPDH FP: 5′-GATATTGTTGACATCAATGAC-3′; GAPDH RP:
5′-TTGATTTTGGAGGGATCTCG-3′; miR-526b RTP:
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACACAGAA-3′; miR-526b
FP: 5′-GCGACTCTTGAGGGAAGCACT-3′; miR-526b RP:
5′-AGTGCAGGGTCCGAGGTATT-3′; U6 RTP:
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCATGCT-3′; U6 FP:
5′-CGGTCCAACGATACAGAGAAG-3′; U6 RP: 5′-AGTGCAGGGTCCGAGGTATT-3′.
Luciferase reporter assay
The entire 3′-UTR of ATG7 was inserted into a
luciferase reporter plasmid (Synthgene). In order to assess the
binding specificity, the sequences that interacted with miR-526a
were mutated, and the mutant ATG7-1 3′-UTR was inserted into an
equivalent luciferase reporter plasmid. For the luciferase reporter
assay, cells were plated in 24-well plates, and each well was
transfected with 1 µg of luciferase reporter plasmid, 1 µg of
β-galactosidase plasmid (internal control), and 100 pmole of
miR-526a mimic, and control mimic using Lipofectamine 2000 (Thermo
Fisher Scientific, Inc.). After 48 h, luciferase signals were
measured using a luciferase assay kit according to the
manufacture's protocol (Promega Corporation).
Statistical analysis
All experiments were repeated three times and the
data are presented as the mean ± standard deviation using SPSS 18.0
(SPSS, Inc.). One-way ANOVA and post hoc Dunnett's T3 test were
performed in order to compare the differences among and between
groups, respectively. P<0.05 was considered to indicate a
statistically significant result.
Results
Excessive autophagy is evident in an
in vitro sepsis-induced kidney injury model
In order to simulate the cell model, the present
study used LPS (100 ng/ml) to treat HK2 cells for 12, 24, 48 h and
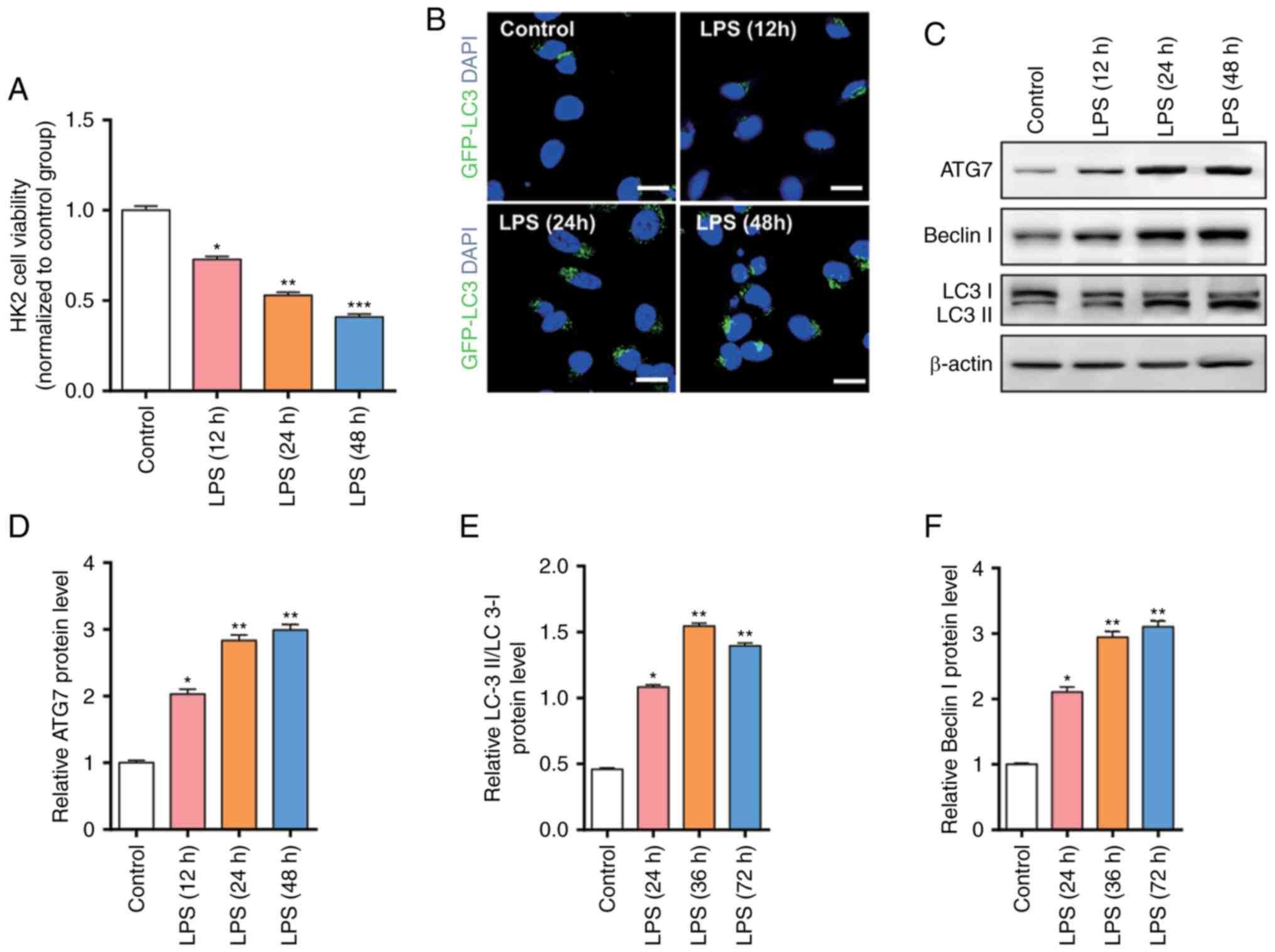
detected the cell viability through an MTT assay. As presented in
Fig. 1A, the cell viability was
significantly decreased following LPS stimulation, and the cell
viability was decreased with prolonged time. In order to further
evaluate the degree of autophagy, the present study used GFP-LC3 to
detect the expression level of autophagy-associated proteins LC3
(Fig. 1B). The results revealed
that excessive autophagy was induced following LPS stimulation
leading to apoptosis, indicating excessive autophagy is involved in
sepsis-induced kidney injury.
The present study demonstrated that significant
autophagy is induced following LPS stimulation; however, the
molecular mechanism remains unknown. The present study further
examined the expression level of autophagy-associated proteins
using western blotting (Fig. 1C).
The results revealed that autophagy-associated proteins, such as
ATG7, Beclin I and LC3, are gradually increased as time increased
(Fig. 1D-F).
ATG7 protein, but not mRNA, is
upregulated in LPS-treated HK2 cells
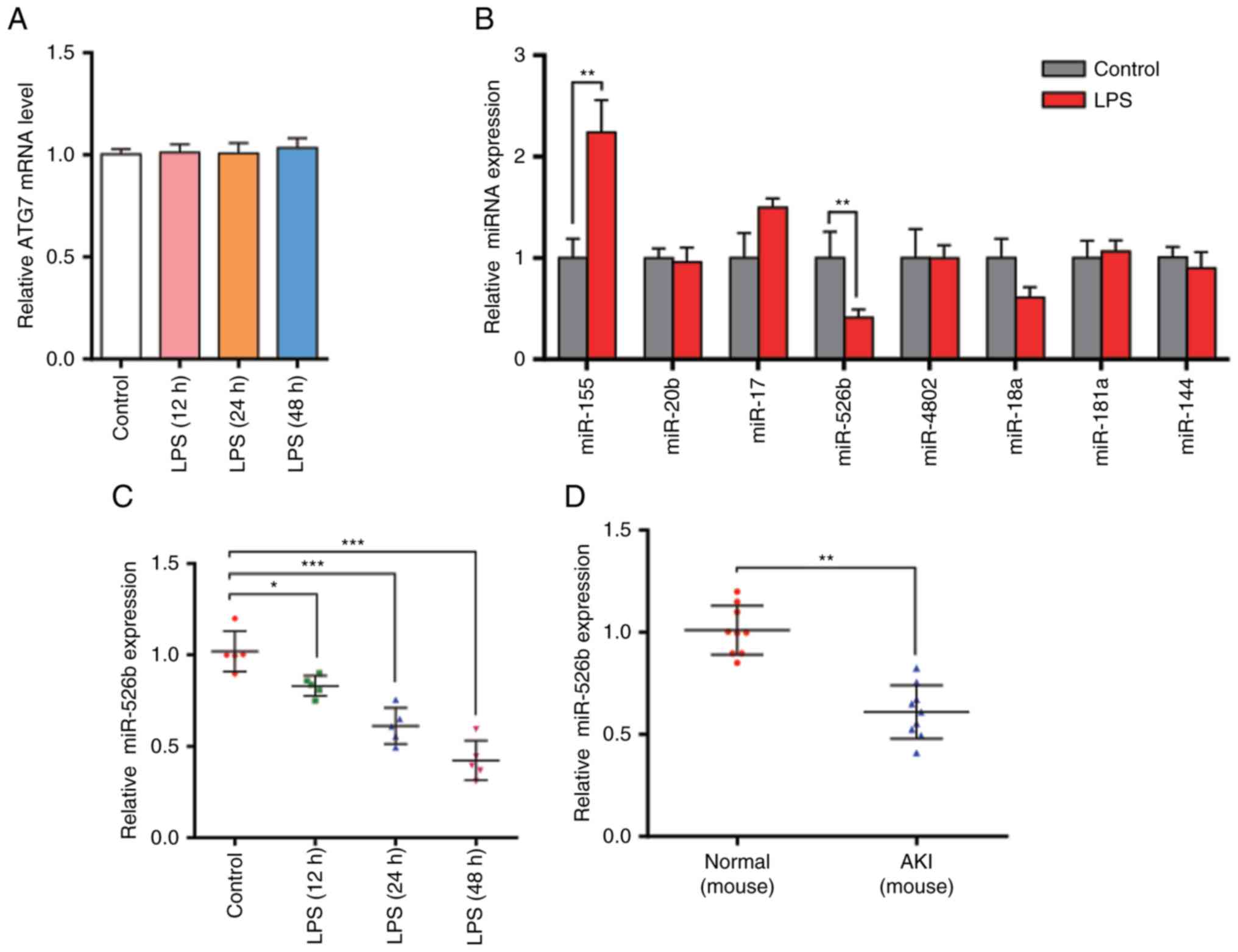
The present study revealed that ATG7 protein levels
were significantly increased in LPS-treated HK2 cells. The mRNA
levels were then examined using RT-qPCR. The results demonstrated
that ATG7 mRNA levels were not significantly different (Fig. 2A). The inconsistency between ATG7
mRNA level and protein expression suggests that there may be a
post-transcriptional mechanism that is involved in the
downregulation of ATG7 protein levels in LPS-treated HK2 cells.
miR-526b is downregulated in
sepsis-induced kidney injury models both in vitro and in vivo
As miRNA is an important and widespread molecule
that post-transcriptionally regulates gene expression, and it has
been reported to be involved in regulating the cell autophagy, the
present study hypothesized that ATG7 may be regulated by miRNA. In
order to test this hypothesis, the present study used
bioinformatics software combined with others' reports to predict
the potential miRNAs that target ATG7. The present study identified
8 miRNAs: miR-155, miR-20b, miR-17, miR-526b, miR-4802, miR-18a,
miR-181a and miR-144. The present study then analyzed the miRNA
levels following LPS treatment (Fig.
2B). As miRNAs should have opposite expression pattern changes
with their targets, and ATG7 protein levels were increased in
LPS-treated HK2 cells, the present study aimed to identify those
miRNAs that were downregulated. The results revealed that only
miR-526b was decreased following LPS treatment, indicating that
miR-526b is involved in the regulation of ATG7 expression. The
present study then checked the expression levels of miR-526b after
treating HK2 cells for 12, 24 and 48 h (Fig. 2C). The results revealed that
miR-526b levels were downregulated with the prolonged time.
Furthermore, the mouse sepsis model was established using cecal
ligation and puncture (CLP). The present study isolated the total
RNA from the mouse kidneys and measured miR-526b levels via RT-qPCR
(Fig. 2D). Decreased miR-526b
levels were observed, which is consistent with the findings in cell
models. Furthermore, the present study demonstrated that ATG7
protein levels in AKI mice were significantly increased compared
with normal mice (Fig. 2E and F),
while the ATG7 mRNA levels revealed no difference (Fig. 2G). The results indicated that
miR-526b has a potential role in the autophagy process of renal
tubular epithelial cells in sepsis-induced kidney injury at the
post-transcriptional level.
miR-526b directly regulates ATG7
expression at the post-transcriptional level
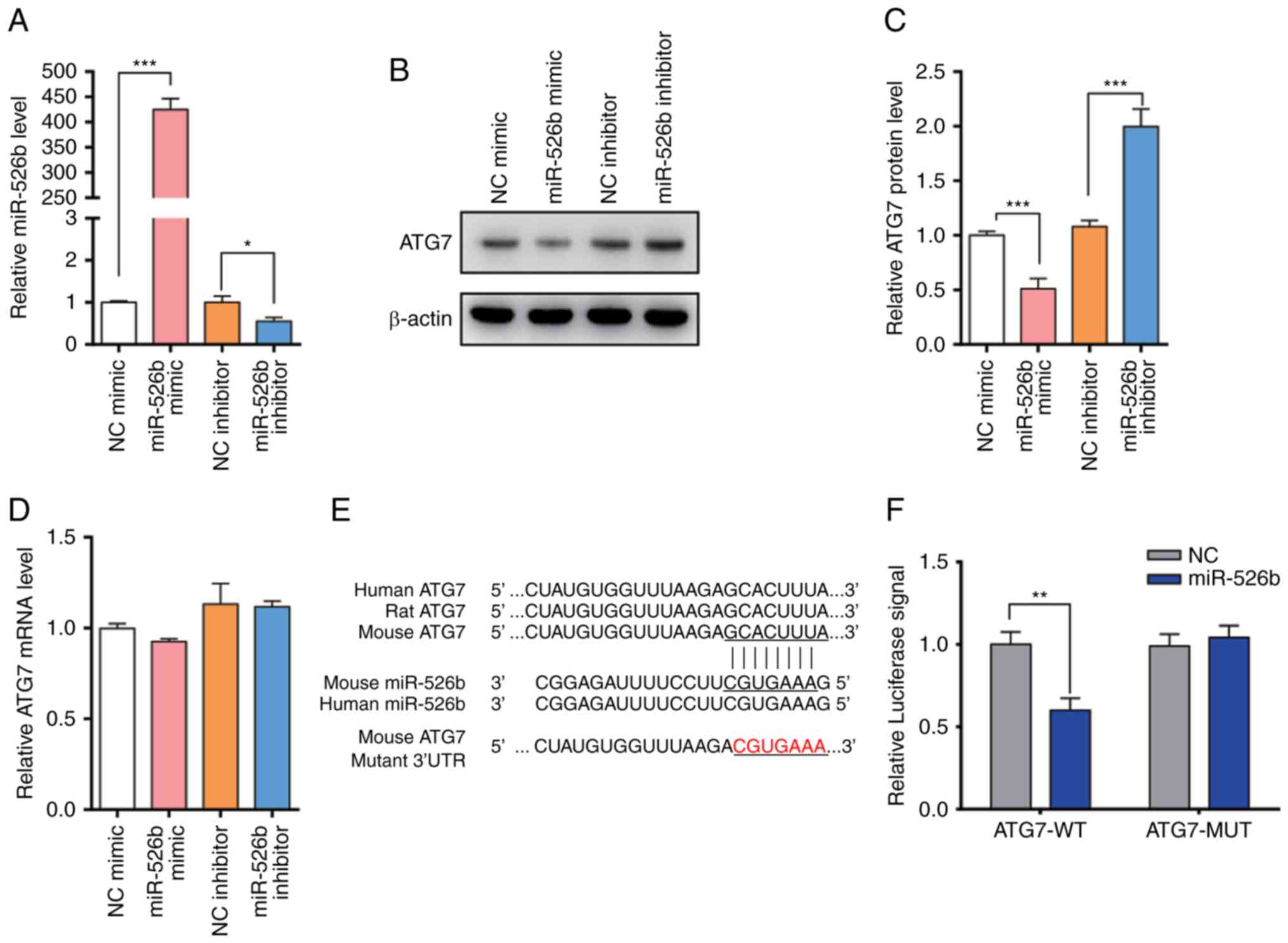
The present study modulated miR-526b levels in order
to investigate whether ATG7 was regulated directly by miR-526b
in vitro. The present study efficiently overexpressed or
knocked-down miR-526b using miR-526b mimic or inhibitor in HK2
cells (Fig. 3A). ATG7 protein
levels dramatically decreased upon miR-526b overexpression, while
ATG7 protein levels were increased when treated with miR-526b
inhibitor (Fig. 3B and C).
Furthermore, the present study detected the ATG7 mRNA level under
different conditions (Fig. 3D).
The alteration of miR-526b had no significant effect on the ATG7
level. The results indicated that miR-526b regulates the expression
of ATG7 through post-transcriptional regulation.
In order to validate that miR-526b suppressed ATG7
expression by directly binding to the 3′-UTR of ATG7 mRNA, the
present study performed a luciferase reporter assay (Fig. 3E). A reporter plasmid was
constructed that contained the 3′-UTR of ATG7 fragment, and the
resulting plasmid was transfected into HK2 cells along with the
miR-526b mimic or scrambled negative control RNAs. miR-526b mimic
decreased the luciferase reporter activity significantly when
compared with cells transfected with the control mimic.
Furthermore, the present study constructed a mutant binding site in
the ATG7 mRNA 3′-UTR on the reporter plasmid. The results revealed
that miR-526b mimic affected WT luciferase activity (Fig. 3F), suggesting that the binding
sites strongly contribute to the interactions of ATG7 mRNA with
miR-526b.
miR-526b inhibits autophagy in HK2
cells through targeting ATG7
The present study hypothesized that miR-526b can
inhibit autophagy through suppressing ATG7. Thus, HK2 cells were
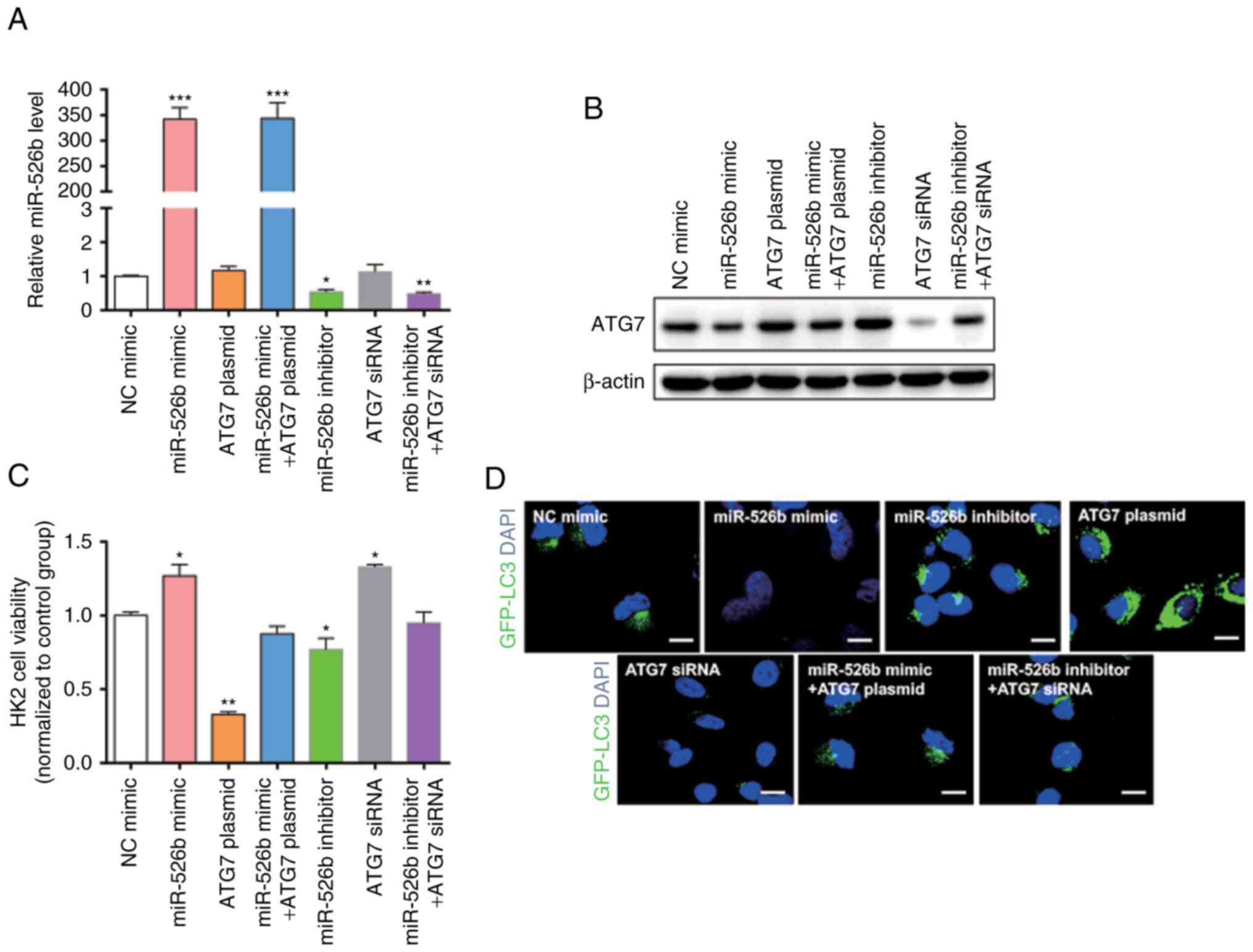
transfected with miR-526b mimic, ATG7 overexpression plasmid,
miR-526b inhibitor, ATG7 siRNA, co-transfected with miR-526b mimic
and ATG7 overexpression plasmid, or co-transfected with miR-526b
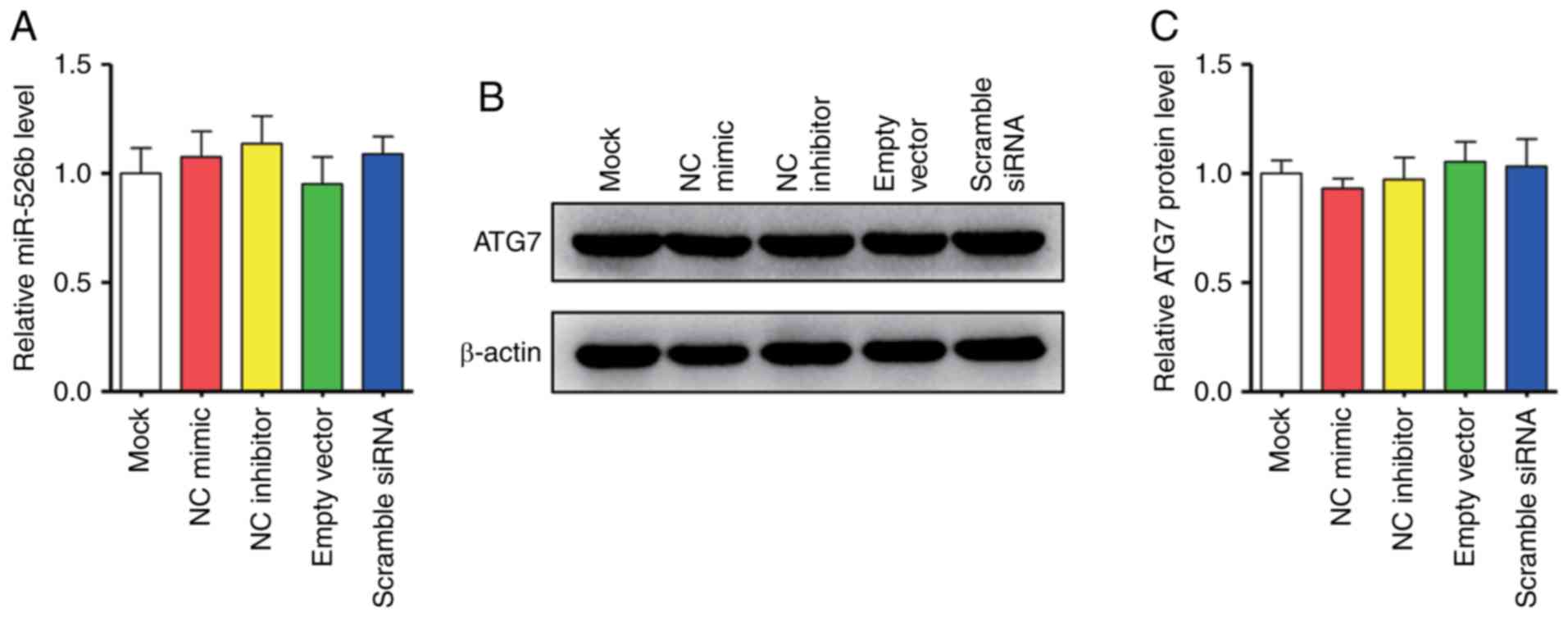
inhibitor and ATG7 siRNA. We first confirmed that the expression of
miR-526b and ATG7 in the cells would not be affected by the empty
vector and the transfection itself by using RT-qPCR and western
blot (Fig. 4A-C). Transfection
efficiency was detected using RT-qPCR (Fig. 5A) and western blot (Fig. 5B). The present study detected the
cell viability through an MTT assay (Fig. 5C); the cell viability exhibited a
significant increase in cells transfected with miR-526b mimic or
ATG7 siRNA only. The cell viability was significantly decreased in
cells transfected with ATG7 overexpression plasmid or miR-526b
inhibitor alone. MiR-526b mimic rescued the decrease in ATG7
overexpression-induced cell viability, while miR-526b inhibitor
rescued the increase in ATG7 siRNA-induced cell viability,
indicating that targeting of ATG7 is one mechanism by which
miR-526b exerts its oncomiR function. Furthermore, the present
study analyzed the autophagy in HK2 cells treated with LPS by
measuring LC3 expression level (Fig.
5D). The results revealed that miR-526b mimic inhibited cell
autophagy, meanwhile, ATG7 overexpression promoted cell autophagy.
MiR-526b mimic rescued ATG7-overexpression-mediated cell autophagy.
Similarly, miR-526b inhibitor promoted cell autophagy and ATG7
knockdown inhibited cell autophagy. Also, miR-526b inhibitor
rescued ATG7 knockdown-inhibited cell autophagy.
Furthermore, the present study measured the Beclin
I, LC3 protein levels using western blotting in HK2 cells (Fig. 5E-H). The results revealed that
Beclin I and LC3 protein levels were decreased following miR-526b
mimic, while Beclin I and LC3 protein levels were increased
following miR-526b inhibitor, suggesting that miR-526b inhibited
cell autophagy. Furthermore, Beclin I and LC3 protein levels were
increased following ATG7 overexpression, and decreased following
ATG7 knockdown, suggesting that ATG7 promoted cell autophagy.
However, Beclin I and LC3 protein levels were recovered when
co-transfected with miR-526b mimic and ATG7 overexpression plasmid
or co-transfected with miR-526b inhibitor and ATG7 siRNA. In
conclusion, miR-526b participated in the process of cell autophagy
by regulating ATG7 at the post-transcriptional level.
Discussion
AKI is a common disease in the adult intensive care
unit (ICU) (38). An increasing
amount of evidence has demonstrated that the incidence of AKI is
increasing. In the last few decades, AKI incidence increased by
2.8% per year in research that included >90,000 patients from
>20 ICUs (39). Furthermore,
effective therapy for AKI has been established. Recent studies
suggest that the origin of most cases of AKI is multifaceted, and
sepsis is considered to be the most common pathogenic factor for
AKI. The pathogenesis of sepsis is associated with various stress
factors. Sharma et al demonstrated that sepsis can induce
autophagy, which led to a negative effect on the body (40). Autophagy, as a highly regulated
lysosomal intracellular degradation pathway, maintains cellular
homeostasis by scavenging damaged organelles, clearance of
intracellular pathogens, innate and adaptive immunity and cell
death. Autophagy is induced by oxidant injury, energy depletion,
cell starvation and other harmful insults, the majority of which
are involved in the pathogenesis of AKI. Autophagy can also affect
the expression levels of autophagy constituent proteins LC3,
autophagy-associated genes (ATGs) and autophagy-associated protein
Beclin I (41,42). The present study demonstrated that
the cell viability is significantly decreased following LPS
stimulation, and upregulated the protein levels of LC3, ATG7 and
Beclin I. These results indicated that autophagy is involved in the
pathological process of sepsis-induced AKI.
In addition, the present study observed that ATG7
mRNA levels did not exhibit significant alterations. This result
suggested that a post-transcriptional mechanism is involved in the
suppression of ATG7 expression. As an important molecule that
post-transcriptionally regulates gene expression, miRNA
participates in the pathological process of AKI. For example, the
miR-17 family has been demonstrated to be involved in the
production of pro-inflammatory cytokines in rodent models of renal
IRI (43,44). Upregulation of miR-21 provide
protective roles in animal models of AKI (45). miR-21 can inhibit autophagy by
targeting LC3, Beclin I and Ras-related proteins in brain 11 a
(Rab-11a). Renal fibrosis and macrophage infiltration are decreased
by blocking miR-21 (46). The
present study measured the miRNA expression levels and revealed
that miR-526b was downregulated in sepsis-induced kidney injury
models both in vitro and in vivo. Then, the present
study used bioinformatics algorithms to predict whether miR-526b
could target ATG7. The results confirmed that ATG7 as a miR-526b
target using HK2 cells. Furthermore, the present study also
revealed the important effects of miR-526b-driven suppression in
ATG7 on the inhibition of expression of LC3 and Beclin I which led
to the inhibition of cell autophagy.
In summary, the present study identified that
autophagy is involved in the pathological process of sepsis-induced
AKI, and that miR-526b is involved in the regulation of autophagy
by targeting ATG7. Further studies on miR-526b and ATG7 will
provide additional knowledge regarding the molecular mechanisms
underlying sepsis-induced AKI and facilitate the development of new
approaches for molecular therapeutics for this disease.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Fundamental
Research Funds for Nanjing City Medical Science and Technology
Development Project (grant no. YKK16123).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YL, JX, JS, WC, SW, RF and HL performed the
experiments and analyzed the data. YL wrote the paper. HB designed
the present study and provided experimental materials. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures performed in the studies involving
animals were in accordance with the ethical standards of the
Institutional Animal Care and Use Committee (IACUC) of Nanjing
Medical University. Animal care and euthanasia were performed with
the approval of the Institutional Animal Care and Use Committee
(IACUC) of Nanjing Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bagshaw SM, George C and Bellomo R; ANZICS
Database Management Committee, : Early acute kidney injury and
sepsis: A multicentre evaluation. Crit Care. 12:R472008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vincent JL, Sakr Y, Sprung CL, Ranieri VM,
Reinhart K, Gerlach H, Moreno R, Carlet J, Le Gall JR and Payen D;
Sepsis Occurrence in Acutely Ill Patients Investigators, : Sepsis
in European intensive care units: Results of the SOAP study. Crit
Care Med. 34:344–353. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Raghavan M and Kellum JA: Acute kidney
injury: What's the prognosis? Nat Rev Nephrol. 7:209–217. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gottlieb RA: Autophagy in Health and
Disease. Elsevier; Amsterdam: pp. 72–78. 2015
|
|
5
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Randow F and Youle RJ: Self and nonself:
How autophagy targets mitochondria and bacteria. Cell Host Microbe.
15:403–411. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maejima I, Takahashi A, Omori H, Kimura T,
Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y and
Yoshimori T: Autophagy sequesters damaged lysosomes to control
lysosomal biogenesis and kidney injury. EMBO J. 32:2336–2347. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lenoir O, Tharaux PL and Huber TB:
Autophagy in kidney disease and aging: Lessons from rodent models.
Kidney Int. 90:950–964. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takabatake Y, Kimura T, Takahashi A and
Isaka Y: Autophagy and the kidney: Health and disease. Nephrol Dial
Transplant. 29:1639–1647. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hartleben B, Gödel M, Meyer-Schwesinger C,
Liu S, Ulrich T, Köbler S, Wiech T, Grahammer F, Arnold SJ,
Lindenmeyer MT, et al: Autophagy influences glomerular disease
susceptibility and maintains podocyte homeostasis in aging mice. J
Clin Inves. 120:1084–1096. 2010. View
Article : Google Scholar
|
|
13
|
Kimura T, Takabatake Y, Takahashi A,
Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T,
Soga T, et al: Autophagy protects the proximal tubule from
degeneration and acute ischemic injury. J Am Soc Nephrol.
22:902–913. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kume S, Thomas MC and Koya D: Nutrient
sensing, autophagy and diabetic nephropathy. Diabetes. 61:23–29.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kume S, Uzu T, Maegawa H and Koya D:
Autophagy: A novel therapeutic target for kidney diseases. Clin Exp
Nephrol. 16:827–832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huber TB, Edelstein CL, Hartleben B, Inoki
K, Jiang M, Koya D, Kume S, Lieberthal W, Pallet N, Quiroga A, et
al: Emerging role of autophagy in kidney function, diseases and
aging. Autophagy. 8:1009–1031. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thomas W and Huber TB: Implications of
autophagy for glomerular aging and disease. Cell Tissue Res.
343:467–473. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mizushima N, Noda T, Yoshimori T, Tanaka
Y, Ishii T, George MD, Klionsky DJ, Ohsumi M and Ohsumi Y: A
protein conjugation system essential for autophagy. Nature.
395:395–398. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boya P, Reggiori F and Codogno P: Emerging
regulation and functions of autophagy. Nat Cell Biol. 15:713–720.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang Z and Klionsky DJ: Eaten alive: A
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mizushima N: The role of the Atg1/ULK1
complex in autophagy regulation. Curr Opin Cell Biol. 22:132–139.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mizushima N and Levine B: Autophagy in
mammalian development and differentiation. Nat Cell Biol.
12:823–830. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu X, Pan J, Li H, Li X, Fang F, Wu D,
Zhou Y, Zheng P, Xiong L and Zhang D: Atg7 mediates renal tubular
cell apoptosis in vancomycin nephrotoxicity through activation of
PKC-δ. FASEB J. 33:4513–4524. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Johnston RJ and Hobert O: A microRNA
controlling left/right neuronal asymmetry in Caenorhabditis
elegans. Nature. 426:845–849. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim VN: MicroRNA biogenesis: Coordinated
cropping and dicing. Nat Rev Mol Cell Biol. 6:376–385. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brennecke J, Hipfner DR, Stark A, Russell
RB and Cohen SM: Bantam encodes a developmentally regulated
microRNA that controls cell proliferation and regulates the
proapoptotic gene hid in Drosophila. Cell. 113:25–36. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kumar S and Reddy PH: Are circulating
microRNAs peripheral biomarkers for Alzheimer's disease? Biochim
Biophys Acta. 1862:1617–1627. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kumar S and Reddy PH: MicroRNA-455-3p as a
potential biomarker for Alzheimer's disease: An update. Front Aging
Neurosci. 10:412018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kumar S, Chawla YK, Ghosh S and
Chakraborti A: Severity of hepatitis C virus (genotype-3) infection
positively correlates with circulating microRNA-122 in patients
sera. Dis Markers. 2014:4354762014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kumar S, Reddy AP, Yin X and Reddy PH:
Novel MicroRNA-455-3p and its protective effects against abnormal
APP processing and amyloid beta toxicity in Alzheimer's disease.
Biochim Biophys Acta Mol Basis Dis. 1865:2428–2440. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qadir MI and Faheem A: miRNA: A diagnostic
and therapeutic tool for pancreatic cancer. Crit Rev Eukaryot Gene
Expr. 27:197–204. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Armand-Labit V and Pradines A: Circulating
cell-free microRNAs as clinical cancer biomarkers. Biomol Concepts.
8:61–81. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kumarswamy R, Volkmann I and Thum T:
Regulation and function of miRNA-21 in health and disease. RNA
Biol. 8:706–713. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mishra S, Yadav T and Rani V: Exploring
miRNA based approaches in cancer diagnostics and therapeutics. Crit
Rev Oncol Hematol. 98:12–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Baker MA, Davis SJ, Liu P, Pan X, Williams
AM, Iczkowski KA, Gallagher ST, Bishop K, Regner KR, Liu Y and
Liang M: Tissue-specific MicroRNA expression patterns in four types
of kidney disease. J Am Soc Nephrol. 28:2985–2992. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Y, Zheng ZJ, Jia YJ, Yang YL and Xue
YM: Role of p53/miR-155-5p/sirt1 loop in renal tubular injury of
diabetic kidney disease. J Transl Med. 16:1462018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
de Mendonça A, Vincent JL, Suter PM,
Moreno R, Dearden NM, Antonelli M, Takala J, Sprung C and Cantraine
F: Acute renal failure in the ICU: Risk factors and outcome
evaluated by the SOFA score. Intensive Care Med. 26:915–921. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bagshaw SM, George C and Bellomo R; ANZICS
Database Management Committee, : Changes in the incidence and
outcome for early acute kidney injury in a cohort of Australian
intensive care units. Crit Care. 11:R682007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sharma A, Simonson TJ, Jondle CN, Mishra
BB and Sharma J: Mincle-mediated neutrophil extracellular trap
formation by regulation of autophagy. J Infect Dis. 215:1040–1048.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Crowell KT, Soybel DI and Lang CH:
Inability to replete white adipose tissue during the recovery phase
of sepsis is associated with increased autophagy, apoptosis, and
proteasome activity. Am J Physiol Regul Integr Comp Physiol.
312:R388–R399. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Park SY, Shrestha S, Youn YJ, Kim JK, Kim
SY, Kim HJ, Park SH, Ahn WG, Kim S, Lee MG, et al: Autophagy primes
neutrophils for neutrophil extracellular trap formation during
sepsis. Am J Respir Crit Care Med. 196:577–589. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kaucsár T, Révész C, Godó M, Krenács T,
Albert M, Szalay CI, Rosivall L, Benyó Z, Bátkai S, Thum T, et al:
Activation of the miR-17 family and miR-21 during murine kidney
ischemia-reperfusion injury. Nucleic Acid Ther. 23:344–354. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ma L, Wu K, Liu K, Gu S, Wang Y, Xu Z, Yu
X and Meng J: Changes of miRNA-17-5p, miRNA-21 and miRNA-106a level
during rat kidney ischemia-reperfusion injury. Zhonghua Yi Xue Za
Zhi. 95:1488–1492. 2015.(In Chinese). PubMed/NCBI
|
|
45
|
Hu H, Jiang W, Xi X, Zou C and Ye Z:
MicroRNA-21 attenuates renal ischemia reperfusion injury via
targeting caspase signaling in mice. Am J Nephrol. 40:215–223.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chau BN, Xin C, Hartner J, Ren S, Castano
AP, Linn G, Li J, Tran PT, Kaimal V, Huang X, et al: MicroRNA-21
promotes fibrosis of the kidney by silencing metabolic pathways.
Sci Transl Med. 4:121ra182012. View Article : Google Scholar : PubMed/NCBI
|