Introduction
Ischemia/reperfusion injury (IRI) is caused by
restricted blood flow to organs and the subsequent restoration of
blood flow and oxygen; it usually occurs following sepsis,
infarction or organ transplantation, and damages tissues through
triggering inflammatory cascades involving cytokines, chemokines
and reactive oxygen species (ROS) (1,2). IRI
within the kidneys is a major factor contributing to acute kidney
injury, which is a pathological syndrome characterized by rapid
progressive renal dysfunction and a high rate of mortality
(1,3,4).
Pathophysiological investigations have revealed that IRI in kidney
tissue is closely associated with complex interactions among
various molecules and pathogenic events, including cytokines,
inflammatory factors, adhesion molecules, neutrophil activation,
and ROS production and release (5–7).
Moreover, the aberrant apoptosis of renal cells, such as tubular
cells, due to elevated oxidative stress and enhanced mitochondrial
pathway activity has been demonstrated to occur during the
pathogenesis of renal IRI (8).
Several therapeutic reagents and methods that inhibit renal cell
apoptosis have demonstrated promising potential as novel treatments
for renal IRI in animal models, which require further validation;
these include hyperbaric oxygen therapy, hydrogen-rich saline and
hypothermic machine perfusion (9–11).
Investigating the molecular mechanisms that underlie the
pharmacological effects of anti-IRI drugs may enable the
development and clinical application of novel therapies for
IRI.
Penehyclidine hydrochloride (PHC) is a novel
selective anti-cholinergic drug derived from scopolamine, which
demonstrates various pharmacological effects, including
anti-muscarinic, anti-inflammatory, anti-oxidative stress,
anti-apoptotic and anti-nicotinic activity (12). PHC has previously been used in the
clinic to treat pulmonary dysfunction associated with chronic
obstructive pulmonary disease, and organophosphate and soman
poisoning, through its regulation of the microcirculation, lysosome
release and lipid peroxidation (12). In addition, PHC has been reported
to protect against lipopolysaccharide-, and cecal ligation and
puncture-induced acute lung injury through regulating the
mitogen-activated protein kinase (MAPK) and NF-κB signaling
pathways (13,14). PHC has demonstrated promise in
preventing cardiopulmonary bypass-induced cardiopulmonary liver
damage, limb ischemia/reperfusion (I/R)-associated small intestinal
damage and middle cerebral artery occlusion-induced cerebral IRI
(15,16). Notably, PHC significantly
suppressed the histological damage and dysfunction of renal tissue
induced by I/R, and substantially altered the activity of MAPK,
caspase-3 and NF-κB proteins, in a rat model (17). Regulation of the
apoptosis-associated protein caspase-3 by PHC suggested that PHC
may be able to modulate apoptosis during I/R-induced damage to
renal tissues; however, the underlying mechanisms, particularly the
cellular and molecular events that may enable PHC-induced
inhibition of renal IRI remain to be investigated.
Autophagy is the cellular process of disassembling
unnecessary and dysfunctional components, in order to facilitate
lysosome-mediated degradation and the recycling of these useless
cellular components (18). In
certain pathogenic processes, autophagy serves as an adaptive
response to adverse stress and it is a method of maintaining
cellular energy that is required to promote cell survival and
inhibit disease progression (19,20).
For example, the induction of autophagy has been demonstrated to be
an important mechanism for protecting against IRI in liver tissue
and it was also involved in inhibiting ischemic liver damage
produced by cisplatin (21). The
activation of autophagy is linked to various pharmacological
effects, such as the inhibition of d-galactosamine-induced acute
liver injury by schisandrin A (22) and the suppression of hepatic IRI by
hydrogen sulfide (23). Notably,
autophagy was revealed to be important for kidney tubule
maintenance and it contributed to the mechanism that protects
against IRI in kidney tissues (24,25).
The target of rapamycin (TOR) is a highly conserved
serine/threonine protein kinase that acts as a central sensor for
growth factors, nutrient signals and energy status. For example,
TOR expression can be effectively promoted in response to nutrient
signals, which finally modulates cell growth by regulation of
nutrient uptake, protein synthesis and metabolic processes
(26). In addition, the mammalian
(m)TOR signaling pathway serves as a master regulator of autophagy
(27). It has been demonstrated
that Beclin-1, sequestosome 1 (p62) and microtubule-associated
protein light chain 3B (LC3B) are key proteins involved in the
stepwise process of autophagy (28,29).
LC3B is a central player in the autophagy pathway, which is
responsible for autophagy substrate selection and autophagosome
biogenesis (30); the LC3B-II/I
ratio is widely used as a marker of autophagy, as LC3B-I is
converted to LC3B-II during autophagy (31). Beclin-1, as the mammalian
orthologue of yeast autophagy-related gene (ATG)6, promotes
autophagy and programmed cell survival through interacting with
various cofactors, such as ATG14 and survivin proteins (32). p62 is associated with both cell
apoptosis and autophagy processes (33); however, it is unclear whether
autophagy is a mechanism induced by PHC to inhibit renal IRI and if
so, the possible signaling pathway that occurs remains
undiscovered.
The present study aimed to analyze the effects of
PHC treatment on renal tissue damage, cell apoptosis and autophagy
in a rat IRI model and a cellular hypoxia/oxygenation (H/R) model
to determine the molecular mechanisms underlying PHC-induced renal
IRI inhibition. The findings of the present study provided novel
insights into the tissue injury-suppressive roles of PHC.
Materials and methods
Animal studies
All procedures in this study were approved by the
Ethics Committee for Experimental Animals of the Jinshan Branch
Hospital of Shanghai Sixth People's Hospital (Shanghai, China) and
were carried out in strict accordance with the Guide for the Care
and Use of Laboratory Animals, 8th edition (National Institutes of
Health) (34). A total of 50
Sprague-Dawley rats (age, 6 weeks; male; weight, 180–200 g) were
purchased from the Laboratory Animal Center of the Jinshan Branch
Hospital of Shanghai Sixth People's Hospital. The rats were housed
in a room maintained at 27°C, 50% relative humidity, under a 12-h
light/dark cycle, with rodent chow and tap water available ad
libitum. The rats were randomly assigned to five groups (10
rats/group): i) Sham group, which was subjected to the same
surgical operation as the I/R group without occlusion of the renal
artery; ii) I/R group; iii) I/R + PHC group; iv) I/R + PHC +
3-methyladenine (3-MA; an autophagy inhibitor) group; and, v) I/R +
Rapamycin (Rapa; a common autophagy inducer) group.
PHC (0.45 mg/kg; Chengdu List Pharmaceutical, Co.,
Ltd.), 3-MA (0.5 mg/100 g; cat. no. S2767; Selleck Chemicals) and
Rapa (1 mg/100 g; cat. no. AY22989; MedChemExpress) were
intravenously administered to rats 30 min prior to I/R induction,
as previously described (17).
Briefly, the rats were anesthetized by intraperitoneal
administration of sodium pentobarbital (50 mg/kg), and their neck
and abdomen were shaved and washed with povidone iodine. The right
external jugular vein was isolated and catheterized with a catheter
secured with a sterile suture and connected to a T-branch pipe for
administration of PHC, 3-MA and Rapa.
For induction of I RI, the rats were subjected to
removal of the right kidney out of the abdominal cavity, artery
clamping and reperfusion after receiving the specified drug
treatments, as previously described (35). Briefly, the rat kidneys were
exposed and removed by a midline laparotomy, followed by occlusion
of the left renal artery using a microvascular clamp. The left
renal artery occlusion lasted for 40 min and removing the clamp
induced reperfusion.
After reperfusion for 6 h, the rats were sacrificed
and the kidneys were collected for subsequent evaluation and
analysis. Rat blood samples (2 ml) were collected from the inferior
vena cava, centrifuged (3,000 × g; 18 min) at 4°C to remove the
plasma, and serum was stored for serum creatinine (SCr)
measurements performed with a Roche Cobas C111 analyzer (Roche
Diagnostics). For purposes of quantification and statistical
analysis, the SCr measurements in each group were repeated ≥3
times.
ELISA
To evaluate the liver injuries induced by renal IRI,
serum from each group was analyzed for serum aspartate
aminotransferase (ASAT; cat. no. E-EL-R0076; Elabscience
Biotechnology Co., Ltd.) and alanine aminotransferase (ALAT)
expression levels using commercial ELISA kits (cat. no. ZK-R4034;
Shenzhen Ziker Biological Technology Co., Ltd.), according to the
manufacturer's instructions.
Histological analysis
Histological damage in rat renal tubules was
evaluated by hematoxylin & eosin (H&E) staining, Masson's
trichrome staining and Periodic acid-Schiff (PAS) staining, as
previously described (36). The
rat kidney tissues were fixed in 4% paraformaldehyde for 24 h at
room temperature, embedded in paraffin and cut into thin sections
(4-µm). H&E staining was performed according to a standard
protocol; briefly, sections of rat kidney tissue were mounted on
slides and stained with 2% hematoxylin at room temperature for 5
min, followed by staining with 0.5% eosin at room temperature for 2
min. For Masson's trichrome staining, sections of rat renal tissue
were first stained with Ponceau S acid fuchsin for 10 min at room
temperature; after which, they were incubated with
molybdophosphoric acid for 3 min at room temperature, and finally
stained with 0.5% aniline blue for 5 min. For detection of glycogen
by PAS staining, the mounted sections were oxidized in 0.5 periodic
acid solution for 5 min, incubated in Schiff reagent for 12 min,
and then counterstained with hematoxylin for 1 min at room
temperature. Histological images were captured using a light
microscope (all magnifications, ×200 and ×400).
Cell culture and reagents
The human proximal tubular human kidney 2 (HK-2)
cell line (#CRL-2190) was obtained from the American Type Culture
Collection and used to establish a cellular H/R model in
vitro. The cells were cultured in DMEM (Gibco; Thermo Fisher
Scientific, Inc.), supplemented with 0.05 mg/ml bovine pituitary
extract (cat. no. abs9119; AbSin) and 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.), and maintained in a humidified atmosphere at
37°C containing 5% CO2. To establish the cellular H/R
model, HK-2 cells were cultured in low-glucose DMEM without FBS
under hypoxic conditions (1% oxygen) for 4 h, and subsequently
cultured under normal conditions for 24 h, as previously described
(37). Subsequently, HK-2 cells
were treated with 0.1 µM PHC, 4 mM 3-MA or 2 µM Rapa at room
temperature for 2 h, and analyzed for their proliferative,
apoptotic and autophagic ability.
Immunofluorescence analysis
For analysis of the protein expression levels in
renal tissue or HK-2 cells, the paraffin-embedded tissue sections
(4 µm) or cells (1×106/ml) were fixed in 4% formaldehyde
solution at room temperature for 15 min, rinsed three times with
PBS for 5 min and incubated with the BLOT-QuickBlocker blocking
buffer (cat. no. C006011; Sangon Biotech Co., Ltd.) for 60 min at
room temperature. The tissues and cells were subsequently incubated
with anti-LC3B primary antibodies (cat. no. 3868; 1:200; Cell
Signaling Technology, Inc.) overnight at 4°C. Following the primary
antibody incubation, the sections or cells were washed three times
with PBS (5 min/wash), incubated with fluorescence-labeled
secondary antibodies (cat. no. 4414; 1:500; Cell Signaling
Technology, Inc.) for 2 h in the dark, washed with PBS and
subsequently mounted with Prolong® Gold Antifade Reagent
(cat. no. 9071; Cell Signaling Technology, Inc.). The protein
expression levels in rat tissues or cells were determined using a
fluorescence microscope (magnification, ×100).
Western blotting
Total protein was extracted from rat kidney tissues
or HK-2 cells using 0.5 ml lysis buffer (cat. no. C500035; Sangon
Biotech Co., Ltd.) containing 1% phenylmethylsulfonyl fluoride and
a proteinase inhibitor cocktail (Sigma-Aldrich; Merck KGaA).
Following centrifugation (14,000 × g; 10 min; 4°C), the supernatant
was collected and total protein was quantified using a
bicinchoninic acid assay kit (cat. no. 23227; Pierce; Thermo Fisher
Scientific, Inc.). Following quantification, ~30 µg protein was
boiled at 100°C for 5 min and separated by SDS-PAGE on 10% gels.
The separated proteins were subsequently transferred onto PVDF
membranes (EMD Millipore) and blocked for 2 h at room temperature
with 5% non-fat milk solution. The membranes were incubated with
the following primary antibodies (Abcam) diluted in TBS-Tween 20
(0.5%) for 2 h: Anti-Beclin-1 (cat. no. ab62557; 1:2,000), anti-p62
(cat. no. ab56416; 1: 2,500), anti-LC3B (cat. no. ab63817;
1:2,000), anti-Bcl-2 (cat. no. ab196495; 1:2,000), anti-Bax (cat.
no. ab53154; 1:3,000), anti-cleaved caspase-3 (cat. no. ab2302;
1:2,000) and anti-β-actin (cat. no. ab8227; 1:2,500). Following the
primary antibody incubation, the membranes were washed three times
with PBS at room temperature and subsequently incubated with
anti-rabbit IgG (cat. no. ab205718; 1:2,000; Abcam) or anti-mouse
IgG (cat. no. ab205719; 1:2,000; Abcam) secondary antibodies for
1–2 h at room temperature. Protein bands were visualized using the
Pierce™ ECL Western Blotting Substrate (Pierce; Thermo
Fishier Scientific, Inc.). Protein expression was semi-quantified
using Image-Pro Plus version 6.0 software (Media Cybernetics, Inc.)
with β-actin as the loading control.
Cell counting kit-8 (CCK-8) assay
Following treatment as specified, the proliferation
of HK-2 cells was determined using a CCK-8 assay (cat. no. C0038;
Beyotime Institute of Biotechnology), according to the
manufacturer's protocol. Briefly, HK-2 cells (1×106/ml)
were cultured in 96-well plates and incubated with CCK-8 solution
for 24 h at 37°C under normal cell culture conditions. Cell
proliferation was quantified by measuring the absorbance of each
well at 450 nm using a microplate reader. Each CCK-8 assay was
repeated ≥3 times.
Transmission electron microscopy
(TEM)
Autophagic processes in rat renal cells were
evaluated by TEM, as previously described (38). Briefly, sections of renal tissue (4
µm) were fixed with 10% neutral buffered formalin (cat. no. G2240;
Beijing Solarbio Science & Technology Co., Ltd.) at room
temperature for 2 h, embedded with LR white resin (cat. no. 62661;
Sigma-Aldrich; Merck KGaA) overnight at 55°C, mounted on slides and
stained with 0.5% toluidine blue at room temperature for 20 min.
Slides were subsequently rinsed with TBS solution and blocked with
5% BSA solution (cat. no. ST023; Beyotime Institute of
Biotechnology) at room temperature for 30 min. The sections were
washed with TBS solution and incubated with diluted primary
antibodies (cat. no. 3868; 1:200; Cell Signaling Technology, Inc.)
for 1 h at room temperature. Following the primary antibody
incubation, sections were washed with PBS and incubated with a
gold-conjugated secondary antibody (cat. no. GA1004; Boster
Biological Technology) for 1 h at room temperature. Autophagy in
renal cells was analyzed using a transmission electron microscope
(magnification, ×100).
Apoptosis analysis
The apoptotic rates of HK-2 cells after respective
treatments were determined by Hoechst 33258 staining and flow
cytometry, following fixation with 4% paraformaldehyde for 10 min
at room temperature. For apoptosis evaluation by Hoechst staining,
HK-2 cells (2×106/cell) were stained with Hoechst 33258
(5 µg/ml; Sigma-Aldrich; Merck KGaA) for 20 min in the dark at room
temperature, washed three times with PBS (5 min each) and analyzed
using a fluorescence microscope (Canon, Inc.; magnification, ×100).
For flow cytometric analysis of apoptosis, HK-2 cells
(2×106/cell) were stained with 5 µl Annexin V-FITC and 5
µl propidium iodide for 5 min in the dark at room temperature using
the Annexin V-FITC Apoptosis Staining Detection kit (cat. no.
ab14085; Abcam). Cell apoptosis was finally detected using a BD
Accuri C6 flow cytometry (BD Biosciences) and analyzed using FlowJo
7.6.1 software (FlowJo, LLC). Each apoptosis assay was repeated ≥3
times.
Statistical analysis
All data, based on at least three biological
repeats, were analyzed using PASW Statistics version 18.0 software
(SPSS Inc.) and data are presented as the mean ± SD. Statistical
differences between groups were compared using one-way ANOVA
followed by Tukey's post hoc test for multiple comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
PHC suppresses renal injury in the rat
IRI model through autophagy activation
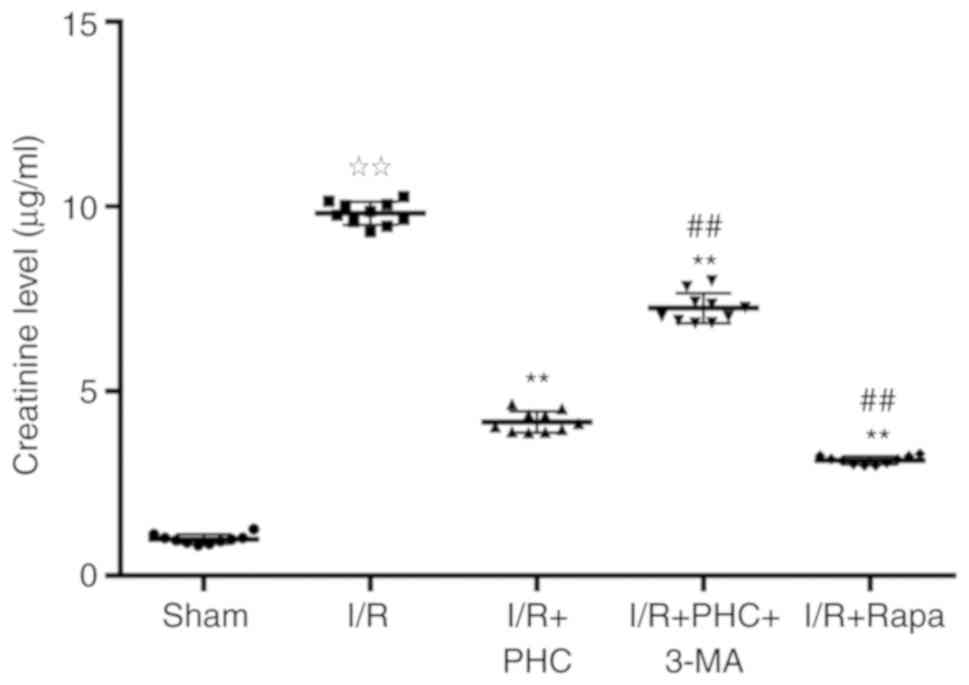
Sprague-Dawley rats subjected to I/R were used as an
animal model for studying the effects of PHC on renal injury. SCr
levels were significantly increased in the I/R group compared with
the sham group (Fig. 1); PHC
treatment significantly decreased this increase in SCr levels
observed in the I/R group (Fig.
1). Notably, treatment with the autophagy inhibitor 3-MA (I/R +
PHC + 3-MA group) induced a significant increase in SCr levels
compared with the I/R + PHC group, suggesting a potential role for
autophagy as a mechanism of action underlying PHC-induced renal IRI
inhibition (Fig. 1). The
involvement of autophagy in PHC-induced renal IRI suppression was
further demonstrated through the significantly decreased SCr levels
observed in the I/R + Rapa group compared with the I/R + PHC group
(Fig. 1).
Similar changes were observed in the liver enzymes
ALAT and ASAT (Fig. S1), the
expression levels of serum ALAT and ASAT in the I/R group were
significantly increased compared with the sham group, whereas
pretreatment with PHC or Rapa (I/R + PHC and I/R + Rapa groups,
respectively) significantly decreased IRI-induced increases in
serum ALAT and ASAT levels compared with the I/R group. However,
treatment with 3-MA (I/R + PHC + 3-MA group) significantly
attenuated the PHC-induced inhibition of ALAT and ASAT observed in
the I/R + PHC group (Fig. S1).
These data indicated that remote liver injury had occurred
following renal IRI and suggested that PHC or Rapa may prevent such
liver injury.
H&E staining of rat kidney tissues demonstrated
markedly aberrant morphological alterations in the I/R group,
including swollen renal tubular epithelial cells, clear necrosis
and cytolysis, and renal interstitial congestion (Fig. 2A). The damage to renal tissues in
the I/R group was suppressed by PHC treatment, promoted to a
certain extent by 3-MA and inhibited by Rapa (Fig. 2A). Masson's trichrome staining of
the collagenous fibers demonstrated inhibition of renal tubular
damage by PHC treatment in the I/R + PHC group; conversely, the
renal tubular damage was exaggerated by 3-MA treatment in the I/R +
PHC group but alleviated by rapamycin treatment in the I/R + Rapa
group (Fig. 2B). PAS staining
revealed mesangial stromal hyperplasia and basement membrane
thickening in renal tissues the I/R group; these pathological
changes were inhibited by PHC treatment (I/R + PHC group), promoted
by combined 3-MA and PHC treatment (I/R + PHC + 3-MA group), and
suppressed by Rapa treatment (I/R + Rapa group) (Fig. 2C). The effects of the autophagy
inhibitor 3-MA and autophagy inducer Rapa on renal tubular damage
in an rat model of IRI suggested that autophagic processes may
serve important roles in renal IRI.
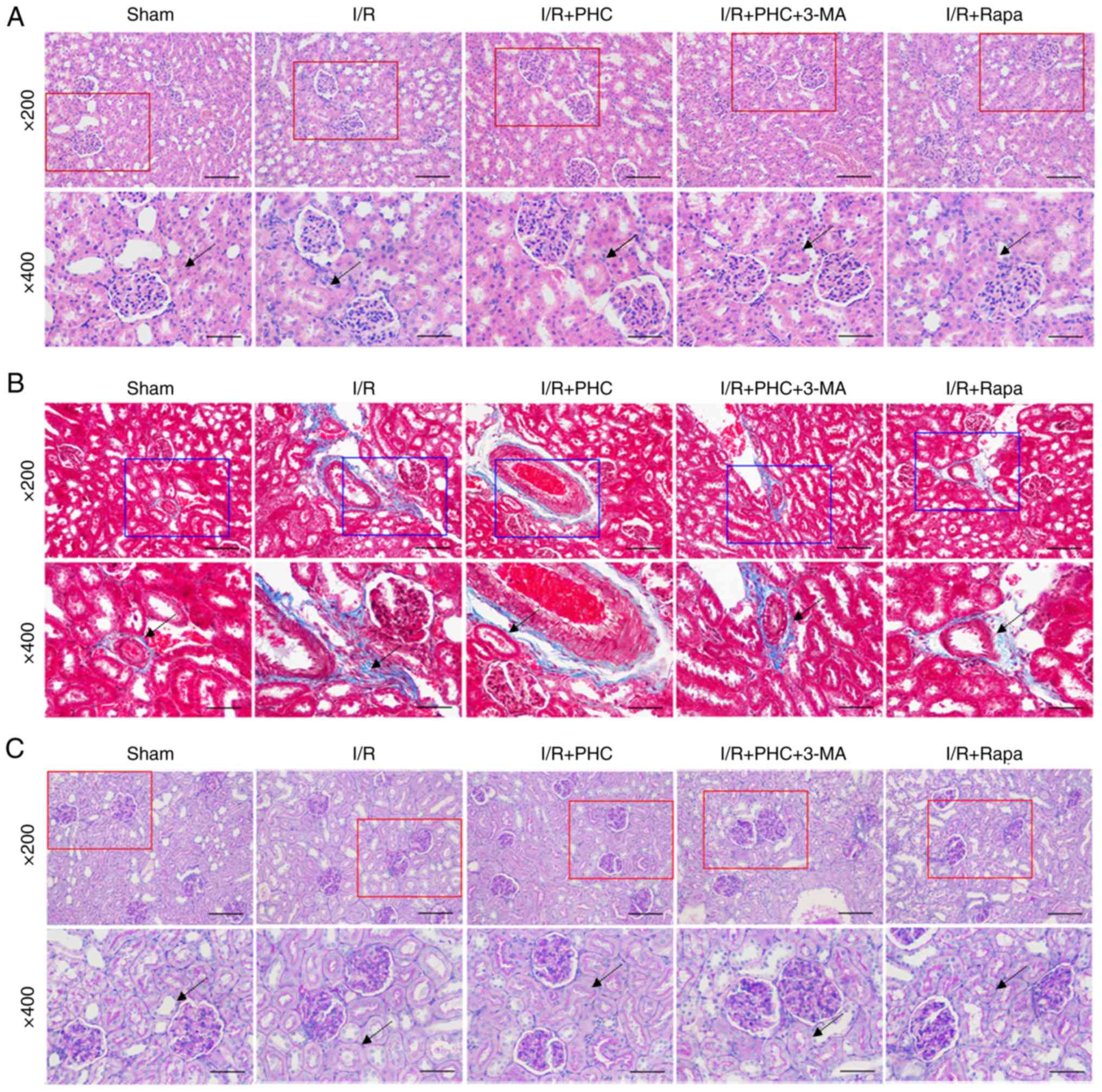 | Figure 2.Modulation of renal IRI by PHC, Rapa
and a combination of 3-MA and PHC. (A) Hematoxylin & eosin
staining of renal tubular injury in the IRI model rats following
treatment with PHC, 3-MA or Rapa (magnifications, ×200 or ×400).
(B) Masson's trichrome staining of collagenous fibers in the IRI
model rats following treatment with PHC, 3-MA or Rapa
(magnifications, ×200 or ×400). (C) Periodic acid-Schiff staining
of mesangial stromal and basement membrane thickening in renal
tissues of the IRI model rats following treatment with PHC, 3-MA or
Rapa (magnifications, ×200 or ×400). Pathogenic alterations are
indicated by black arrows. All scale bars, 50 or 100 µm. 3-MA,
3-methyladenine; I/R, ischemia/reperfusion; IRI, I/R injury; PHC,
penehyclidine hydrochloride; Rapa, rapamycin. |
PHC promotes autophagy processes
during IRI inhibition in vivo
To further investigate the role of autophagy in
PHC-induced IRI inhibition, the expression of autophagy markers in
the renal tissues of IRI model rats were investigated. Expression
levels of the autophagy marker LC3B were markedly increased in the
I/R + PHC group compared with the I/R group (Fig. 3A); however, 3-MA treatment (I/R +
PHC + 3-MA group) decreased LC3B expression levels in the renal
tissues of rats, despite receiving PHC treatment. In contrast, the
I/R + Rapa group demonstrated markedly increased expression levels
of LC3B, in comparison with the I/R group (Fig. 3A). In addition, western blotting
demonstrated that the protein expression levels of Beclin-1 were
significantly increased in the I/R + PHC group compared with the
I/R group; however, expression levels of Beclin-1 were
significantly decreased in the I/R + PHC + 3-MA group compared with
the I/R + PHC group, whereas the I/R + Rapa group demonstrated
significantly increased Beclin-1 expression levels compared with
the I/R group rats (Fig. 3B and
C). Similar alterations in LC3BII expression in rat renal
tissues were also validated by western blotting, whereas LC3BI
expression exhibited no significant changes between these groups
(Fig. 3B and C). The expression
levels of p62 in the renal tissues of all rat groups exhibited
inverse alterations compared with the expression levels of Beclin-1
and LC3B (Fig. 3B and C). These
alterations in the protein expression levels of LC3B, Beclin-1 and
p62 expression suggested that PHC treatment may activate autophagic
processes during the inhibition of renal IRI.
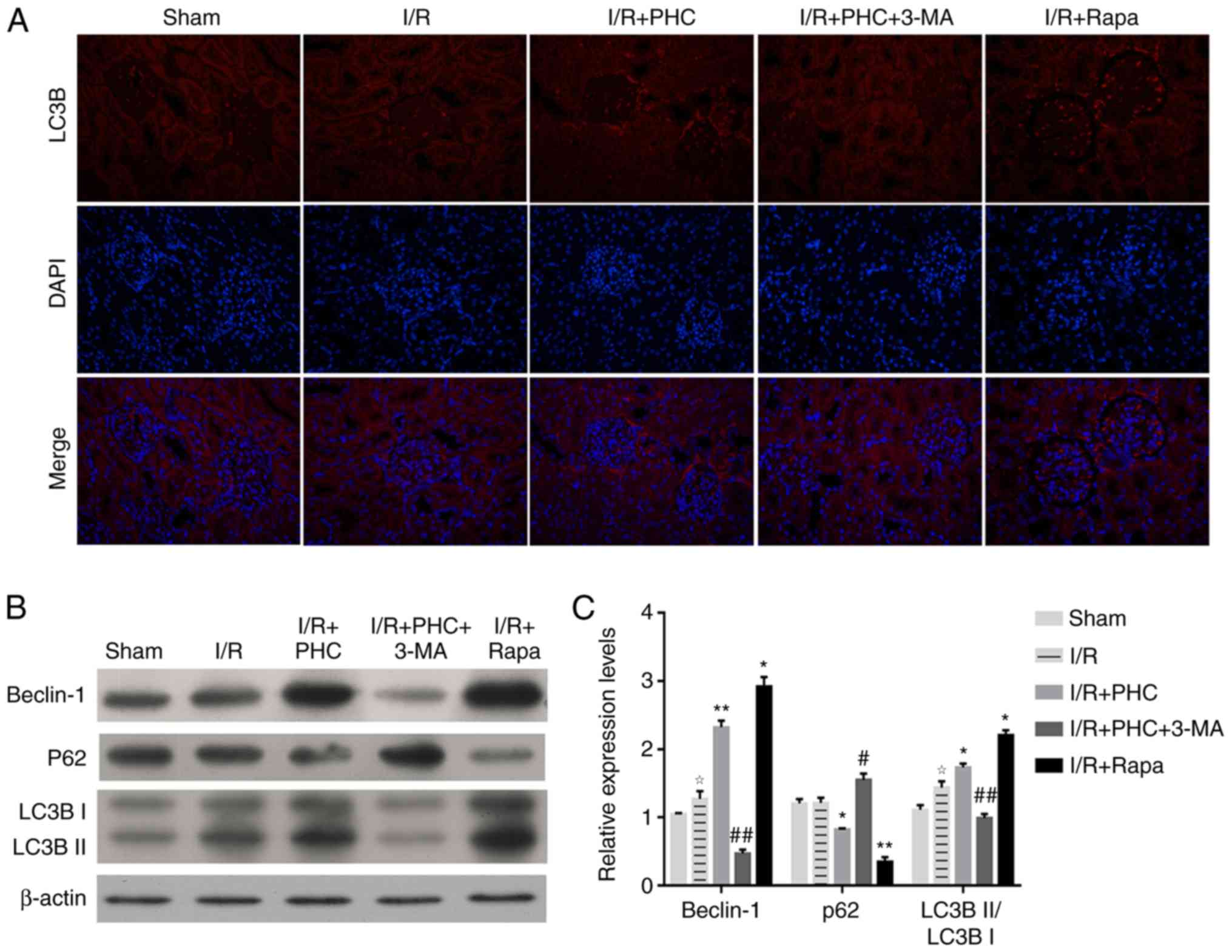 | Figure 3.PHC promotes autophagic processes
during inhibition of renal IRI. (A) Immunofluorescence micrographs
of LC3B protein expression levels in IRI model rats following
treatment with PHC, 3-MA or Rapa. Magnification, ×100. (B)
Expression levels of Beclin-1, p62 and LC3B in the renal tissues of
IRI model rats following treatment with PHC, 3-MA or Rapa, as
determined by western blotting. β-actin served as the internal
loading control. (C) Semi-quantitative analysis of relative
expression levels of Beclin-1, p62 and LC3B analyzed in (B). Fold
change to β-actin expression is presented. ✰P<0.05 vs. sham
group; *P<0.05 and **P<0.01 vs. I/R group; #P<0.05 and
##P<0.01 vs. I/R + PHC group. 3-MA, 3-methyladenine; I/R,
ischemia/reperfusion; IRI, I/R injury; LC3B, microtubule-associated
protein light chain 3B; p62, sequestome 1; PHC, penehyclidine
hydrochloride; Rapa, rapamycin. |
Autophagy promotes PHC-induced
proliferation in the in vitro H/R model
To provide evidence of the involvement of autophagy
PHC-induced IRI inhibition, a cellular H/R model was established
using the human proximal tubular HK-2 cell line. The proliferative
rates of HK-2 cells following H/R (H/R group) were decreased
compared with the control group (Fig.
4A); this proliferative rate was increased by PHC treatment
(H/R + PHC group), whereas treatment with 3-MA (H/R + PHC + 3-MA
group) markedly decreased the proliferation of HK-2 cells compared
with all other groups (Fig. 4A).
Notably, Rapa treatment increased the proliferative rate of H/R
cells (Fig. 4A).
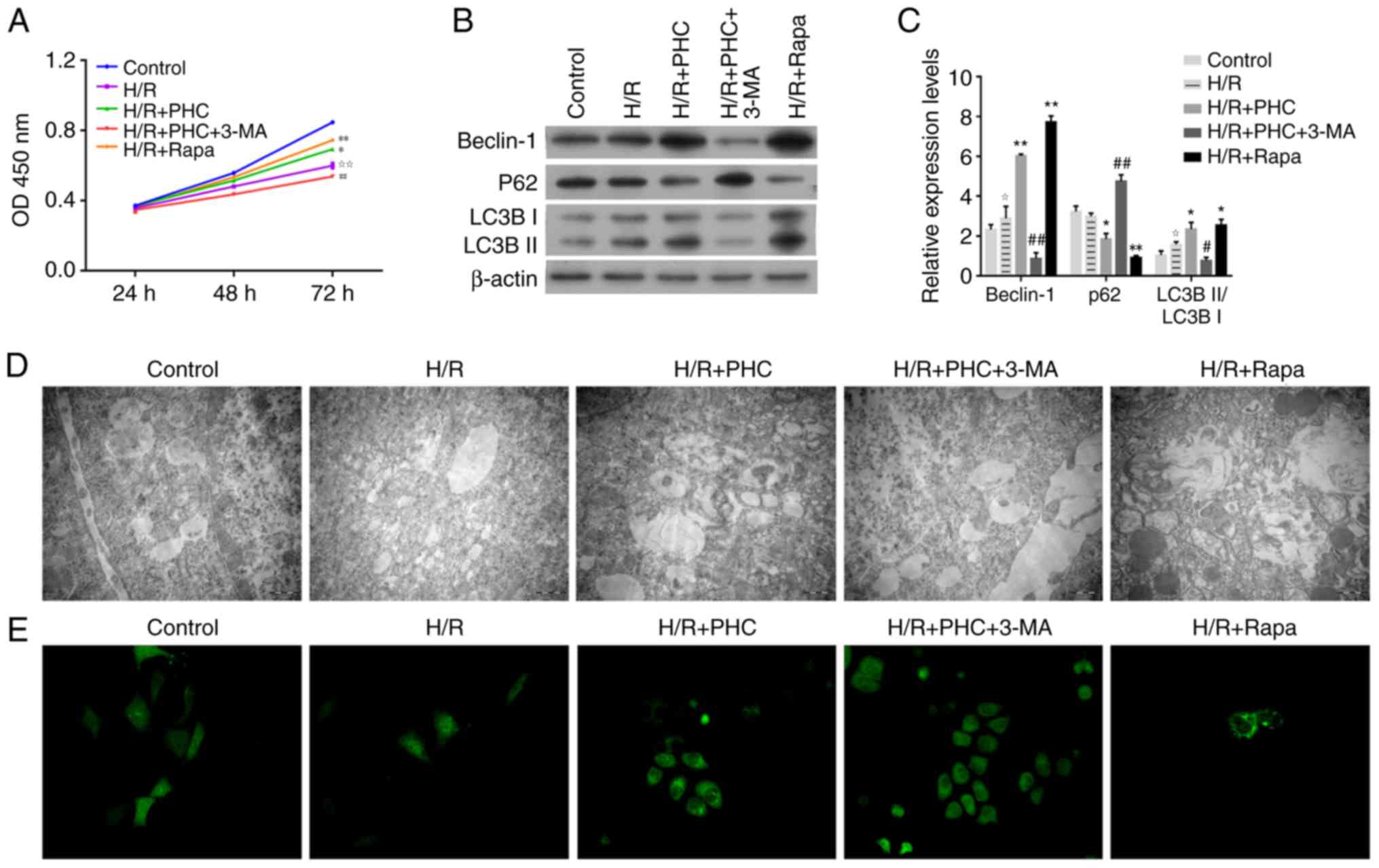 | Figure 4.PHC promotes the proliferation of H/R
model cells through activating autophagy. The cellular H/R model
was established by treating human proximal tubular HK-2 cells with
hypoxia, followed by reoxygenation. (A) Proliferation rates of
human proximal tubular HK-2 cells following treatment with PHC,
3-MA or Rapa. Cell proliferation was determined using the Cell
Counting kit-8 assay. (B) Expression levels of Beclin-1, p62 and
LC3B in H/R model cells following treatment with PHC, 3-MA or Rapa.
β-actin served as the internal loading control. (C)
Semi-quantitative analysis of relative expression levels of
Beclin-1, p62 and LC3B analyzed in (B). Fold change to β-actin
expression is presented. (D) Evaluation of autophagosome formation
in H/R model cells using transmission electron microscopy
(magnification, ×200). (E) LC3B expression levels in H/R model
cells following treatment with PHC, 3-MA or Rapa (magnification,
×200). ✰P<0.05 and ✰✰P<0.01 vs. control group; *P<0.05 and
**P<0.01 vs. H/R group; #P<0.05 and ##P<0.01 vs. H/R + PHC
group. 3-MA, 3-methyladenine; H/R, hypoxia and reoxygenation; LC3B,
microtubule-associated protein light chain 3B; OD, optical density;
p62, sequestome 1; PHC, penehyclidine hydrochloride; Rapa,
rapamycin. |
The protein expression levels of LC3BI, LC3BII and
Beclin-1 were detected in the H/R model cells using western
blotting. LC3BII/LC3BI ratio and Beclin-1 protein levels were
significantly increased in the H/R + PHC and H/R + Rapa groups
compared with the H/R group; however, expression levels were
significantly decreased in the H/R + PHC + 3-MA group compared with
the H/R + PHC group (Fig. 4B and
C). p62 expression levels demonstrated significant changes that
were opposite to the changes in the observed expression levels of
LCB and Beclin-1; p62 expression levels were significantly
decreased by PHC or Rapa treatment compared with H/R cells, but
were significantly increased in the H/R + PHC + 3-MA group compared
with the H/R + PHC group (Fig. 4B and
C).
TEM revealed that PHC or Rapa treatment (H/R + PHC
and H/R + Rapa groups, respectively) promoted autophagosome
formation in the cellular H/R model; however, PHC-induced autophagy
in the HK-2 H/R model was prevented by 3-MA treatment (H/R + PHC +
3-MA group; Fig. 4D). Notably,
immunofluorescence studies demonstrated that LC3B expression levels
were increased in the H/R + PHC group and were markedly decreased
in the H/R + PHC + 3-MA group (Fig.
4E). Furthermore, Rapa treatment markedly increased LC3B
expression levels in the H/R + Rapa group (Fig. 4E). These assays conducted on the
in vitro H/R model suggested that PHC treatment may promote
renal cell proliferation during IRI through the activation of
autophagic processes.
Autophagy mediates PHC-induced
inhibition of apoptosis in the in vitro H/R model
To further investigate the underlying cellular
mechanisms, the apoptotic rate of H/R model cells treated with PHC
was investigated. Flow cytometric analysis and Hoechst staining
revealed a significantly increased number of apoptotic HK-2 cells
in the H/R group compared with the control group (Fig. 5A-C). Treatment with PHC or Rapa
(H/R + PHC and H/R + Rapa groups, respectively) significantly
decreased the rate of cell apoptosis compared with the H/R group;
however, the decrease in apoptosis caused by PHC was ameliorated by
treatment with 3-MA, with the H/R + PHC + 3-MA group demonstrating
significantly enhanced levels of apoptosis compared with the H/R +
PHC group (Fig. 5A-C). In terms of
molecular pathways, Bcl-2 expression levels were significantly
decreased in the H/R group compared with the control group
(Fig. 5D and E). Treatment with
PHC or Rapa significantly increased Bcl-2 expression levels
compared with the H/R group, whereas 3-MA treatment significantly
reduced the increase in Bcl-2 expression levels induced by PHC
(Fig. 5D and E). The protein
expression levels of Bax and caspase-3 in the H/R group exhibited
opposite trends to those exhibited by Bcl-2; Bax and caspase-3
protein expression levels were increased by H/R and 3-MA treatment,
but decreased by PHC or Rapa treatment (Fig. 5D and E). Taken together, these data
suggested that PHC may suppress the apoptosis of renal cells
induced by H/R through promoting autophagy.
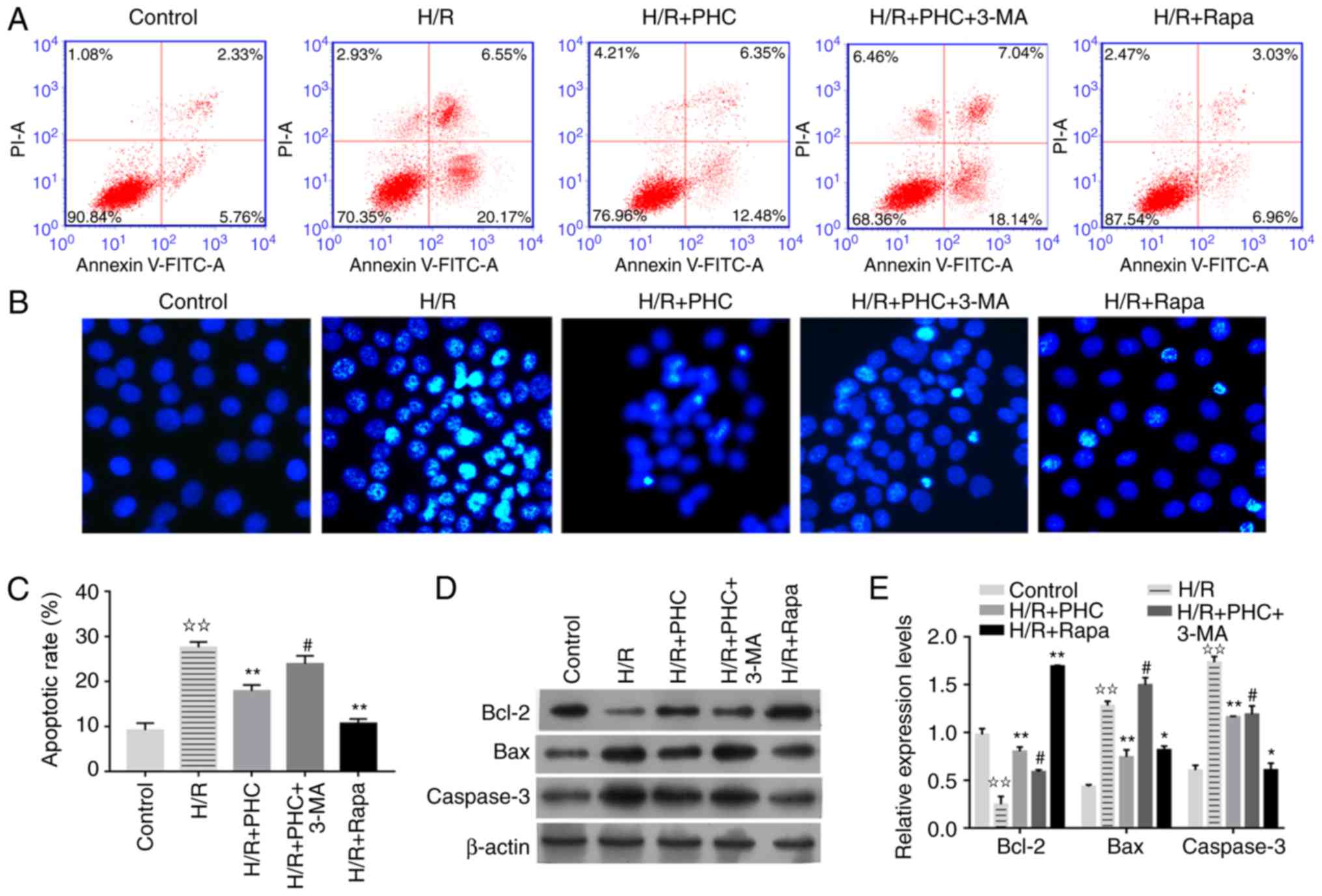 | Figure 5.PHC suppresses the apoptosis of H/R
model cells through activating autophagy. The cellular H/R model
was established by hypoxia and subsequent reoxygenation. (A) Flow
cytometric analysis of apoptosis in the cellular H/R model
following treatment with PHC, 3-MA or Rapa. (B) Analysis of
apoptosis in the cellular H/R model following treatment with PHC,
3-MA or Rapa using Hoechst 33258 staining (magnification, ×400).
(C) Semi-quantitative analysis of apoptosis analyzed in (A). (D)
Expression levels of Bcl-2, Bax and caspase-3 in the cellular H/R
model following treatment with PHC, 3-MA or Rapa using western
blotting. (E) Semi-quantitative analysis of Bcl-2, Bax and
caspase-3 expression levels from (D). Fold change to β-actin
expression is presented. ✰✰P<0.01 vs. control group; *P<0.05
and **P<0.01 vs. H/R group; #P<0.05 vs. H/R + PHC group.
3-MA, 3-methyladenine; H/R, hypoxia and reoxygenation; PHC,
penehyclidine hydrochloride; PI, propidium iodide; Rapa,
rapamycin. |
Discussion
Autophagy is an important cellular event that
protects against various tissue injuries induced by I/R;
significantly increased numbers of autophagosomes, the key
intracellular structures in autophagy, have been observed in the
tubular cells of I/R model mice, as well as in tissue specimens
collected from patients with transplanted kidneys (39). Jiang et al (25) observed that the inhibition of
autophagy led to increased numbers of apoptotic renal cells and
autophagy inhibition in I/R model mice, which promoted more severe
renal injuries, suggesting that autophagy may serve as a protective
mechanism for renal cell survival during IRI (25). When monitoring autophagy and
apoptosis in I/R model mice for a period of 0 to 7 days
post-reperfusion, autophagy was induced in a time-dependent manner
and earlier than cell apoptosis (24). Considering the significance of
autophagy in the pathogenesis of renal IRI, the potential
pharmacological effects of autophagy during PHC-induced renal IRI
inhibition was investigated. Treatment with PHC or the autophagy
inducer Rapa effectively suppressed renal tissue damage in rats in
the I/R group, whereas treatment with the autophagy inhibitor 3-MA
greatly reduced the inhibitory effect of PHC. These results
suggested a critical role for autophagy during PHC-induced
suppression of renal IRI. Rapa is an inhibitor of mTOR that can be
activated by hypoxia or ROS directly or indirectly through the
activation of AKT signaling (40–43).
The activation of mTOR inhibits the expression of ATGs and the
formation of autophagosomes (40,41);
hypoxia-induced autophagy has been reported in IRI (42,43).
Wang et al (17)
demonstrated that PHC significantly suppressed renal IRI through
suppressing MAPK, caspase-3 and NF-κB protein activity.
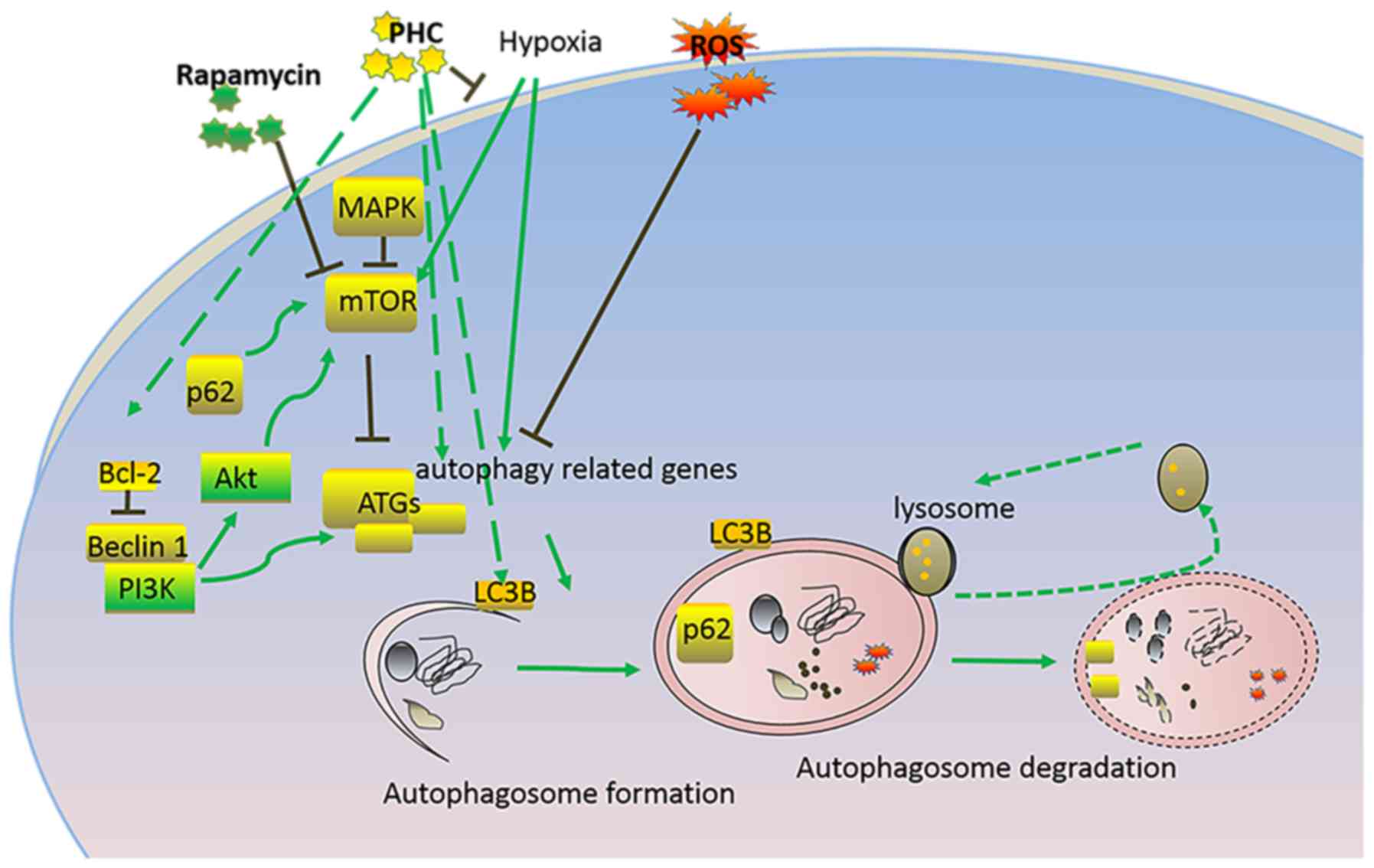
The present study demonstrated that autophagy
processes were enhanced in renal tissue following IRI. The
microtubule-associated protein LC3B is a central coordinator in the
autophagy pathway and it is responsible for autophagy substrate
selection and autophagosome biogenesis (Fig. 6) (30); it is widely used as a protein
marker of autophagosome formation. The present study demonstrated
that PHC treatment significantly increased LC3BII expression levels
in the IRI model rats and in vitro H/R model, suggesting
that PHC may induce autophagy. Beclin-1, as the mammalian
orthologue of yeast ATG6, promotes autophagy and programmed cell
survival through interacting with various cofactors, such as ATG14
and survivin proteins (32). Loss
of function mutations or polymorphisms of autophagy genes, such as
autophagy-related 16-like 1, ATG5 and immunity-related GTPase
family M disrupt cellular autophagy processes and induce cellular
injury, and are therefore relevant to many chronic inflammatory
diseases (44–47). PHC treatment also significantly
increased Beclin-1 expression levels in the in vivo and
in vitro models of renal IRI, whereas the adaptor protein
p62 demonstrated opposite trends in expression levels compared with
Beclin-1 expression. p62 was initially identified as an interacting
partner of atypical protein kinase C, and is associated with both
cell apoptosis and autophagy processes (33). The negative regulation or
elimination of p62 expression by autophagy has been demonstrated to
be an important mechanism of tumorigenesis and other biological
processes (33,48). In the present study, it was
observed that p62 protein expression levels were significantly
suppressed by PHC treatment and autophagy, providing further
evidence that PHC may induce autophagy in renal cells during IRI.
The involvement of autophagy in PHC-induced inhibition of renal IRI
in this study is consistent with other studies, which reported that
the activation of autophagy protected against tissue damage under
various conditions (49–51), and further highlights the
significance of autophagy in suppressing tissue damage.
Suppressed cell proliferation has been frequently
observed during renal IRI and the promotion of tubular cell
proliferation is the natural response during recovery from renal
dysfunction (52,53); for example, the transcription
factor transcriptional repressor GATA-binding 1, which was
initially characterized as an essential regulator of cell
morphogenesis during kidney development, promoted tubular cell
proliferation following IRI through regulating cyclic AMP-specific
3′,5′-cyclic phosphodiesterase activity and AKT signaling, and it
is thus viewed as a potential target for treating kidney IRI
(53). In the present study, it
was demonstrated that PHC treatment significantly promoted renal
cell proliferation, alongside increasing Bcl-2 expression levels
(Fig. 6), which suggested the
importance of tubular cell regulation during an I/R-induced renal
tissue injury. It was also observed that the autophagy inhibitor
3-MA inhibited PHC-induced renal cell proliferation, which further
indicated the mediating role that PHC-induced activation of
autophagy may serve in renal cell proliferation. These findings
suggested that other autophagy and proliferation-promoting reagents
may be used as therapeutic treatments for IRIs.
Autophagy is viewed as a process of programmed cell
survival, which may counteract apoptosis through mediating
interactions among Bcl-2, caspases and Beclin-1 proteins (Fig. 6) (33). The present study demonstrated that
PHC treatment significantly suppressed the apoptosis of renal cells
during I/R and this effect was attributed to PHC-induced autophagy
activation. The suppression of apoptosis by PHC-induced autophagy
was also supported through the decreased expression levels of Bax
and caspase-3, which are two major apoptosis-promoting proteins
(54), as well as through the
increased expression levels of Bcl-2 protein, which is a key
pro-survival factor associated with cell apoptosis regulation
(55). The regulation of cell
proliferation and apoptosis by autophagy has been demonstrated in
various biological and pathogenic processes, such as
hepatocarcinoma development (56).
By observing the synergistic modulation of renal cell proliferation
and apoptosis through PHC-induced autophagy activation, the present
study has contributed to the further understanding of renal IRI
pathogenesis and treatment. Thus, the interactions between changes
in cell apoptosis and proliferation levels associated with tissue
injuries of other origins, as well as their involvement in
treatment with other therapeutic reagents, requires further
investigation.
In conclusion, it is well known that autophagy is
mediated by mTOR-regulated activation of ATGs, which further
promote LC3B activation and autophagy progression, and autophagy
could also be regulated by Bcl-2, Beclin-1 and p62 (Fig. 6). Our present study demonstrated
that PHC may suppress renal IRI through inducing autophagy in an
in vivo rat model and an in vitro cellular model;
this effect was mediated through enhanced renal cell proliferation
and suppressed renal cell apoptosis. These findings may provide
novel insight into the therapeutic effects of PHC on renal IRI
treatment, and may also provide a basis for developing novel
anti-IRI drugs.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by The Jinshan District of
Shanghai Science and Technology Innovation Fund Project: The
Program of Effect of Penehyclidine Hydrochloride on Renal Autophagy
in Ischemia-reperfusion Rats (grant no. 2016-3-08).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JuZ conceived and designed the project. YK, YL, HW,
JiZ and ZF performed the experiments. YK and HW analyzed and
interpreted the data. YK and YL wrote the paper.
Ethics approval and consent to
participate
All procedures in this study were approved by The
Ethics Committee for Experimental Animals of the Jinshan Branch
Hospital of Shanghai Sixth People's Hospital and were carried out
in strict accordance with The Guidelines for the Care and Use of
Laboratory Animals (National Institutes of Health).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Malek M and Nematbakhsh M: Renal
ischemia/reperfusion injury; from pathophysiology to treatment. J
Renal Inj Prev. 4:20–27. 2015.PubMed/NCBI
|
|
2
|
Sharfuddin AA and Molitoris BA:
Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol.
7:189–200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Danobeitia JS, Djamali A and Fernandez LA:
The role of complement in the pathogenesis of renal
ischemia-reperfusion injury and fibrosis. Fibrogenesis Tissue
Repair. 7:162014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kellum JA, Unruh ML and Murugan R: Acute
kidney injury. BMJ Clin Evid. 2011(pii): 20012011.PubMed/NCBI
|
|
5
|
Ling H, Chen H, Wei M, Meng X, Yu Y and
Xie K: The effect of autophagy on inflammation cytokines in renal
ischemia/reperfusion injury. Inflammation. 39:347–356. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu D, Shang H and Liu Y: Stanniocalcin-1
protects a mouse model from renal ischemia-reperfusion injury by
affecting ROS-mediated multiple signaling pathways. Int J Mol Sci.
17(pii): E10512016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wen J, Shu Y and Zhang W: ROS, P53, and
ischemic acute kidney injury in diabetic models. Kidney Int.
88:198–199. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qiao X, Chen X, Wu D, Ding R, Wang J, Hong
Q, Shi S, Li J, Xie Y, Lu Y and Wang Z: Mitochondrial pathway is
responsible for aging-related increase of tubular cell apoptosis in
renal ischemia/reperfusion injury. J Gerontol A Biol Sci Med Sci.
60:830–839. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Migita H, Yoshitake S, Tange Y,
Choijookhuu N and Hishikawa Y: Hyperbaric oxygen therapy suppresses
apoptosis and promotes renal tubular regeneration after renal
ischemia/reperfusion injury in rats. Nephrourol Mon. 8:e344212016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jie L, Hong Z, Hong L, Zhou J, Cui L, Yuan
S, Chu X and Yu P: Hydrogen-rich saline promotes the recovery of
renal function after ischemia/reperfusion injury in rats via
anti-apoptosis and anti-inflammation. Front Pharmacol.
7:1062016.PubMed/NCBI
|
|
11
|
Zhang Y, Fu Z, Zhong Z, Wang R, Hu L,
Xiong Y, Wang Y and Ye Q: Hypothermic machine perfusion decreases
renal cell apoptosis during ischemia/reperfusion injury via the
Ezrin/AKT pathway. Artif Organs. 40:129–135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu C and Wang J: Neuroprotective effect of
penehyclidine hydrochloride on focal cerebral ischemiareperfusion
injury. Neural Regen Res. 8:622–632. 2013.PubMed/NCBI
|
|
13
|
Shen W, Gan J, Xu S, Jiang G and Wu H:
Penehyclidine hydrochloride attenuates LPS-induced acute lung
injury involvement of NF-kappaB pathway. Pharmacol Res. 60:296–302.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhan J, Liu Y, Zhang Z, Chen C, Chen K and
Wang Y: Effect of penehyclidine hydrochloride on expressions of
MAPK in mice with CLP-induced acute lung injury. Mol Biol Rep.
38:1909–1914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cai DS, Jin BB, Pei L and Jin Z:
Protective effects of penehyclidine hydrochloride on liver injury
in a rat cardiopulmonary bypass model. Eur J Anaesthesiol.
27:824–828. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Y, Leng YF, Xue X, Zhang Y, Wang T
and Kang YQ: Effects of penehyclidine hydrochloride in small
intestinal damage caused by limb ischemia-reperfusion. World J
Gastroenterol. 17:254–259. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang YP, Li G, Ma LL, Zheng Y, Zhang SD,
Zhang HX, Qiu M and Ma X: Penehyclidine hydrochloride ameliorates
renal ischemia-reperfusion injury in rats. J Surg Res. 186:390–397.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Caramés B, Taniguchi N, Otsuki S, Blanco
FJ and Lotz M: Autophagy is a protective mechanism in normal
cartilage, and its aging-related loss is linked with cell death and
osteoarthritis. Arthritis Rheum. 62:791–801. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cursio R, Colosetti P and Gugenheim J:
Autophagy and liver ischemia-reperfusion injury. Biomed Res Int.
2015:4175902015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cardinal J, Pan P and Tsung A: Protective
role of cisplatin in ischemic liver injury through induction of
autophagy. Autophagy. 5:1211–1212. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu Y, Wang WJ, Song YZ and Liang ZQ: The
protective mechanism of schisandrin A in d-galactosamine-induced
acute liver injury through activation of autophagy. Pharm Biol.
52:1302–1307. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang D, Ma Y, Li Z, Kang K, Sun X, Pan S,
Wang J, Pan H, Liu L, Liang D and Jiang H: The role of AKT1 and
autophagy in the protective effect of hydrogen sulphide against
hepatic ischemia/reperfusion injury in mice. Autophagy. 8:954–962.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guan X, Qian Y, Shen Y, Zhang L, Du Y, Dai
H, Qian J and Yan Y: Autophagy protects renal tubular cells against
ischemia/reperfusion injury in a time-dependent manner. Cell
Physiol Biochem. 36:285–298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jiang M, Liu K, Luo J and Dong Z:
Autophagy is a renoprotective mechanism during in vitro hypoxia and
in vivo ischemia-reperfusion injury. Am J Pathol. 176:1181–1192.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Weisman R: Target of rapamycin (TOR)
regulates growth in response to nutritional signals. Microbiol
Spectr. 4:2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cao QH, Liu F, Yang ZL, Fu XH, Yang ZH,
Liu Q, Wang L, Wan XB and Fan XJ: Prognostic value of autophagy
related proteins ULK1, Beclin 1, ATG3, ATG5, ATG7, ATG9, ATG10,
ATG12, LC3B and p62/SQSTM1 in gastric cancer. Am J Transl Res.
8:3831–3847. 2016.PubMed/NCBI
|
|
29
|
Hamacher-Brady A: Autophagy regulation and
integration with cell signaling. Antioxid Redox Signal. 17:756–765.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Abeliovich H: Guidelines for the use and
interpretation of assays for monitoring autophagy. Haematologica.
27:151–175. 2012.
|
|
31
|
Tiwari RV, Parajuli P and Sylvester PW:
Synergistic anticancer effects of combined γ-tocotrienol and
oridonin treatment is associated with the induction of autophagy.
Mol Cell Biochem. 408:123–137. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moscat J and Diaz-Meco MT: p62 at the
crossroads of autophagy, apoptosis, and cancer. Cell.
137:1001–1004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals, . Guide for the Care and Use of Laboratory Animals. 8th.
National Academies Press (US); Washington (DC): 2011
|
|
35
|
Zheng Y, Lu M, Ma L, Zhang S, Qiu M and
Wang Y: Osthole ameliorates renal ischemia-reperfusion injury in
rats. J Surg Res. 183:347–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shimizu S, Saito M, Kinoshita Y, Ohmasa F,
Dimitriadis F, Shomori K, Hayashi A and Satoh K: Nicorandil
ameliorates ischaemia-reperfusion injury in the rat kidney. Br J
Pharmacol. 163:272–282. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jia X, Zhang L and Mao X: S-propranolol
protected H9C2 cells from ischemia/reperfusion-induced apoptosis
via downregultion of RACK1 gene. Int J Clin Exp Pathol.
8:10335–10344. 2015.PubMed/NCBI
|
|
38
|
Swanlund JM, Kregel KC and Oberley TD:
Investigating autophagy: Quantitative morphometric analysis using
electron microscopy. Autophagy. 6:270–277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Suzuki C, Isaka Y, Takabatake Y, Tanaka H,
Koike M, Shibata M, Uchiyama Y, Takahara S and Imai E:
Participation of autophagy in renal ischemia/reperfusion injury.
Biochem Biophys Res Commun. 368:100–106. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu ZZ, Zhang JJ, Gao CC, Zhao M, Liu SY,
Gao GM and Zheng ZH: Expression of autophagy related genes mTOR,
Becline-1, LC3 and p62 in the peripheral blood mononuclear cells of
systemic lupus erythematosus. Am J Clin Exp Immunol. 6:1–8.
2017.PubMed/NCBI
|
|
41
|
Cerni S, Shafer D, To K and Venketaraman
V: Investigating the role of everolimus in mTOR inhibition and
autophagy promotion as a potential host-directed therapeutic target
in mycobacterium tuberculosis infection. J Clin Med. 8(pii):
E2322019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rouschop K and Wouters BG: Regulation of
autophagy through multiple independent hypoxic signaling pathways.
Curr Mol Med. 9:417–424. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ma S, Wang Y, Chen Y and Cao F: The role
of the autophagy in myocardial ischemia/reperfusion injury. Biochim
Biophys Acta. 1852:271–276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nuij VJAA, Peppelenbosch MP, van der Woude
CJ and Fuhler GM: Genetic polymorphism in ATG16L1 gene is
associated with adalimumab use in inflammatory bowel disease. J
Transl Med. 15:2482017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cucu MG, Streața I, Riza AL and Lilin A:
Polymorphisms in autophagy genes and active pulmonary tuberculosis
susceptibility in Romania. Rev Romana Med Lab. 25:47–53. 2017.
|
|
46
|
Kabat AM, Harrison OJ, Riffelmacher T,
Moghaddam AE, Pearson CF, Laing A, Abeler-Dörner L, Forman SP,
Grencis RK, Sattentau Q, et al: The autophagy gene Atg16l1
differentially regulates Treg and TH2 cells to control intestinal
inflammation. Elife. 5:e124442016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lin YC, Chang PF, Lin HF, Liu K, Chang MH
and Ni YH: Variants in the autophagy-related gene IRGM confer
susceptibility to non-alcoholic fatty liver disease by modulating
lipophagy. J Hepatol. 65:1209–1216. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mathew R, Karp CM, Beaudoin B, Vuong N,
Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al:
Autophagy suppresses tumorigenesis through elimination of p62.
Cell. 137:1062–1075. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pu D, Lianos EA, Ma J and Lin PH:
Autophagy, innate immunity and tissue repair in acute kidney
injury. Int J Mol Sci. 17(pii): E6622016.PubMed/NCBI
|
|
50
|
Mukhopadhyay P, Eid N, Abdelmegeed MA and
Sen A: Interplay of oxidative stress, inflammation, and autophagy:
Their role in tissue injury of the heart, liver, and kidney. Oxid
Med Cell Longev. 2018:20908132018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sekiguchi A, Kanno H, Ozawa H, Yamaya S
and Itoi E: Rapamycin promotes autophagy and reduces neural tissue
damage and locomotor impairment after spinal cord injury in mice. J
Neurotrauma. 29:946–956. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nguan CYC, Guan Q, Gleave ME and Du C:
Promotion of cell proliferation by clusterin in the renal tissue
repair phase after ischemia-reperfusion injury. Am J Physiol Renal
Physiol. 306:F724–F733. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ju-Rong Y, Ke-Hong C, Kun H, Bi-Qiong F,
Li-Rong L, Jian-Guo Z, Kai-Long L and Ya-Ni H: Transcription factor
Trps1 promotes tubular cell proliferation after
ischemia-reperfusion injury through cAMP-specific 3′,5′-cyclic
phosphodiesterase 4D and AKT. J Am Soc Nephrol. 28:532–544. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Peiró G, Diebold J, Baretton GB, Kimmig R
and Löhrs U: Cellular apoptosis susceptibility gene expression in
endometrial carcinoma: Correlation with Bcl-2, Bax, and caspase-3
expression and outcome. Int J Gynecol Pathol. 20:359–367. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Adams JM and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Guo XL, Li D, Hu F, Song JR, Zhang SS,
Deng WJ, Sun K, Zhao QD, Xie XQ, Song YJ, et al: Targeting
autophagy potentiates chemotherapy-induced apoptosis and
proliferation inhibition in hepatocarcinoma cells. Cancer Lett.
320:171–179. 2012. View Article : Google Scholar : PubMed/NCBI
|