Introduction
Congenital hypothyroidism (CH) is the most common
congenital endocrine disorder with an incidence of approximately
1/2,000-4,000 newborns (1).
According to the locations of lesions, CH can be classified into
primary, central and peripheral hypothyroidism (1,2).
Primary hypothyroidism accounts for >95% of CH cases (3), the majority of which (80-85%) are
caused by alterations occurring during gland organogenesis. These
alterations result in thyroid dysgenesis (TD) (1,4). The
remaining cases (15-20%) are attributed to inborn defects in
thyroid hormone synthesis. These defects are collectively known as
dyshormonogenesis (DH) and are generally characterized by either
goiter or normal thyroid glands (1,5,6). By
contrast, central and peripheral hypothyroidisms are rare
disorders.
Considerable progress has been made in the
understanding of CH pathophysiology. Although most cases of CH
occur sporadically, approximately 20% are familial and caused by
genetic abnormalities (1,2). For TD, approximately 2–5% of reported
cases have a genetic origin. Genes associated with TD (PAX8,
NKX2-1/TTF-1, FOXE1/TTF-2, NKX2-5 and
TSHR) play important roles during thyroid morphogenesis
(1,4). The molecular mechanism of DH has been
well characterized and most of the cases have been linked to
mutations in genes involved in thyroid hormone synthesis (TG,
TPO, DUOX1/2, DUOXA2, SLC5A5, SLC26A4/PDS,
IYD/DEHAL1 and SECISBP2). These mutations are
usually transmitted in an autosomal recessive mode (1,5,7). The
underlying molecular basis of central and peripheral hypothyroidism
remains unclear, although genetic ascertainment is possible in some
cases (1,6). Mutations have been reported in genes
controlling the biosynthetic pathway of thyroid stimulating hormone
(TSH; TSHB, TRHR and IGSF1), pituitary
development (POU1F1, PROP1, HESX1, LHX3, LHX4 and
SOX3) (1,6) and thyroid hormone transport or action
(SLC16A2/MCT8, THRB and THRA) (1). Other genes (FOXI1, GLIS3, UBR1
and ZNF252P) have been reported in cases with syndromic
hypothyroidism or transient CH and may be involved in CH (8–13).
These causative genes and their functions are described in Table SI.
Although CH can be classified as a disease with a
strong genetic component, many issues remain unresolved. One is the
commonly observed variable phenotype-genotype correlations in
patients (5,14). This phenotypic or genetic
heterogeneity suggests that mono- and polygenic factors and
environmental modulators have roles in the determination of disease
severity (4,5). Some cases have oligogenic mutations
apart from single-gene mutations and demonstrate heterogeneous
phenotypes to those carrying monogenic mutations (15–17).
These cases may not be inherited in a monogenic manner; that is, a
digenic or oligogenic inheritance may be considered, or mutations
may occur, acting as a genetic modifiers (18,19).
However, no definite evidence is able to prove this phenomenon.
Next-generation sequencing (NGS) can be used for the simultaneous
sequencing of multiple genes in a single sample and is useful in
determining mutations in multiple genes that are potentially
associated with diseases (20,21).
Thus, NGS is a powerful tool for unraveling the pathogeneses of
complex diseases. Given the genetic complexity and heterogeneity of
CH, all known causative genes should be comprehensively screened
for mutations for the proper understanding of CH pathogenesis.
According to the largest national newborn screening
program between 2013 and 2015, the total incidence rate of CH in
China is 4.13 per 10,000 live births, which is higher than the
worldwide level (22,23). To date, the comprehensive screening
of the known pathogenic genes in Chinese patients is limited. The
present study designed a targeted NGS panel including 29 CH-related
genes to screen mutations in a Chinese patient cohort from Shaanxi
Province, China.
Materials and methods
Subjects
A total of 43 patients with CH from 42 families were
recruited in Xi'an Children's Hospital and Chang'an Hospital,
Xi'an, China, between October 2015 and August 2016. The age of the
patients at the time of the study was 3 months-13 years. The
inclusion criteria were: Positive neonatal screening with a
diagnosis of CH confirmed by serum thyroid function tests at 2–4
weeks of age. Neonatal screening for CH was taken from 72 h to 7
days after birth. Blood samples were collected from the heel to
determine TSH levels by using time-resolved fluorescence assay
(PerkinElmer, Inc.). Newborns (2-4 weeks) with TSH levels of >10
µIU/ml were recalled for the re-examination of serum TSH and FT4
levels by electrochemiluminescence assay (Cobas 6000, Roche
Diagnostics). CH diagnosis was based on elevated serum TSH (>7.5
µIU/ml; normal: 0.27–3.20 µIU/ml) and decreased FT4 levels (0.5–3.1
pmol/l; normal: 12–22 pmol/l). Levothyroxine (L-T4) treatment was
initiated when elevated TSH level (>10 µIU/ml) was confirmed.
All the patients had a phenotypical classification by thyroid
ultrasonography performed during the neonatal period prior to
treatment. Additional information on the possible existence of
thyroid disease in family members was collected in all cases. The
neonatal screening, diagnosis and follow-up of each patient were
conducted in the same hospital: Xi'an Children's or Chang'an
Hospital. In addition, 100 subjects with normal FT4 and TSH levels
and undergoing neonate thyroid screening were included in the
normal control group. They were all Han Chinese from Shaanxi, China
and consisted of 45 males and 55 females with a mean age of 5 days
(4–10 days) on the day of sample collection for neonatal screening.
Blood samples from the fathers and mothers of 20 patients were
collected for segregation analysis. At the time of the study, the
mean age of these fathers and mothers was 30.7 years (22–42 years)
and 29.3 years (20–45 years), respectively. The parents of all the
participants gave written informed consent in accordance with the
Declaration of Helsinki. The study was approved by the Medical
Ethics Committees of Xi'an Children's Hospital and Chang'an
Hospital.
DNA extraction and sequencing
Blood samples were collected from recruited
patients, their family members and the control subjects and stored
in EDTA tubes. Genomic DNA was extracted and analyzed as previously
described (24).
According to the previous findings described in
published literature (1,4–6) and
the retrieval results of the Human Gene Mutation Database (HGMD
Professional 2016, http://www.hgmd.cf.ac.uk/ac/index.php), 29 causative
genes (PAX8, FOXE1, NKX2-5, TSHR, NKX2-1, DUOX2, DUOXA2, TPO,
SLC26A4, FOXI1, TG, SLC5A5, IYD, SECISBP2, TSHB, IGSF1, TRHR,
HESX1, LHX3, LHX4, POU1F1, PROP1, SOX3, THRB, THRA, SLC16A2, GLIS3,
UBR1 and ZNF252P) associated with CH were selected. The
recruited patients were genetically screened with a customized
AmpliSeq panel (Thermo Fisher Scientific, Inc.) that included 29
CH-associated genes. The primers for the customized panel were
designed with Ion AmpliSeq Designer (https://www.ampliseq.com/browse.action) for the
inclusion of coding exons and the 20 flanking base pairs of the
splice junctions surrounding the exons of the targeted genes. A
total of 457 amplified amplicons were obtained at each sequencing
run. Amplicon length was 125–374 bp (median 358 bp; Table SII). The amplicon library
preparation and DNA template preparation and enrichment were
conducted according to the manufacturers' protocols. DNA sequencing
was performed with an Ion Torrent PGM instrument. An Ion PGM 200
sequencing kit and Ion 316™ Chip (Thermo Fisher Scientific, Inc.)
were used, according to the manufacturers' protocols.
Variant detection and
prioritization
Raw data were processed by the Torrent Suite
software (version 5.0.4; Thermo Fisher Scientific, Inc.) for the
generation of sequence reads. Each read was aligned to the hg19
human reference genome for the detection of variants. Called
variants were functionally annotated with Ion Reporter (https://ionreporter.lifetechnologies.com/ir/secure/home.html)
and ANNOVAR package (http://wannovar.wglab.org/). Identified variants were
filtered as follows: i) Synonymous variants and nonsplice variants
in the intronic region were excluded; ii) variants with minor
allele frequencies (MAF) of ≤0.01 or no MAF values in the dbSNP
database (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000
Genomes Project (http://ftp.ncbi.nih.gov/), Exome Sequencing Project
(http://evs.gs.washington.edu/EVS/)
and the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/) were selected; iii)
variants without rs numbers in the dbSNP database were considered
novel rare variants; iv) CH-associated variants reported in the
published literature or by the HGMD database (HGMD Professional
2019.3) were selected even if ii) or iv) was not met; v) all
selected variants were validated through Sanger sequencing with
ABI3500 ×L Dx (Applied Biosystems, Thermo Fisher Scientific, Inc.);
and vi) all validated novel variants were determined in the normal
control by Sanger sequencing and were filtered by MAF ≤0.01. The
present study was conducted on the basis of these prioritization
criteria. The prioritized variants were detected in the parental
samples of patients for the verification of the inheritance of
variants and segregation with phenotype.
Bioinformatics and statistical
analysis
The possible functional effects of detected variants
were assessed by in silico programs. For missense or indel
variants, five in silico tools were used, including
Polymorphism Phenotyping v2 (http://genetics.bwh.harvard.edu/pph2/
index.shtml), MutationTaster (http://www.mutationtaster.org/), Rare Exome Variant
Ensemble Learner (https://sites.google.com/site/revelgenomics/) and
Mendelian Clinically Applicable Pathogenicity (http://bejerano.stanford.edu/MCAP/). For the
splicing variants, the deleterious effect on RNA splicing was
predicted with MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html),
Berkeley Drosophila Genome Project (http://www.fruitfly.org/seq_tools/ splice.html)
and NetGene2 (http://www.cbs.dtu.dk/services/ NetGene2/).
The evolutionary conservation analysis was performed
using the CLC Sequence Viewer 6.5.2 software (CLC bio, Qiagen AB).
Protein domains and structures were obtained from UniProt and
InterPor Knowledgebase (http://www.uniprot. orssswq333 g/; http://www.ebi.ac.uk/interpro/).
The pathogenicity of each variant was assessed
according to the standards described by the American College of
Medical Genetics (ACMG) (25).
The clinical features of the two groups (such as
patients with DUOX2 mutation and patients without
DUOX2 mutation) were compared through a Mann-Whitney test
for nonparametric values. P<0.05 was considered to indicate a
statistically significant difference.
Results
Demographic and clinical
characteristics of the patients
The present study included 43 Chinese Han patients
diagnosed with primary CH. The recorded demographic data and
clinical features of the patients are presented in Table I: The enrolled patients consisted of
25 females and 18 males, aged 3 months to 13 years. All of the
cases were from unrelated families with no histories of thyroid
diseases, apart from patients 32 and 33, who were monozygotic
twins. Thyroid ultrasonography suggested that 11 patients had TD,
of which 6 had athyreosis, 4 had hypoplasia and 1 had ectopy. A
total of 32 cases had eutopically located glands in situ
(GIS), 18 of which had normal thyroid sizes and 14 had goiter
(Table I).
 | Table I.Clinical Information, detected
variants, and results of family segregation analysis of studied
patients with CH. |
Table I.
Clinical Information, detected
variants, and results of family segregation analysis of studied
patients with CH.
|
|
|
|
|
| Screening | Neonatal
period |
|
|
|
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|
|---|
| Patients ID | Agea, sex | Birth
weight(g) | Gestational age
(week+day) | Thyroid gland | TSH (uIU/ml) | Age | TSH (uIU/ml) | FT4 (pmol/l) | Detected
variant | Father | Mother |
Solved/ambiguous/unsolved |
|---|
| 1 | 7y10m, F | 3050 | 32+5 | Hypoplasia | 14.1 | 39d | 15 | 6.7 | SLC5A5
p.Q639* (CT) | NA | NA | Ambiguous |
| 2 | 3y9m, F | 3000 | Full term | Athyreosis
(53d) | 35.4 | 69d | >100 | 1.2 | TPO p.R361L
(GT) | GG | GT | Ambiguous |
| 3 | 1y1m, M | 4000 | Full term | Hypoplasia | 20.3 | 76d | 14.8 | 6.7 | SLC26A4
p.A434T (GA), TRHR p.I168M (TG) | NA | NA | Ambiguous |
| 4 | 1y3m, F | 3400 | Full term | Normal | 28 | 25d | 35.2 | 4.9 | DUOX2
p.A1206T (GA) | GA | GG | Ambiguous |
| 5 | 3m, M | 3600 | 41 | Normal | 21.2 | 20d | 92.8 | 3.6 | DUOX2
p.E879K (GA) | NA | NA | Ambiguous |
| 6 | 4y6m, M | 3300 | Full term | Goiter | 29 | 74d | >100 | 2.6 | DUOX2
p.K530* (AT), DUOX2 p.R1110Q (GA) | AA;GA | AT;GG | Solved |
| 7 | 5m, M | 3800 | Full term | Hypoplasia | 9.61 | 43d | 20 | 5.7 | DUOX2
p.T803fs (c.2406_2407insCCTG) | NA | NA | Ambiguous |
| 8 | 4y, F | 4000 | 40+5 | Athyreosis
(1y) | 20 | 58d | 36.5 | 4.8 |
| NA | NA | Unsolved |
| 9 | 7m, M | 2750 | 36+5 | Goiter | 20.5 | 30d | 30.2 | 5.1 | DUOX2
p.R434* (CT) | CC | CT | Ambiguous |
| 10 | 8m, F | 3000 | 33+2 | Normal | 7.5 | 56d | 16.9 | 6.6 |
| NA | NA | Unsolved |
| 11 | 1y9m, M | 3800 | Full term | Normal | 19 | 60d | 25 | 6.2 | TSHR p.R528S
(CA), TSHR p.R450H (GA) | CC;GA | CA;GG | Solved |
| 12 | 3y, F | 3400 | Full term
(Suspected) | Ectopy | >100 | 33d | >100 | 0.8 | DUOX2
p.V779M (GA), SLC26A4 p.Y578H (TC) | GG;TC | GA;TT | Ambiguous |
| 13 | 1y3m, F | 3200 | Full term | Normal | 15.3 | 63d | 20 | 6.4 |
| NA | NA | Unsolved |
| 14 | 2y9m, M | 3400 | Full term | Goiter | 24.2 | 33d | 32.8 | 5 |
| NA | NA | Unsolved |
| 15 | 2y4m, M | 3900 | Full term | Normal | 21 | 57d | 28 | 5.3 | DUOXA2
p.R94C (CT), DUOXA2 p.Y246* (CG), TG p.N212S (AG) | CC;CG; AG | CT;CC, AA | Solved |
| 16 | 11m10d, M | 3200 | Full term | Goiter | 100 | 62d | >100 | 1.1 |
DUOX2 p.R376W (CT),
DUOX2 p.R434_S440del (del/wt), DUOX2 p.R1110Q
(GA) | CT;del/wt;GG | CC;wt/wt;GA | Solved |
| 17 | 11m, M | 3100 | Full term | Normal | 14.6 | 65d | 19.54 | 6.5 | DUOX2
p.R885L (GT), DUOX2 p.Y1415C (AG) | GT;AA | GG; AG | Solved |
| 18 | 2y2m, F | 2600 | Full term | Normal | 77 | 67d | 77 | 4.1 | TG p.R2585W
(CT), DUOX2 p.A1206T (GA) | NA | NA | Ambiguous |
| 19 | 1y, M | 3050 | full term | Hypoplasia | 18 | 32d | 21 | 6.3 |
| NA | NA | Unsolved |
| 20 | 9y, F | 3500 | Full term | Normal | >100 | 20d | >100 | 0.7 | TPO p.E757*
(dupT/wt), TG p.I1931V (AG), DUOX2 p.K530* (AT) | wt/wt;AA; AA | dupT/wt; AG;AT | Ambiguous |
| 21 | 9y, F | 3000 | Full term | Athyreosis | >100 | 30d | >100 | 0.5 |
| NA | NA | Unsolved |
| 22 | 4y7m, M | 3500 | Full term | Normal | 31 | 49d | 40 | 4.8 |
| NA | NA | Unsolved |
| 23 | 3y, F | 3000 | Full term | Goiter | 40 | 74d | 40 | 4.8 | TG p.R896Q
(GA) | NA | NA | Ambiguous |
| 24 | 1y6m, M | 3500 | Full term | Normal | 19 | 61d | 58.21 | 4.5 | TPO p.R846W
(CT), TG p.E955fs (c.2864delA), TG p.I1931V (AG),
TG p.L2282fs (c.6840_6843delTTGT) | CT;del/wt; AG,
wt/wt | CC;wt/wt;AA,
del/wt | Solved |
| 25 | 8m, F | 2400 | Full term | Normal | 58.21 | 32d | >100 | 6.7 | TG p.I2394M
(CG) | CC | CG | Ambiguous |
| 26 | 13y, F | 3900 | Full term | Goiter | 16.5 | 28d | >100 | 6.5 |
| NA | NA | Unsolved |
| 27 | 2y6m, F | 3100 | 38+1 | Goiter | 25.6 | 35d | 35 | 6.3 |
| NA | NA | Unsolved |
| 28 | 2y, F | 3000 | 40 | Goiter | 26 | 72d | 46.5 | 4.7 | TG p.V1738I
(GA), TG p.S1912N (GA), DUOX2 p.D137E (CA),
DUOX2 p.R432H (GA), DUOXA2 p.Y246* (CG) | GA;GA; CA;GG;
CG | GG;GG; CC;GA;
CC | Solved |
| 29 | 3y3m, F | 3300 | Full term | Goiter | >100 | 58d | >100 | 2 | DUOX2
p.R625* (CT), DUOX2 p.R1110Q (GA) | CC;GA | CT;GG | Solved |
| 30 | 6y, M | 3000 | 37 | Goiter | 18 | 58d | 20 | 6.4 | TPO p.S571R
(CG), PROP1 p.G51V (GT) | CG;GG | CC;GT | Ambiguous |
| 31 | 5y6m, F | 2900 | 40 | Normal | 20.6 | 53d | 18.3 | 6.5 |
| NA | NA | Unsolved |
| 32 | 5y11m, M | 2200 | 37+1 | Normal | 20 | 65d | 18.2 | 6.5 | DUOX2
p.F591S (TC), DUOX2 p.E879K (GA), DUOX2 p.G1521*
(GT) | TC;GA; GG | TT;GG; GT | Solved |
| 33 | 5y11m, M | 2600 | 37+1 | Normal | 22.5 | 65d | 20.3 | 6.3 | DUOX2
p.F591S (TC), DUOX2 p.E879K (GA), DUOX2 p.G1521*
(GT) | TC;GA; GG | TT;GG; GT | Solved |
| 34 | 1y4m, F | 3300 | 40+1 | Goiter | 16.5 | 71d | 15.8 | 6.7 | DUOX2
p.R885L (GT), DUOX2 p.R1110Q (GA) | GG;GA | GT;GG | Solved |
| 35 | 2y10m, F | 3650 | Full term | Athyreosis | >100 | 52d | >100 | 0.8 |
| NA | NA | Unsolved |
| 36 | 1y11m, F | 3400 | 39+4 | Athyreosis | >100 | 47d | >100 | 0.9 |
| NA | NA | Unsolved |
| 37 | 1y, M | 3750 | Full term | Goiter | 35.1 | 78d | 35 | 4.9 | TSHR p.C176R
(TC), TSHR p.K618* (AT), TPO p.P883S (CT) | TT;AT; CC | TC;AA; CT | Solved |
| 38 | 1y2m, F | 3300 | Full term | Athyreosis | 46.1 | 64d | >100 | 1.9 | TPO p.P883S
(CT), GLIS3 p.A753V (CT) | NA | NA | Ambiguous |
| 39 | 9m, F | 3600 | 39+2 | Normal | >100 | 47d | >100 | 2.1 | DUOX2
p.E389K (GA) | NA | NA | Ambiguous |
| 40 | 8m, F | 1900 | 38 | Normal | 15 | 33d | 15 | 7.1 | TPO p.S309P
(TC) | NA | NA | Ambiguous |
| 41 | 4y9m, F | 2800 | Full term | Normal | 21 | 90d | 18 | 6.5 | DUOX2
p.G624fs (c.1871delG) | NA | NA | Ambiguous |
| 42 | 1y3m, F | 2700 | 41+2 | Goiter | >100 | 68d | >100 | 0.7 | SLC26A4
p.Y78H (TC), DUOX2 p.R885L (GT), DUOX2 p.R1110Q
(GA) | TT, GG, GA | TC, GT, GG | Solved |
| 43 | 1y1m, M | 3800 | 40 | Goiter | >100 | 74d | >100 | 0.6 | TPO p.R846W
(CT), TG p.R896Q (GA), c.3693+1G>T (GT) DUOX2
p.V407F (GT), DUOX2 | NA | NA | Ambiguous |
| Normal |
|
| <10 |
| 0.27–3.2 | 12-22 |
|
|
|
|
|
|
Sequencing data analysis
NGS analysis was performed on the 43 patients with
CH and the result showed that the number of mapped reads for
individual samples was 149,698-1,458,751 (median: 394571, n=43).
The percentages of the on-target sequences in each sample all had a
median of 99%, with an average base coverage depth ranging from
215.9–3× in individual samples. The average total coverages of all
the targeted bases were 94.80% at 20×, 87.06% at 100× and 57.51% at
500×. Coverage was also uniform across all samples. On average,
94.8% of the called bases had a quality score of ≥Q20 (Table SIII).
In 25 genes (86.2%), all of the targeted amplicons
were covered by 20 reads or more (Fig.
S1). Exons that were covered less than 20× or missed by the NGS
were subjected to Sanger sequencing (Table SIV).
Variant detection and mutation
spectrum
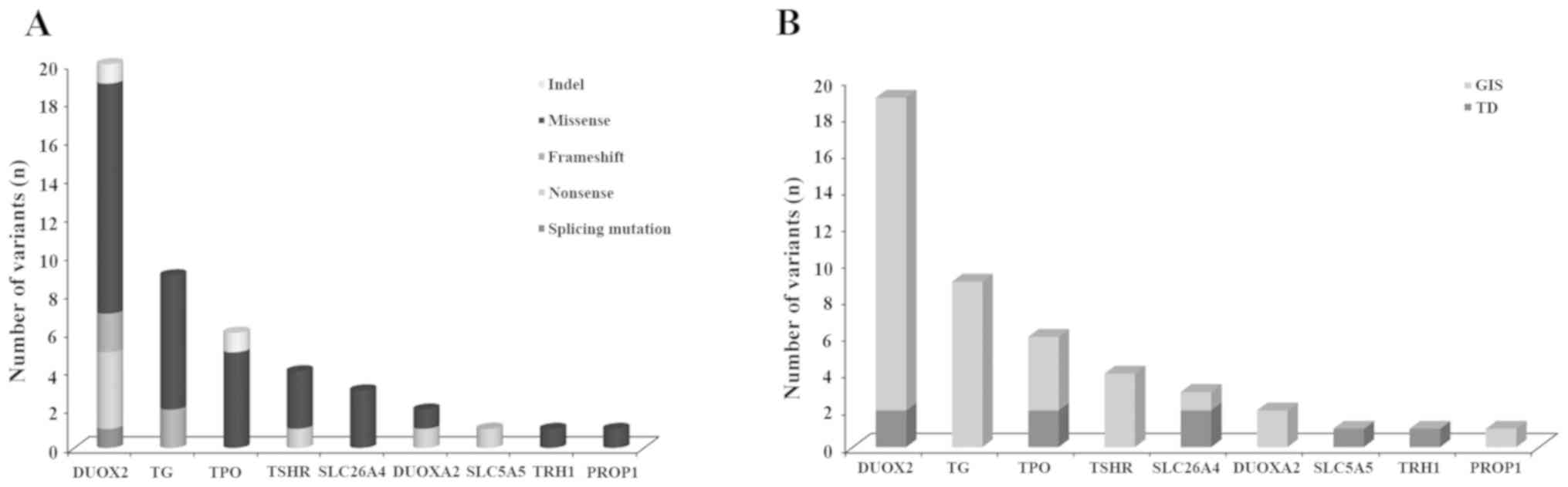
Following functional filtering, 47 rare
nonpolymorphic variants were identified in 31 of 43 patients (71%).
These variants were distributed in 9 genes, including 6
DH-associated genes (DUOX2, TG, TPO, SLC26A4, DUOXA2 and
SLC5A5), 1 gene associated with TD (TSHR) and 2 genes
associated with central hypothyroidism (PROP1 and
TRHR, Fig. 1A). Notably, all
variants were detected in heterozygous status in patients. Various
types of mutations have been detected and most of them were
missense variants (Table II;
Fig. 1A). A total of 8 novel
variants were identified (Table
II; Fig. S2); these were
absent in the local control samples. The 39 remaining variants had
been reported in HGMD, dbSNP, gnomAD and/or 1000 human genome
databases.
| Table II.Potential pathological variants
detected in the present study. |
Table II.
Potential pathological variants
detected in the present study.
|
|
|
|
| Minor allele
frequency |
|
|
|
|---|
|
|
|
|
|
|
|
|
|
|---|
| Gene | Amino Acids
change | cDNA change | Exon/Intron
position | rs ID | Patients
(n=43a) | GnomAD east
asian | 1000 Genome
CHB | Statusb | ACMG
classification |
|---|
| TSHR | p.C176R | c.526T>C | 6 |
| 0.012 | 0 | 0 | Novel | LP |
| TSHR | p.R450H | c.1349G>A | 10 | rs189261858 | 0.012 | 0.002597 | 0.0049 | Knownd, DM | P |
| TSHR | p.R528S | c.1582C>A | 10 |
| 0.012 | 0 | 0 | Knownd, DM | LP |
| TSHR | p.K618* | c.1852A>T | 10 |
| 0.012 | 0 | 0 | Novel | P |
| DUOX2 | p.D137E | c.411C>A | 5 |
| 0.012 | 0 | 0 | Novel | LP |
| DUOX2 | p.R376W | c.1126C>T | 10 | rs119472029 | 0.012 | 0 | 0 | Knownd, DM | LP |
| DUOX2 | p.E389K | c.1165G>A | 11 |
| 0.012 | 0 | 0 | Novel | VUS |
| DUOX2 | p.V407F | c.1219G>T | 11 |
| 0.012 | 0 | 0 | Knownd, DM | VUS |
| DUOX2 | p.R432H | c.1295G>A | 12 | rs530736554 | 0.012 | 0.0004769 | 0 | Knownd, DM | LP |
| DUOX2 | p.R434* | c.1300C>T | 12 | rs119472026 | 0.012 | 0.000116 | 0 | Knownd, DM | P |
| DUOX2 | p.R434_ | c.1300_1320 |
|
|
|
|
|
|
|
|
| S440del | delCG | 12 |
| 0.012 | 0 | 0 | Knownd, DM | P |
|
|
| AGATATGGGG |
|
|
|
|
|
|
|
|
|
| CTGCCCAGC |
|
|
|
|
|
|
|
| DUOX2 | p.K530* | c.1588A>T | 14 | rs180671269 | 0.024 | 0.009274 | 0.0095 | Knownd, DM | P |
| DUOX2 | p.F591S | c.1772T>C | 15 |
| 0.024 | 0.00007081 | 0 | Knownc | VUS |
| DUOX2 | p.G624fs | c.1871delG | 16 | rs769258094 | 0.012 | 0.0004638 | 0 | Knownd, DM | P |
| DUOX2 | p.R625* | c.1873C>T | 16 | rs770083296 | 0.012 | 0 | 0 | Knownd, DM | P |
| DUOX2 | p.V779M | c.2335G>A | 19 | rs145061993 | 0.012 | 0.004094 | 0.0049 | Knownd, DM? | VUS |
| DUOX2 | p.T803fs | c.2406_2407 | 19 |
| 0.012 | 0 | 0 | Novel | P |
|
|
| insCCTG |
|
|
|
|
|
|
|
| DUOX2 | p.E879K | c.2635G>A | 20 | rs774556391 | 0.036 | 0.000954 | 0 | Knownd, DM | P |
| DUOX2 | p.R885L | c.2654G>T | 20 | rs181461079 | 0.036 | 0.005777 | 0.0049 | Knownd, DM? | LP |
| DUOX2 | p.R1110Q | c.3329G>A | 25 | rs368488511 | 0.06 | 0.002597 | 0.0049 | Knownd, DM | P |
| DUOX2 | p.A1206T | c.3616G>A | 28 | rs762588205 | 0.024 | 0.0001739 | 0 | Knownd, DM? | LP |
| DUOX2 | IVS28+1G>T | c.3693+1G>T | intron 28 | rs200717240 | 0.012 | 0.001537 | 0 | Knownd, DM | P |
| DUOX2 | p.Y1415C | c.4244A>G | 32 | rs757012152 | 0.012 | 0.00008334 | 0 | Knownc | LP |
| DUOX2 | p.G1521* | c.4561G>T | 34 | rs765781255 | 0.024 | 0.001044 | 0 | Knownd | LP |
| DUOXA2 | p.R94C | c.280C>T | 3 |
| 0.012 | 0 | 0 | Knownd, DM | VUS |
| DUOXA2 | p.Y246* | c.738C>G | 5 | rs4774518 | 0.024 | 0.00188 | 0.0291 | Knownd, DM | P |
| SLC5A5 | p.Q639* | c.1915C>T | 15 |
| 0.012 | 0 | 0 | Novel | VUS |
| TPO | p.S309P | c.925T>C | 8 |
| 0.012 | 0 | 0 | Novel | VUS |
| TPO | p.R361L | c.1082G>T | 8 | rs201781919 | 0.012 | 0.009273 | 0.0194 | Knownd, DM | VUS |
| TPO | p.S571R | c.1713C>G | 10 |
| 0.012 | 0 | 0 | Novel | VUS |
| TPO | p.E757* | c.2268dupT | 13 | rs770781635 | 0.012 | 0.00159 | 0 | Knownd, DM | P |
| TPO | p.R846W | c.2536C>T | 15 | rs28913014 | 0.024 | 0.00159 | 0.0049 | Knownc | VUS |
| TPO | p.P883S | c.2647C>T | 16 | rs190968346 | 0.024 | 0.005409 | 0.0146 | Knownd, DM | LB |
| PROP1 | p.G51V | c.152G>T | 2 | rs2233783 | 0.012 | 0.002151 | 0 | Knownc | VUS |
| SLC26A4 | p.Y78H | c.232T>C | 3 | rs760794201 | 0.012 | 0 | 0 | Knownd, DM | VUS |
| SLC26A4 | p.A434T | c.1300G>A | 11 | rs757552791 | 0.012 | 0 | 0 | Knownd, DM | VUS |
| SLC26A4 | p.Y578H | c.1732T>C | 16 | rs781728302 | 0.012 | 0 | 0 | Knownc | VUS |
| TRHR | p.I168M | c.504T>G | 1 | rs13306060 | 0.012 | 0.00212 | 0.0049 | Knownc | VUS |
| TG | p.N212S | c.635A>G | 5 | rs187737243 | 0.012 | 0.002122 | 0 | Knownc | VUS |
| TG | p.R896Q | c.2687G>A | 10 | rs374707675 | 0.024 | 0.0007431 | 0.0049 | Knownd, DM | VUS |
| TG | p.E955fs | c.2864delA | 11 | rs767858769 | 0.012 | 0.00005798 | 0 | Knownc | P |
| TG | p.V1738I | c.5212G>A | 26 | rs115053637 | 0.012 | 0.001325 | 0.0097 | Knownc | VUS |
| TG | p.S1912N | c.5735G>A | 31 | rs762807254 | 0.012 | 0 | 0 | Knownc | VUS |
| TG | p.I1931V | c.5791A>G | 31 | rs115877910 | 0.024 | 0.002391 | 0.0146 | Knownd, DM? | VUS |
| TG | p.L2282fs | c.6840_6843 | 39 | rs774153375 | 0.012 | 0 | 0 | Knownc | P |
|
|
| delTTGT |
|
|
|
|
|
|
|
| TG | p.I2394M | c.7182C>G | 41 |
| 0.012 | 0.00005798 | 0 | Knownc | VUS |
| TG | p.R2585W | c.7753C>T | 44 | rs114211101 | 0.012 | 0.005379 | 0.0049 | Knownd, DM? | VUS |
Of the 9 mutated genes, the gene with the highest
number of variants was DUOX2, followed by TG, TPO and
TSHR (Fig. 1A). Twenty
variants in DUOX2 were identified in 19 cases (19/43, 44%),
that is, 17 patients with GIS and 2 patients with TD (Fig. 1B). Of these patients, 10 (10/43,
23%) carried ≥2 different heterozygous variants in DUOX2. A
total of 6 patients carried DUOX2 mutation(s) in association
with mutation(s) in TG (n=4) or SLC26A4 (n=2). The
most common mutation was p.R1110Q (DUOX2: c.3329G>A),
which was found in 5 patients, accounting for 11% of all the cases.
Of the 3 novel variants in DUOX2, p.T803fs was a frameshift
mutation and had a potential deleterious effect on protein function
and p.D137E and p.E389K were missense mutations located in the
peroxidase-like domain (Fig.
S3A).
A total of 9 variants in TG were identified
in 8 CH patients (8/43, 18.6%), 2 of which had ≥2 TG
variants. Apart from carrying TG mutation(s), 6 cases also
had mutation(s) in genes associated with DH (SLC26A4, DUOX2,
DUOXA2 and TPO).
A total of 6 TPO variants were separately
found in 6 patients (6/43, 14%) in heterozygous status. All but 1
patient had a TPO mutation in association with mutation(s)
in different genes. A total of 2 novel variants, p.S309P and
p.S571R, were located in a myeloperoxidase-like domain, the
catalytic site of the enzyme (Fig.
S3B).
A total of 4 TSHR variants were found in 2
patients and were compound heterozygotes for 2 different
TSHR mutations. The TSHR variant p.R450H was a
recurrent inactivating mutation (26) and p.C176R and p.K618 were novel.
p.C176R is located in the leucine-rich repeat region of the
extracellular domain and responsible for high-affinity hormone
binding and p.R528S and p.K618* are located in the cytoplasmic
loops (Fig. S3C).
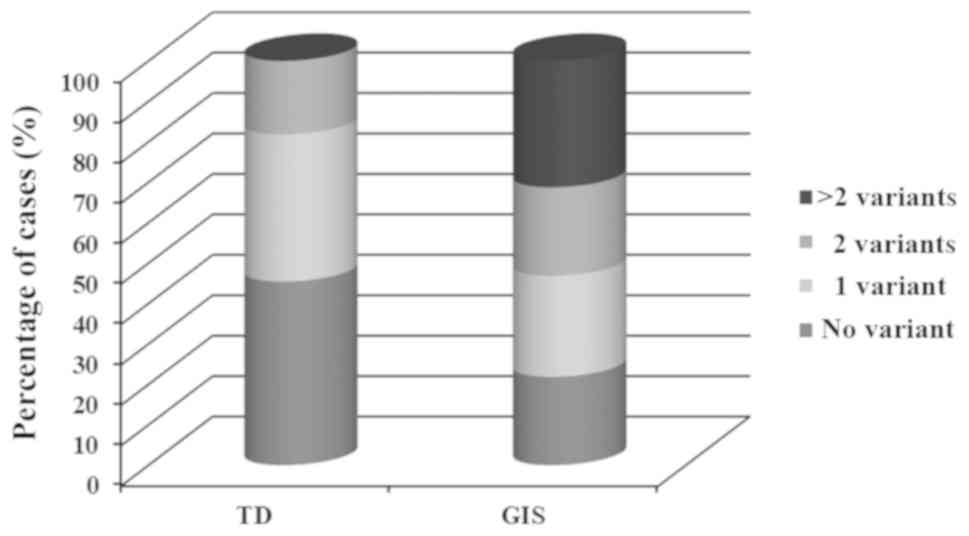
Patients with GIS had a higher tendency to be
affected with mutations than patients with TD [25/32 (78%) vs. 6/11
(54%), Fig. 2]. Variants in TG,
TSHR, DUOXA2, SLC5A5 and PROP1 genes were found
exclusively in patients with GIS, and 1 variant in TRHR was
found in patients with TD. Other genes, including DUOX2, TPO
and SLC26A4, were associated with either dysgenesis or GIS
phenotype (Table II and Fig. 1B). The variants detected in the 6
patients with TD (two athyreosis, two hypoplasia and one ectopy)
were all located in genes associated with DH. A total of 12
patients (12/43, 28%) carried only 1 heterozygous variant and 19
cases (19/43, 44%) had ≥2 variants, 8 of which were monogenic
(having mutations in the same gene) and 11 were oligogenic (having
mutations in different genes, Table
III).
| Table III.Mutation spectrum of ‘solved’ and
‘ambiguous’ cases with CH. |
Table III.
Mutation spectrum of ‘solved’ and
‘ambiguous’ cases with CH.
| Solved (n=13), all
were CH with GIS |
|---|
|
|---|
|
| Monogenic
(n=8) | Oligogenic
(n=5) |
|
|
|
|
| Gene | Number of
variants | Number of
patients | Gene1 | Number of
variants | Gene2 | Number of
variants | Gene3 | Number of
variants | Number of
patients |
|---|
| TSHR | 2 | 1 | TSHR | 2 | TPO | 1 |
|
| 1 |
| DUOX2 | 2 | 4 | DUOXA2 | 2 | TG | 1 |
|
| 1 |
| DUOX2 | 3 | 3 | DUOX2 | 2 | SLC26A4 | 1 |
|
| 1 |
|
|
|
| DUOX2 | 2 | TG | 2 | DUOXA2 | 1 | 1 |
|
|
|
| TG | 3 | TPO | 1 |
|
| 1 |
|
| Ambiguous
(n=18) |
|
|
Monogenic(n=12) | Oligogenic
(n=6) |
|
|
| Gene | Number of
variants | Number of
patients | Gene1 | Number of
variants | Gene2 | Number of
variants | Gene3 | Number of
variants | Number of
patients |
|
| DUOX2 | 1 | 6 | SLC26A4 | 1 | TRH1 | 1 |
|
| 1 |
| TG | 1 | 2 | DUOX2 | 1 | SLC26A4 | 1 |
|
| 1 |
| TPO | 1 | 3 | DUOX2 | 1 | TG | 1 |
|
| 1 |
| SLC5A5 | 1 | 1 | DUOX2 | 1 | TG | 1 | TPO | 1 | 1 |
|
|
|
| DUOX2 | 2 | TG | 1 | TPO | 1 | 1 |
|
|
|
| TPO | 1 | PROP1 | 1 |
|
| 1 |
Pathogenicity assessment
The pathogenicity of the detected variants was
classified in accordance with ACMG standards and guidelines
(Table II, Tables SV and SVI). Among the 47 variants, 25 were
classified as pathogenic (P) or likely pathogenic (LP), namely, 16
in DUOX2, 4 in TSHR, 2 in TPO, 2 in TG
and 1 in DUOXA2 gene. A total of 21 variants were classified
as variants of uncertain significance (VUS) and 1 variant in
TPO was classified as likely benign. Among the 8 novel
variants, 4 were classified as P (p.C176R and p.K618* in
TSHR, p.T803fs in DUOX2) or LP (p.D137E in
DUOX2), the other were classified as VUS.
Genotype and phenotype
relationship
Through family segregation and pathogenicity
assessment, 13 cases (patients 6, 11, 15,17, 16, 28, 29, 32, 33,
34, 37 and 42) were considered ‘solved,’ reaching a diagnosis
detection rate of 30% (Tables I and
III, and Fig. S1). These ‘solved’ cases all carried
at least 2 pathogenic variants in the same gene, which were of
either paternal or maternal origin, but not from a single parent,
following the identification of a decisive link between genotype
and phenotype. A total of 18 cases (41.9%) were considered
‘ambiguous’ owing to the weak link between genotype and phenotype.
In addition, 12 cases were considered ‘unsolved’ because they
carried no mutations in any of the listed genes.
Among the solved cases, 8 were monogenic and 5 were
oligogenic. The main pathogenic genes were DUOX2 (n=9),
TSHR (n=2), DUOXA2 (n=1) and TG (n=1).
Notably, all of the ‘solved’ cases were patients with GIS (Tables I and III). Therefore, the diagnosis rate in
patients with GIS patients was 40.6% (13/32; Table III).
According to the number of variants carried, the
studied cases were classified into different groups and the serum
levels of TSH and FT4 were compared among these groups (Fig. SIV). The results showed that only
the average serum TSH level of patients with TG mutation at
diagnosis were significantly higher than those without TG
mutation (49.54 µIU/ml vs. 68.71 µIU/ml, P=0.037, Fig. S4A-b). The average serum levels of
FT4 of patients with monogenic mutation at diagnosis were higher
compared with patients with oligogenic mutations, but the
difference was not statistically significant (P=0.05, Fig. S4D-c).
Discussion
To the best of our knowledge, the present study is
the first in which the currently largest targeted NGS panel
containing 29 known causative genes was used for the comprehensive
examination of the mutation spectrum of Han Chinese CH patients.
The present study found a high mutation rate (44%) in primary CH
patients and most of the mutations (91.5%) were identified in genes
associated with DH. In addition, mutations in genes associated with
thyroid development or transcription were rarely identified. The
majority of CH were caused by TD and <20% of cases showed strong
genetic predisposition (1,2). However, in the patient cohort, DH
(n=32) was more common than TD (n=11). This result is in agreement
with the data reported in China (27–31).
Given that DH is largely caused by genetic defects and considered a
hereditary disease, a majority of CH cases in Chinese are
hereditary and have a strong genetic origin. In the present study,
mutation detection rate in CH patients with DH was 78% and the
diagnosis rate in DH was 40.6%. Notably, the 6 patients with TD
harbored mutations that were all associated with DH, 2 of whom had
athyreosis. Patients with TD, especially those with athyreosis, are
unlikely to carry variants associated with DH. However, in the
present study, patients were subjected to thyroid ultrasonography
rather than to whole-body nuclear magnetic resonance scanning for
the examination of thyroid morphology. Thus, those examined as
athyreosis could not be excluded for the likelihood of ectopy.
Similarly, other studies have found variants associated with DH in
patients with athyreosis (30,32).
In addition, 2 individual variants in genes associated with
pituitary development or central CH (PROP1 and TRHR)
respectively were found in 1 CH patient with GIS and 1 patient with
TD, and both variants co-occurred with genetic variants associated
with DH. Currently, it is difficult to be sure that the genetic
defects associated with DH contribute to the development of TD or
the potential effect of variants associated with pituitary
development on primary CH. However, the findings of the present
study validated the complicated pathological mechanism of CH. Thus,
studies on the genetic origin of TD or DH diseases should not be
limited to well-known causative genes (4,33).
In the present study, DUOX2 was the most
common genetic alteration identified in CH patients. The detection
rate of DUOX2 mutation in the studied CH cohort and the GIS
patients were 44% (19/43) and 53.1% (17/32), respectively, most of
which carried ≥2 DUOX2 mutations. This finding was in
accordance with those previously reported showing that DUOX2
mutation is the leading genetic cause of CH in Asian populations,
including other Han Chinese, Japanese and Koreans; the detection
rate of DUOX2 in patients from Asian populations is 16.5–3%
and ≤83% in patients with DH (27,29,31,34–40).
TG and TPO mutations were also commonly found in the
studied cohort. However, a majority of these mutations were either
separately presented at a heterozygous status or detected with
mutations in different genes. Thus, TG and TPO may
act as contributing genetic factors apart from being the main
genetic causes of CH in the studied cohort. In some Caucasian
cohorts, TPO has been identified as the main genetic cause
of CH (41–43). In addition, loss-of-function (LOF)
mutations in the TSH receptor (TSHR) gene were identified as
the most frequent cause of TSH resistance, leading to a wide
spectrum of phenotypes ranging from severe CH to mild euthyroid
hyperthyrotropinemia (26,44). In the present study, 2 patients with
CH who were compound heterozygotes for 2 different TSHR
mutations (1 for p.R528S and p.R450H and 1 for p.C176R and p.K618*)
demonstrated mild clinical phenotypes (5 pmol/l≤FT4<10 pmol/l)
(45). This finding is in agreement
with previous studies reporting that compound heterozygotes of less
severe LOF mutations are usually associated with mild/borderline
forms of hypothyroidism, wherein an appropriate increase in TSH
serum levels can compensate for the reduced sensitivity of the
thyroid (partially or fully compensated TSH resistance) (26,44,46,47).
Previously reported clinical cases with TSHR mutations are
always characterized by normal-sized or hypoplastic thyroid gland
(26,46,47).
However, in the present study, patient 37 suffered from goiter. In
addition to TSHR mutations, this patient also carried a
heterozygous variant in TPO, which may be the reason leading
to this phenotypic variability.
At present, patients with CH caused by genetic
defects are considered to be inherited in a monogenic manner.
However, phenotypic variability observed in patients with same
mutations indicated the influence of other factors, such as genetic
heterogeneity (15,34,42,43,48–50).
The present study found a high percentage (25.6%) of involvement of
oligogenic mutations in studied cases, which is similar to that of
previous studies simultaneously assessing multiple genes (15,30–32).
These studies reported frequent oligogenic involvement in CH, with
oligogenic mutations in 20–43.5% of patients with CH and GIS and/or
patients with TD in different ethnic populations. In addition,
among the 13 ‘solved’ cases through family segregation and
pathogenicity assessment, 5 cases carried oligogenic mutations and
none of the mutations was inherited from a single parent. These
findings suggested that, not only monogenic inheritance, but also
digenic or oligogenic inheritance is involved in the pathogenesis
of CH. However, available evidence is insufficient for oligogenic
inheritance verification, and protein-protein interaction for the
two proteins or genes, pedigree data, animal models or very
specific functional experiments are key factors (18). At present, only 1 study performed in
mice demonstrated a multigenic origin of CH with TD (51). Therefore, further studies are
required for the validation of the role and mechanism of
oligogenicity in CH pathogenesis.
The data of the present study were compared with
those of several similar studies that analyzed the mutation
spectrum of CH patients in China by using NGS (29–31).
The investigated patients in the present study were from
northwestern China (Shaanxi Province), whereas those in the
previous studies were mainly from southern China (Jiangsu and
Guangxi Provinces). The general mutation profiles of patients with
CH demonstrated by these studies were similar. For example, the
total mutation rate in CH patients reported in these studies was
relatively high, i.e., 48.5% (29),
65.09% (31), 80.9% (30) and 72% (the present study).
DUOX2 mutations were the prevalent genetic alterations in
these studies, with a mutation rate of 31.8% (29), 31.3% (31), 60% (30) and 44% (the present study).
Therefore, the region (or population)-specific characteristics in
patients with CH from these studies could not be ascertained.
However, some different findings in the present study were
observed. For example, DUOX2 and TG mutations were
the first and second most common mutations detected in all these
studies. However, the third and fourth most common genetic
mutations were different. In the present study, the third and
fourth most common genetic mutation were TPO and TSHR
mutations, respectively. in addition, the third and fourth most
common genetic mutations reported by Long et al (31) were TSHR and GNAS
mutations, respectively and those by Sun et al (30) were TPO and DUOXA1
mutations, respectively. These discrepancies may be caused by the
relatively small sample size, sampling criteria and/or different
targeted genes determined.
Several limitations were observed in the present
study. The sample size was relatively small, and most patients were
too young to exhibit clinical phenotypes. Thus, determining the
clinical significance of the detected mutations was difficult.
Pedigree analysis was not performed in all the cases carrying
mutations and evidence to support the pathogenicity of detected
variants was insufficient. The diagnosis detection rate in the
present study would be >30% were these requirements met.
Finally, in vitro functional study of novel variants
identified in the current study should be carried out.
In conclusion, using the currently largest targeted
NGS panel containing 29 known genes, the mutation spectrum of 43
Han Chinese patients with CH was comprehensively determined. The
main findings showed that DH other than TD is the common cause of
CH in Chinese populations and genetic alterations associated with
thyroid hormone biosynthesis, especially DUOX2 mutations,
are the main genetic causes of CH. In addition, a high percentage
of involvement of oligogenic mutation in the studied cases
confirmed the potential role of oligogenicity or non-Mendelian
inheritance in CH pathogenesis.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81702483) and Shaanxi
Innovative Talents Promotion Plan (grant no. 2017KJXX-33).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
HW and XY conceived the project. YW, LirZ performed
the experiment. XK and LixZ analyzed and interpreted the data. CC
designed the study and revised the article. HW reviewed the
literature and wrote the article. YP, YZ, XC and ZH were involved
in sample and medical record recruitment. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The parents of all participants gave written
informed consent in accordance with the Declaration of Helsinki.
The study was approved by the Medical Ethics Committees of Xi'an
Children's Hospital and Chang'an Hospital.
Patient consent for publication
Written informed consent was obtained from the
parents of all the participants for the publication of their
data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rastogi MV and Lafranchi SH: Congenital
hypothyroidism. Orphanet J Rare Dis. 5:172010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wassner AJ and Brown RS: Congenital
hypothyroidism: Recent advances. Curr Opin Endocrinol Diabetes
Obes. 22:407–412. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nygaard B: Hyperthyroidism (primary). Bmj
Clin Evid. 2010:06112010.PubMed/NCBI
|
|
4
|
Nettore IC, Cacace V, De Fusco C, Colao A
and Macchia PE: The molecular causes of thyroid dysgenesis: A
systematic review. J Endocrinol Invest. 36:654–664. 2013.PubMed/NCBI
|
|
5
|
Grasberger H and Refetoff S: Genetic
causes of congenital hypothyroidism due to dyshormonogenesis. Curr
Opin Pediatr. 23:421–428. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schoenmakers N, Alatzoglou KS, Chatterjee
VK and Dattani MT: Recent advances in central congenital
hypothyroidism. J Endocrinol. 227:R51–R71. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park SM and Chatterjee VK: Genetics of
congenital hypothyroidism. J Med Genet. 42:379–389. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hulander M, Kiernan AE, Blomqvist SR,
Carlsson P, Samuelsson EJ, Johansson BR, Steel KP and Enerbäck S:
Lack of pendrin expression leads to deafness and expansion of the
endolymphatic compartment in inner ears of Foxi1 null mutant mice.
Development. 130:2013–2025. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zenker M, Mayerle J, Lerch MM, Tagariello
A, Zerres K, Durie PR, Beier M, Hülskamp G, Guzman C, Rehder H, et
al: Deficiency of UBR1, a ubiquitin ligase of the N-end rule
pathway, causes pancreatic dysfunction, malformations and mental
retardation (Johanson-Blizzard syndrome). Nat Genet. 37:1345–1350.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang T, Vidarsson H, Rodrigo-Blomqvist S,
Rosengren SS, Enerback S and Smith RJ: Transcriptional control of
SLC26A4 is involved in Pendred syndrome and nonsyndromic
enlargement of vestibular aqueduct (DFNB4). Am J Hum Genet.
80:1055–1063. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Secchi LA, Mazzeu JF, Córdoba MS, Ferrari
I, Ramos HE and Neves Fde A: Transient neonatal hypothyroidism in a
boy with unbalanced translocationt (8;16). Arq Bras Endocrinol
Metabol. 56:564–569. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lichti-Kaiser K, ZeRuth G and Jetten AM:
Transcription factor Gli-Similar 3 (GLIS3): Implications for the
development of congenictal hypothyroidism. J Endocrinol Diabetes
Obes. 2:10242014.PubMed/NCBI
|
|
13
|
Kang HS, Kumar D, Liao G, Lichti-Kaiser K,
Gerrish K, Liao XH, Refetoff S, Jothi R and Jetten AM: GLIS3 is
indispensable for TSH/TSHR-dependent thyroid hormone biosynthesis
and follicular cell proliferation. J Clin Invest. 127:4326–4337.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Muzza M, Rabbiosi S, Vigone MC, Zamproni
I, Cirello V, Maffini MA, Maruca K, Schoenmakers N, Beccaria L,
Gallo F, et al: The clinical and molecular characterization of
patients with dyshormonogenic congenital hypothyroidism reveals
specific diagnostic clues for DUOX2 defects. J Clin Endocrinol
Metab. 99:E544–E553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nicholas AK, Serra EG, Cangul H, Alyaarubi
S, Ullah I, Schoenmakers E, Deeb A, Habeb AM, Almaghamsi M, Peters
C, et al: Comprehensive screening of eight known causative genes in
congenital hypothyroidism with gland-in-situ. J Clin Endocrinol
Metab. 101:4521–4531. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoshizawa-Ogasawara A, Abe K, Ogikubo S,
Narumi S, Hasegawa T and Satoh M: Transient congenital
hypothyroidism caused by compound heterozygous mutations affecting
the NADPH-oxidase domain of DUOX2. J Pediatr Endocrinol Metab.
29:363–371. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng X, Ma SG, Qiu YL, Guo ML and Shao
XJ: A novel c.554+5C>T mutation in the DUOXA2 gene combined with
p.R885Q mutation in the DUOX2 gene causing congenital
hypothyroidism. J Clin Res Pediatr Endocrinol. 8:224–227. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schäffer AA: Digenic inheritance in
medical genetics. J Med Genet. 50:641–652. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kousi M and Katsanis N: Genetic modifiers
and oligogenic inheritance. Cold Spring Harb Perspect Med.
5:a0171452015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu JT, Campeau PM and Lee BH:
Genotype-phenotype correlation-promiscuity in the era of
next-generation sequencing. N Engl J Med. 371:593–596. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Manolio TA, Collins FS, Cox NJ, Goldstein
DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR,
Chakravarti A, et al: Finding the missing heritability of complex
diseases. Nature. 461:747–753. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Deng K, He C, Zhu J, Liang J, Li X, Xie X,
Yu P, Li N, Li Q and Wang Y: Incidence of congenital hypothyroidism
in China: Data from the national newborn screening program,
2013–2015. J Pediatr Endocrinol Metab. 31:601–608. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hinton CF, Harris KB, Borgfeld L,
Drummond-Borg M, Eaton R, Lorey F, Therrell BL, Wallace J and Pass
KA: Trends in incidence rates of congenital hypothyroidism related
to select demographic factors: Data from the United States,
California, Massachusetts, New York, and Texas. Pediatrics. 125
(Suppl 2):S37–S47. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen X, Kong X, Zhu J, Zhang T, Li Y, Ding
G and Wang H: Mutational spectrum analysis of seven genes
associated with thyroid dyshormonogenesis. Int J Endocrinol.
2018:89864752018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cassio A, Nicoletti A, Rizzello A,
Zazzetta E, Bal M and Baldazzi L: Current loss-of-function
mutations in the thyrotropin receptor gene: When to investigate,
clinical effects, and treatment. J Clin Res Pediatr Endocrinol. 5
(Suppl 1):29–39. 2013.PubMed/NCBI
|
|
27
|
Jiang H, Wu J, Ke S, Hu Y, Fei A, Zhen Y,
Yu J and Zhu K: High prevalence of DUOX2 gene mutations among
children with congenital hypothyroidism in central China. Eur J Med
Genet. 59:526–531. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Albert BB, Cutfield WS, Webster D, Carll
J, Derraik JG, Jefferies C, Gunn AJ and Hofman PL: Etiology of
increasing incidence of congenital hypothyroidism in New Zealand
from 1993–2010. J Clin Endocrinol Metab. 97:3155–3160. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fan X, Fu C, Shen Y, Li C, Luo S, Li Q,
Luo J, Su J, Zhang S, Hu X, et al: Next-generation sequencing
analysis of twelve known causative genes in congenital
hypothyroidism. Clin Chim Acta. 468:76–80. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun F, Zhang JX, Yang CY, Gao GQ, Zhu WB,
Han B, Zhang LL, Wan YY, Ye XP, Ma YR, et al: The genetic
characteristics of congenital hypothyroidism in China by
comprehensive screening of 21 candidate genes. Eur J Endocrinol.
178:623–633. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Long W, Lu G, Zhou W, Yang Y, Zhang B,
Zhou H, Jiang L and Yu B: Targeted next-generation sequencing of
thirteen causative genes in Chinese patients with congenital
hypothyroidism. Endocr J. 65:1019–1028. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
de Filippis T, Gelmini G, Paraboschi E,
Vigone MC, Di Frenna M, Marelli F, Bonomi M, Cassio A, Larizza D,
Moro M, et al: A frequent oligogenic involvement in congenital
hypothyroidism. Hum Mol Genet. 26:2507–2514. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
De Felice M and Di Lauro R: Thyroid
development and its disorders: Genetics and molecular mechanisms.
Endocr Rev. 25:722–746. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Narumi S, Muroya K, Asakura Y, Aachi M and
Hasegawa T: Molecular basis of thyroid dyshormonogenesis: Genetic
screening in population-based Japanese patients. J Clin Endocrinol
Metab. 96:E1838–E1842. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Park KJ, Park HK, Kim YJ, Lee KR, Park JH,
Park JH, Park HD, Lee SY and Kim JW: DUOX2 mutations are frequently
associated with congenital hypothyroidism in the Korean population.
Ann Lab Med. 36:145–153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang F, Lu K, Yang Z, Zhang S, Lu W, Zhang
L, Liu S and Yan S: Genotypes and phenotypes of congenital goitre
and hypothyroidism caused by mutations in dual oxidase 2 genes.
Clin Endocrinol (Oxf). 81:452–457. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fu C, Zhang S, Su J, Luo S, Zheng H, Wang
J, Qin H, Chen Y, Shen Y, Hu X, et al: Mutation screening of DUOX2
in Chinese patients with congenital hypothyroidism. J Endocrinol
Invest. 38:1219–1224. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fu C, Luo S, Zhang S, Wang J, Zheng H,
Yang Q, Xie B, Hu X, Fan X, Luo J, et al: Next-generation
sequencing analysis of DUOX2 in 192 Chinese subclinical congenital
hypothyroidism (SCH) and CH patients. Clin Chim Acta. 458:30–34.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Matsuo K, Tanahashi Y, Mukai T, Suzuki S,
Tajima T, Azuma H and Fujieda K: High prevalence of DUOX2 mutations
in Japanese patients with permanent congenital hypothyroidism or
transient hypothyroidism. J Pediatr Endocrinol Metab. 29:807–812.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tan M, Huang Y, Jiang X, Li P, Tang C, Jia
X, Chen Q, Chen W, Sheng H, Feng Y, et al: The prevalence,
clinical, and molecular characteristics of congenital
hypothyroidism caused by DUOX2 mutations: A population-based cohort
study in Guangzhou. Horm Metab Res. 48:581–588. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cangul H, Aycan Z, Olivera-Nappa A, Saglam
H, Schoenmakers NA, Boelaert K, Cetinkaya S, Tarim O, Bober E,
Darendeliler F, et al: Thyroid dyshormonogenesis is mainly caused
by TPO mutations in consanguineous community. Clin Endocrinol
(Oxf). 79:275–281. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Avbelj M, Tahirovic H, Debeljak M,
Kusekova M, Toromanovic A, Krzisnik C and Battelino T: High
prevalence of thyroid peroxidase gene mutations in patients with
thyroid dyshormonogenesis. Eur J Endocrino. 156:511–519. 2007.
View Article : Google Scholar
|
|
43
|
Löf C, Patyra K, Kuulasmaa T, Vangipurapu
J, Undeutsch H, Jaeschke H, Pajunen T, Kero A, Krude H, Biebermann
H, et al: Detection of novel gene variants associated with
congenital hypothyroidism in a Finnish patient cohort. Thyroid.
26:1215–1224. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Persani L, Calebiro D, Cordella D, Weber
G, Gelmini G, Libri D, de Filippis T and Bonomi M: Genetics and
phenomics of hypothyroidism due to TSH resistance. Mol Cell
Endocrinol. 322:72–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Léger J, Olivieri A, Donaldson M,
Torresani T, Krude H, van Vliet G, Polak M and Butler G;
ESPE-PES-SLEP-JSPE-APEG-APPES-ISPAE; Congenital Hypothyroidism
Consensus Conference Group, : European society for paediatric
endocrinology consensus guidelines on screening, diagnosis, and
management of congenital hypothyroidism. Horm Res Paediatr.
81:80–103. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kanda K, Mizuno H, Sugiyama Y, Imamine H,
Togari H and Onigata K: Clinical significance of heterozygous
carriers associated with compensated hypothyroidism in R450H, a
common inactivating mutation of the thyrotropin receptor gene in
Japanese. Endocrine. 30:383–388. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tsunekawa K, Onigata K, Morimura T,
Kasahara T, Nishiyama S, Kamoda T, Mori M, Morikawa A and Murakami
M: Identification and functional analysis of novel inactivating
thyrotropin receptor mutations in patients with thyrotropin
resistance. Thyroid. 16:471–479. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Maruo Y, Takahashi H, Soeda I, Nishikura
N, Matsui K, Ota Y, Mimura Y, Mori A, Sato H and Takeuchi Y:
Transient congenital hypothyroidism caused by biallelic mutations
of the dual oxidase 2 gene in Japanese patients detected by a
neonatal screening program. J Clin Endocrinol Metab. 93:4261–4267.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jin HY, Heo SH, Kim YM, Kim GH, Choi JH,
Lee BH and Yoo HW: High frequency of DUOX2 mutations in rransient
or permanent congenital hypothyroidism with eutopic thyroid glands.
Horm Res Paediatr. 82:252–260. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hu X, Chen R, Fu C, Fan X, Wang J, Qian J,
Yi S, Li C, Luo J, Su J, et al: Thyroglobulin gene mutations in
Chinese patients with congenital hypothyroidism. Mol Cell
Endocrinol. 423:60–66. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Amendola E, De Luca P, Macchia PE,
Terracciano D, Rosica A, Chiappetta G, Kimura S, Mansouri A, Affuso
A, Arra C, et al: A mouse model demonstrates a multigenic origin of
congenital hypothyroidism. Endocrinology. 146:5038–5047. 2005.
View Article : Google Scholar : PubMed/NCBI
|