Introduction
Retinitis pigmentosa (RP; OMIM:226800) is an
inherited retinal dystrophy and a genetically heterogeneous
disease, with a worldwide prevalence of ~1:4,000 (1). The main features of RP are vision
loss, tubular vision, night blindness, retinal ‘bone-spicule’
pigmentation, reduced vascular atrophy and certain complications of
deafness (2,3). The genetic pattern of RP is diverse:
The majority of the genetic mutations are autosomal dominant or
autosomal recessive (ARRP), some are X-linked (4), and other modes of inheritance,
including digenic or mitochondrial inheritance, have also been
reported (5,6). The affected patients generally suffer
poor vision under dim light and progressive visual loss due to the
death of rod cells (7). To date,
there is no effective way to treat this disease.
RP is one of the most genetically heterogeneous
disorders. To date, >90 genes have been associated with ARRP
(RetNet; https://sph.uth.edu/retnet/, accessed
July 2019). In addition, mutations in a single gene can be
associated with a broad phenotypic spectrum, and a specific
phenotype can be caused by mutations in multiple genes (8). At present, known mutations can explain
~60% of RP cases (9,10). Therefore, identifying additional
disease genes involved in RP is important for genetics diagnosis.
Next-generation sequencing (NGS) has become one of the most
important tools for the identification of disease-causing genes
(11–18). In previous studies, a targeted NGS
method has been used successfully to systematically screen coding
regions of known retinal genes to identify pathological mutations
(19–21).
The most common syndromic RP is Usher syndrome,
which accompanies RP with hearing loss, with a prevalence of
~1:20,000 (22). In total, 16 genes
have been identified as causative genes for Usher syndrome
(https://sph.uth.edu/retnet/sum-dis.htm). The gene for
Usher syndrome type 2 (USH2), USH2A, was mapped using
linkage analysis by Kimberling et al (23) and Lewis et al (24) to chromosome 1 with 72 exons. It
encodes usherin, a basement membrane protein with laminin epidermal
growth factor and fibronectin type III domains (25,26)
that is found in numerous tissues, including capillary and
structural basement membranes in the retina and inner ear.
Mutations in the USH2A gene are the most common cause of
Usher syndrome (29% of all cases) and one of the most common
reasons for ARRP (19-23%) (27–29).
Mutations in USH2A were reported to be responsible for 7% of
RP cases in North America (30).
Jiang et al (31) identified
40 USH2A mutations in 32 patients with USH2, and reported
that USH2A mutation severity determined patient clinical
phenotypes.
The aim of the present study was to apply targeted
NGS to a cohort of 62 families with RP and 30 sporadic cases from
China to identify mutations underlying the disease. The current
study focused on mutations in USH2A. In total, 6 novel
mutations and 2 known mutations were identified.
Materials and methods
Subjects and clinical assessments
The present study was approved by the Ethics
Committee of the Hospital of Sichuan Academy of Medical Sciences
and Sichuan Provincial People's Hospital (Chengdu, Sichuan, P.R.
China; approval no. SCPH-2017-076). All patients (age range, 17–65;
55% male patients and 45% female patients) from 62 unrelated Han
Chinese families and 30 sporadic cases, as well as 1,000 healthy
Han Chinese control individuals were recruited from the Sichuan
Provincial People's Hospital between January 2014 and December
2016. All the patients, family members and controls involved in the
study signed written informed consent for the collection of samples
for sequencing and the publication of patient images and data. The
patients and family members were clinically diagnosed at Sichuan
Provincial People's Hospital. Peripheral blood samples were
collected from probands and their family members. Genomic DNA was
extracted using the QIAamp DNA Blood Mini kit (Qiagen, Inc.)
according to the manufacturer's protocols.
Targeted NGS and genetic analysis
A targeted-NGS sequencing method described by Wang
et al (19) was adapted in
the present study. Briefly, genomic DNA samples were randomly
fragmented into 300–500 bp fragments whose ends were end-repaired
using polynucleotide kinase (Thermo Fisher Scientific, Inc.) and
Klenow (New England Biolabs, Inc.). Subsequently, extra ‘adenine’
bases were added to the 3′end of the DNA fragments. Illumina
Y-shaped index adapters (Illumina, Inc.) were then applied to
generate DNA paired-end libraries. Subsequently, each captured
library was loaded on to Illumina HiSeq 2000 (Illumina, Inc.) for
quantification and sequencing. Using Burrows-Wheeler Aligner
(version bwa-0.7.15), sequencing reads were aligned to the
reference human genome (hg19 UCSC assembly; genome.ucsc.edu) (32). SAMtools (version 1.3) was applied to
identify single-nucleotide variants and insertions/deletions
(33). Filtrations and annotations
of the variants were conducted according to a previously described
protocol (34) based on autosomal
recessive inheritance pattern using the following databases: i)
NCBI CCDS (www.ncbi.nlm.nih.gov/projects/CCDS/CcdsBrowse.cgi);
ii) RefSeq (www.ncbi.nlm.nih.gov/RefSeq); iii) Ensembl (www.ensembl.org); and iv) ENCODE (genome.ucsc.edu/ENCODE).
To ensure the accuracy and efficacy of the candidate
mutations, the synonymous, intergenic and intronic variants were
first filtered out, and mutations in any of the following databases
were excluded: i) dbSNP138 (www.ncbi.nlm.nih.gov/projects/SNP); ii) 1000 genomes
Project (ftp.1000genomes.ebi.ac.uk/vol1/ftp); iii) YH database
(yh.genomics.org.cn); iv) HapMap Project
(ftp://ftp.ncbi.nlm.nih.gov/hapmap); and v) an ‘in
house’ database generated from 1,800 samples sequenced by whole
exome sequencing (34).
According to the bioinformatics pipeline,
low-quality variants and variants predicted to be benign by online
tools (SIFT (sift-dna.org), PROVEAN (provean.jcvi.org) and Ensembl Variant Effect Predictor
(grch37.ensembl.org/info/docs/tools/vep) were excluded
(10).
Sanger sequencing analysis
DNA sequencing was performed using the dideoxy
Sanger method by fluorescent automated sequencing on the ABI 3130×l
Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc.). Sanger sequencing analysis was used to verify whether the
remaining variants co-segregated with the RP phenotypes in the
families. PCR primers (Table I)
were designed using the Primer3 online tool (SourceForge.Net) and synthesized by Shanghai Sangong
Pharmaceutical Co., Ltd. to amplify genomic DNA fragments. The PCR
products were purified using FastAP Thermosensitive Alkaline
Phosphatase (Thermo Fisher Scientific, Inc.) and sequenced using a
BigDye™ Terminator v3.1 cycle sequencing kit (Thermo Fisher
Scientific, Inc.). The following thermocycling conditions were
used: 28 cycles of 96°C for 15 sec, 50°C for 10 sec and 60°C for 4
min; followed by maintenance at 4°C. The sequencing data were
analyzed using Sequencing Analysis ABI Software v5.3 (Applied
Biosystems; Thermo Fisher Scientific, Inc.).
 | Table I.Primers used for mutation
analysis. |
Table I.
Primers used for mutation
analysis.
| Amplicon | Primer | Temperature
(°C) | Size (bp) |
|---|
| p.G2752R | F:
ACATTCTGAGGTACGGTGGG | 59 | 543 |
|
| R:
TGTCCTTCAGAACACCCACA | 60 |
|
| p.C577X | F:
ATAGAAGCACACAGGCCTCC | 59 | 427 |
|
| R:
ACCCTACCATGCCCGTAAAT | 60 |
|
| p.G268R | F:
GTCCTACAGTGTCCATGGAGA | 58 | 464 |
|
| R:
TCCTCAAGAGTAGCACTAGTGA | 57 |
|
| p.H4029fs | F:
CTCTGCTGTAGTGTTTGCGC | 59 | 462 |
|
| R:
ACAGGCTGTGAAGGGAGTTT | 59 |
|
| p.G4489S | F:
CCACCTGCAGCCTTACTCTC | 60 | 330 |
|
| R:
AAAATACCCCCTTGGCTGTT | 59 |
|
| p.W3918X | F:
GTGTGCAGCTGTCACTGGTT | 59 | 312 |
|
| R:
AATGCCATTGGGAGATTCTG | 59 |
|
| p.P3110A | F:
AATGAGGGAAGGTGGGATTC | 60 | 501 |
|
| R:
TATGCTCCGCAAAAGGATTC | 60 |
|
| p.W2744C | F:
CAAACCCAGGAAACAGCATT | 60 | 310 |
|
| R:
TGCGGAAGTCACATTGGTTA | 60 |
|
Clinical diagnosis
Ophthalmic examinations were conducted, including
visual acuity, intraocular-pressure, ocular-motility,
pupillary-reaction, slit-lamp, visual field test, dilated-fundus,
optical coherence tomography examination and visual
electrophysiological tests.
Results
Clinical characteristics of the
families with RP
In the present study, the two recruited families who
were initially diagnosed with RP followed the pattern of autosomal
recessive inheritance. Ophthalmic examinations identified 2
affected members in family RP-2148, and 1 affected member in
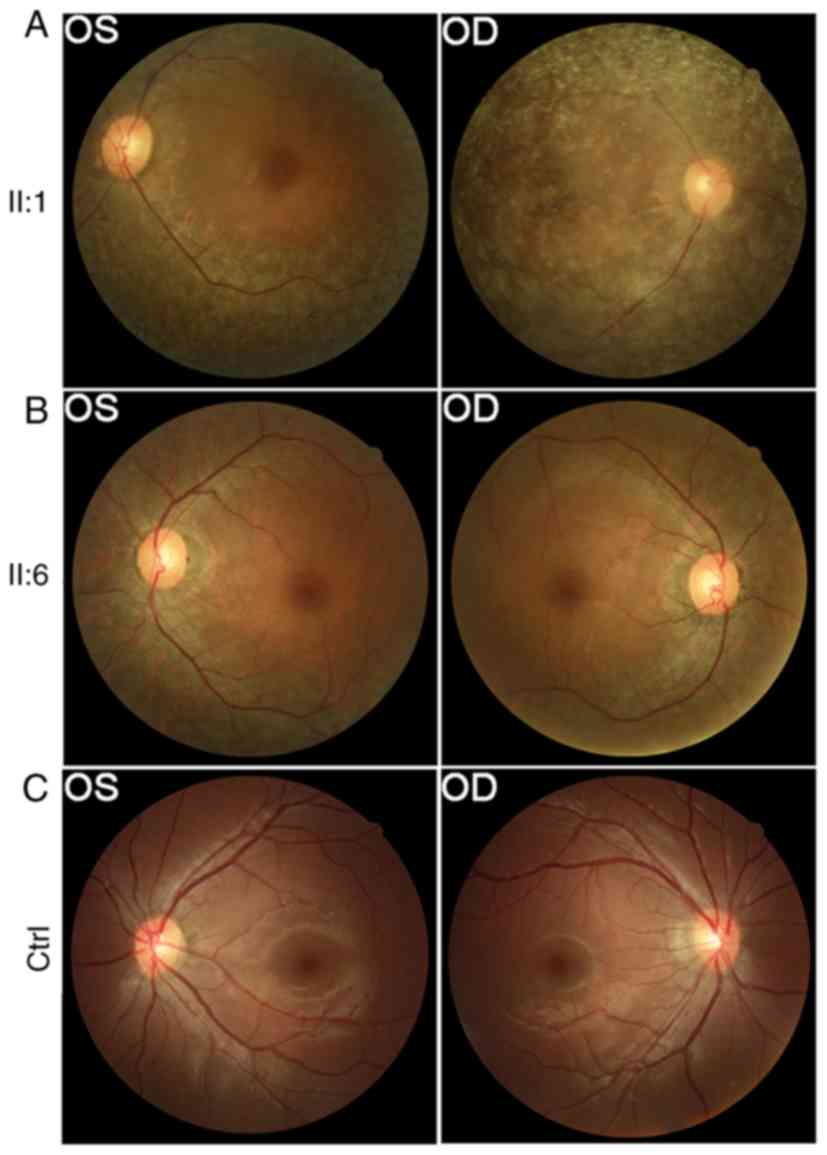
RP-2150. Representative fundus photographs are shown in Fig. 1, which indicate an obvious waxen
appearance of the discs, bone-spicule pigment deposit in the
midperiphery, attenuation of the retinal arteries (Fig. 1A), waxy-pale disc and bone-spicule
pigment (Fig. 1B). The patients'
parents and other unaffected family members did not show any RP
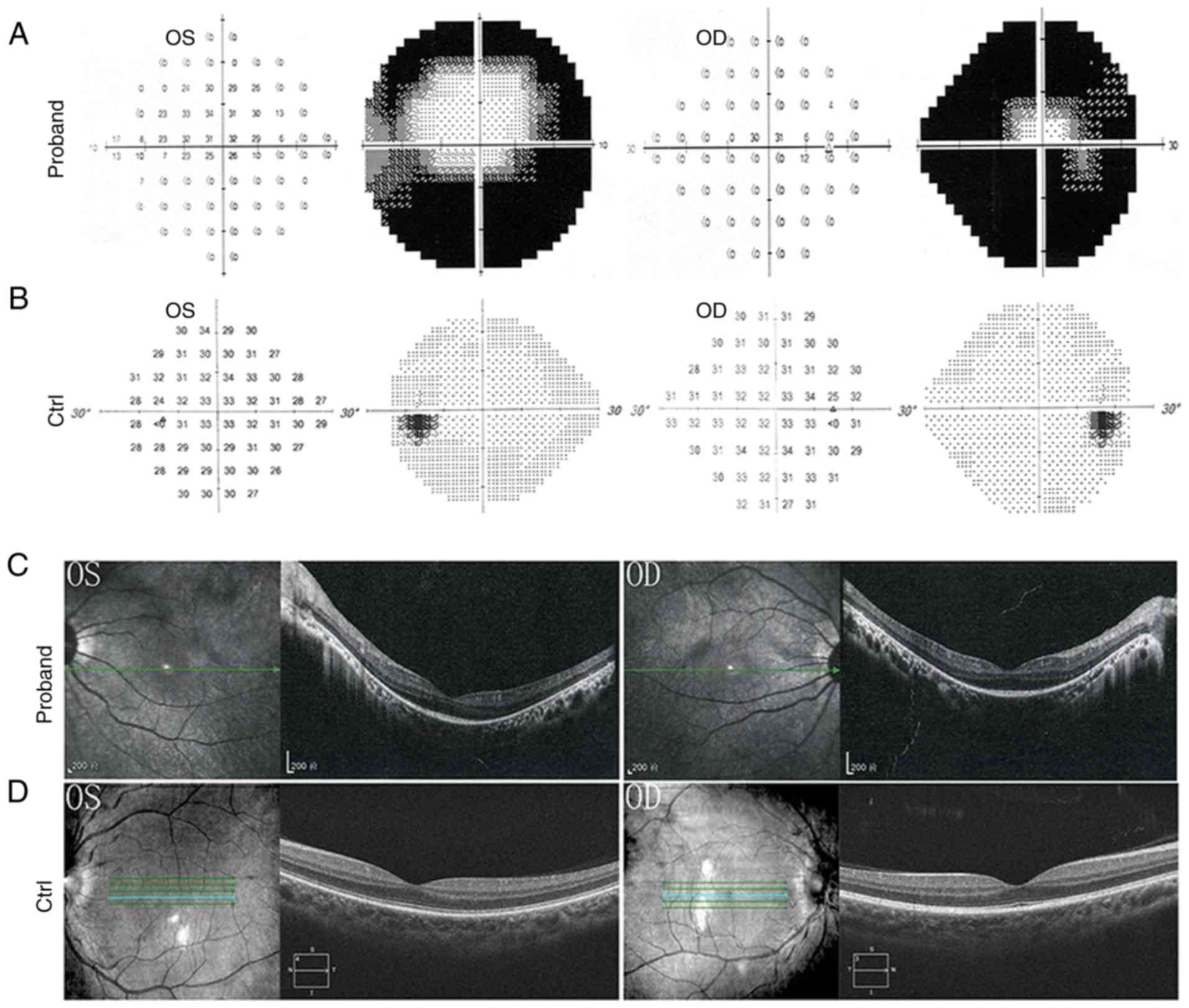
features. A visual field test revealed loss of peripheral vision in
both eyes (Fig. 2A and B). Optical
coherence tomography examination, which provides an indirect
measure of axonal and neuronal injury in the anterior visual
pathways, revealed disorganized photoreceptor layers and thinner
retina (Fig. 2C).
Genetic findings
To reveal pathogenic genetic factors, targeted NGS
was applied to a cohort of patients with RP. Sanger sequencing was
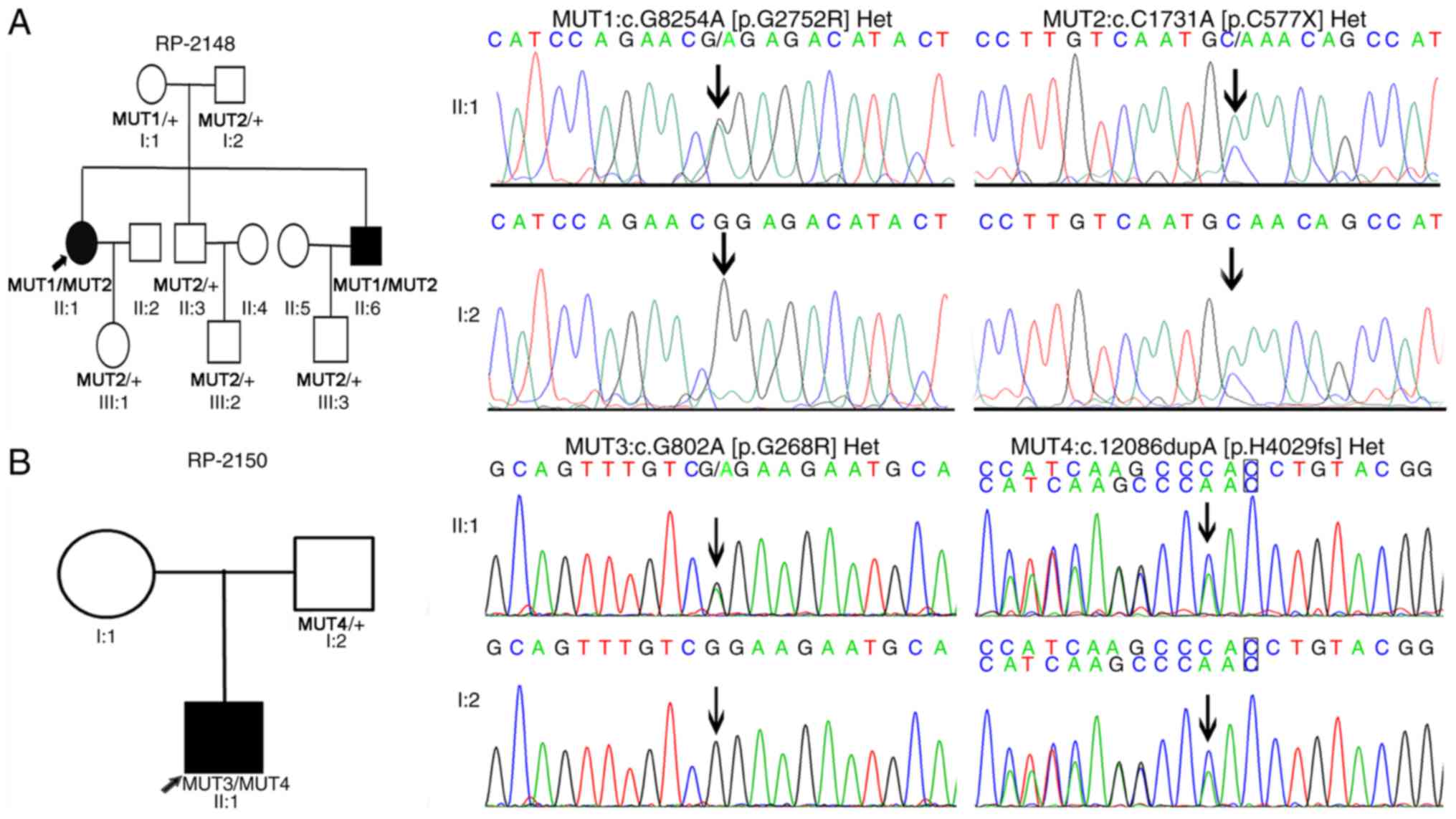
performed to verify the identified mutations (Fig. 3). In the proband of RP-2148, a
stop-gain mutation (c.C1731A:p.C577X) was identified in exon 10 and
1 missense mutation (c.G8254A:p.G2752R) was identified in exon 42
(Table II and Fig. 3A). The proband RP-2148 II:1 carried
compound heterozygous mutations: One allele (c.G8254A) came from
the mother, whereas the other allele (c.C1731A) came from the
father. Both his mother and father were heterozygous carriers. In
RP-2150, the present study identified a missense mutation
(c.G802A:p.G268R) and a frameshift insertion mutation
(c.12086dupA:p.H4029fs) in the proband of RP-2150 II:1, which
carried compound heterozygous mutations (Table II and Fig. 3B). One allele (c.12086dupA) came
from the proband's father. Unfortunately, his mother was not
available for genotyping. The missense mutation p.G268R has been
previously reported in patients with RP (35). Sequence alignment analysis showed
that these mutations affect highly conserved amino acid residues
(Fig. 4). Furthermore, the mutation
c.G8254A:p.G2752R was predicted to be damaging or deleterious by
SIFT or PROVEAN software. None of the aforementioned mutations were
present in 1,000 ethnically matched controls.
| Table II.USHA2 mutations identified in
patients with RP. |
Table II.
USHA2 mutations identified in
patients with RP.
| Family ID | Nucleotide
change | Allele state | Amino acid
change | dbSNP | Type | SIFT | PROVEAN |
|---|
| 2148-II:1 | c.C1731A | Het | p.C577* | Novel | Stop-gain | NA | NA |
| 2148-II:1 | c.G8254A | Het | p.G2752R | Novel | Missense | Damaging | Deleterious |
| 2150-II:1 | c.G802A | Het | p.G268R | Known | Missense | Damaging | Deleterious |
| 2150-II:1 | c.12086dupA | Het | p.H4029fs | Novel | Frameshift | NA | NA |
| rpz05-II:1 | c.G13465A | Het | p.G4489S | Novel | Missense | Damaging | Deleterious |
| rpz05-II:1 | c.G11754A | Het | p.W3918* | Novel | Stop-gain | NA | NA |
| rpz06-II:1 | c.C9328G | Het | p.P3110A | Novel | Missense | Damaging | Deleterious |
| rpz06-II:1 | c.G8232C | Het | p.W2744C | Known | Missense | Damaging | Deleterious |
In an attempt to identify additional USH2A
mutations, 3 novel mutations and 1 known mutation were detected in
two unrelated families. A stop-gain mutation c.G11754A [p.W3918X]
and a missense mutation c.G13465A [p.G4489S] were identified in
family rpz05 (Fig. S1). In family
rpz06, a novel missense mutation c.C9328G [p.P3110A] was detected
in the proband (Fig. S2). The
proband also carried a known missense mutation, c.G8232C:p.W2744C
(36). Both c.G8254A [p.G2752R] and
c.C9328G [p.P3110A] affected highly conserved amino residues
(Fig. 4), and were predicted to be
damaging or deleterious by SIFT or PROVEN software (Table II). In addition, none of the
aforementioned mutations were present in 1,000 ethnically matched
controls. These novel mutations data provide valuable information
for the genetic diagnosis of USH2A-related RP.
Discussion
Mutations in the USH2A gene are the most
common cause of Usher syndrome (29% of all cases) and one of the
most common causes underlying ARRP (19-23%) (25,26).
In North America, USH2A mutations account for 7% of RP cases
(30). In a multiethnic cohort,
Jiang et al (31) identified
40 USH2A mutations in 32 patients with USH2. Huang et
al (37) reported 8 mutations
in USH2A in 4 patients from 75 patients with non-syndromic
RP, and 2 mutations in a family with Usher syndrome by Sanger
sequencing screening. The present study applied targeted NGS
analysis, and identified 8 USH2A mutations in a cohort of 62
patients with non-syndromic RP and 30 simplex cases in northern and
western China. In total, 6 of the identified mutations were novel
and absent from ethnically matched controls. Initially, no hearing
problems were noted in these patients when they were enrolled in
this study, and therefore hearing tests were not performed. Further
follow-up hearing tests on these patients, however, could provide
valuable information regarding the effect of these mutations on
hearing.
Human USH2A encodes a large protein, usherin,
containing 5,202 amino acids. In mammalian photoreceptors, the
usherin protein is localized to the apical inner segment, in the
amphibious photoreceptor (38).
Deficiency in the USH2A gene in mice causes progressive
photoreceptor degeneration and moderate hearing impairment,
mimicking the visual and auditory deficits in patients with
USH2A mutations (38). In
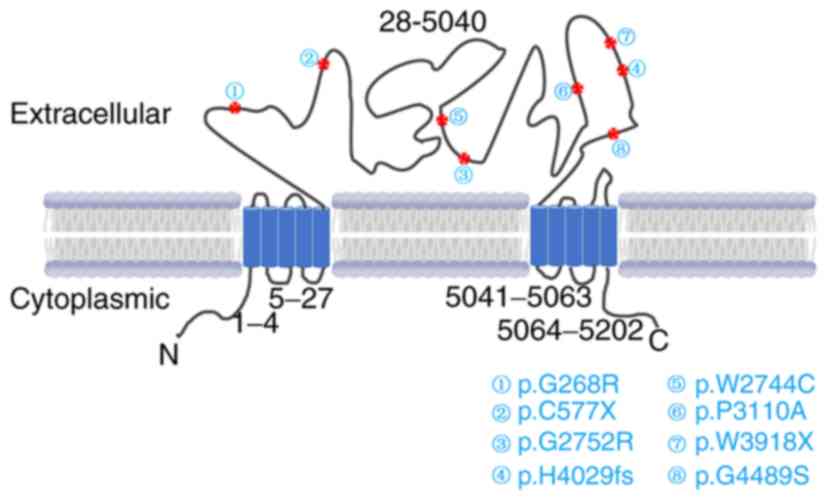
the present study, all the 8 mutations identified were located in
the exoplasmic functional domains of USH2A, and are predicted to be
damaging (Fig. 5).
Taken together, in the present study 8 mutations
have been identified in USH2A in a cohort of patients with
non-syndromic RP from northern and western China. A total of 6
mutations were novel, of which 3 were stop-gain or frameshift
insertions that disrupted the protein's function. In addition, all
3 novel missense mutations were predicted to be harmful, as
determined by prediction software. These data have provided
valuable information for the genetic diagnosis of RP cases caused
by USH2A. Due to the limited scope of the current study,
biological functions of these mutant proteins, however, were not
assessed. Therefore, the effect of these missense mutations
warrants further investigation. In summary, these data expand on
the mutation spectrum of patients with non-syndromic RP in the
Chinese population.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (www.nsfc.gov.cn/; grant no. 81770950) and the
Department of Science and Technology of Sichuan Province
(www.scst.gov.cn; grant no. 2018JZ0019, ,
20YSZH0011). The funders had no role in study design, data
collection and analysis or preparation of the manuscript.
Availabilty of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ, SL, KS and XJZ analyzed and interpreted the
data. XZ, XL, WT and YY performed the sample sequencing and data
analysis. XZ and XJZ wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Hospital of Sichuan Academy of Medical Sciences
and Sichuan Provincial People's Hospital (Chengdu, China; approval
no. SCPH-2017-076). All the patients, family members and controls
involved in the study signed written informed consent for the
collection of samples for sequencing.
Patient consent for publication
All the patients, family members and controls
involved in the study signed written informed consent for the
publication of patient images and data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
den Hollander AI, Black A, Bennett J and
Cremers FP: Lighting a candle in the dark: Advances in genetics and
gene therapy of recessive retinal dystrophies. J Clin Invest.
120:3042–3053. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grover S, Fishman GA and Brown J Jr:
Patterns of visual field progression in patients with retinitis
pigmentosa. Ophthalmology. 105:1069–1075. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang S, Vaccarella L, Olatunji S, Cebulla
C and Christoforidis J: Diagnostic challenges in retinitis
pigmentosa: Genotypic multiplicity and phenotypic variability. Curr
Genomics. 12:267–275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pawlyk BS, Bulgakov OV, Sun X, Adamian M,
Shu X, Smith AJ, Berson EL, Ali RR, Khani S, Wright AF, et al:
Photoreceptor rescue by an abbreviated human RPGR gene in a murine
model of X-linked retinitis pigmentosa. Gene Ther. 23:196–204.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dryja TP, Hahn LB, Kajiwara K and Berson
EL: Dominant and digenic mutations in the peripherin/RDS and ROM1
genes in retinitis pigmentosa. Invest Ophthalmol Vis Sci.
38:1972–1982. 1997.PubMed/NCBI
|
|
6
|
Holt IJ, Harding AE, Petty RK and
Morgan-Hughes JA: A new mitochondrial disease associated with
mitochondrial DNA heteroplasmy. Am J Hum Genet. 46:428–433.
1990.PubMed/NCBI
|
|
7
|
Ayuso C and Millan JM: Retinitis
pigmentosa and allied conditions today: A paradigm of translational
research. Genome Med. 2:342010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Broadgate S, Yu J, Downes SM and Halford
S: Unravelling the genetics of inherited retinal dystrophies: Past,
present and future. Prog Retin Eye Res. 59:53–96. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dan H, Huang X, Xing Y and Shen Y:
Application of targeted panel sequencing and whole exome sequencing
for 76 Chinese families with retinitis pigmentosa. Mol Genet
Genomic Med. 8:e11312020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Q, Xu M, Verriotto JD, Li Y, Wang H,
Gan L, Lam BL and Chen R: Next-generation sequencing-based
molecular diagnosis of 35 Hispanic Retinitis Pigmentosa Probands.
Sci Rep. 6:327922016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu JH, Liu JH, Ko YC, Wang CT, Chung YC,
Chu KC, Liu TT, Chao HM, Jiang YJ, Chen SJ and Chung MY:
Haploinsufficiency of RCBTB1 is associated with Coats disease and
familial exudative vitreoretinopathy. Hum Mol Genet. 25:1637–1647.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Panagiotou ES, Sanjurjo Soriano C, Poulter
JA, Lord EC, Dzulova D, Kondo H, Hiyoshi A, Chung BH, Chu YW, Lai
CHY, et al: Defects in the cell signaling mediator β-catenin cause
the retinal vascular condition FEVR. Am J Hum Genet. 100:960–968.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gong B, Zhang H, Huang L, Chen Y, Shi Y,
Tam PO, Zhu X, Huang Y, Lei B, Sundaresan P, et al: Mutant RAMP2
causes primary open-angle glaucoma via the CRLR-cAMP axis. Genet
Med. 21:2345–2354. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou Y, Li S, Huang L, Yang Y, Zhang L,
Yang M, Liu W, Ramasamy K, Jiang Z, Sundaresan P, et al: A splicing
mutation in aryl hydrocarbon receptor associated with retinitis
pigmentosa. Hum Mol Genet. 27:2563–2572. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu M, Xie YA, Abouzeid H, Gordon CT,
Fiorentino A, Sun Z, Lehman A, Osman IS, Dharmat R, Riveiro-Alvarez
R, et al: Mutations in the Spliceosome component CWC27 cause
retinal degeneration with or without additional developmental
anomalies. Am J Hum Genet. 100:592–604. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
El Shamieh S, Neuille M, Terray A, Orhan
E, Condroyer C, Démontant V, Michiels C, Antonio A, Boyard F,
Lancelot ME, et al: Whole-exome sequencing identifies KIZ as a
ciliary gene associated with autosomal-recessive rod-cone
dystrophy. Am J Hum Genet. 94:625–633. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nikopoulos K, Farinelli P, Giangreco B,
Tsika C, Royer-Bertrand B, Mbefo MK, Bedoni N, Kjellström U, El
Zaoui I, Di Gioia SA, et al: Mutations in CEP78 cause cone-rod
dystrophy and hearing loss associated with primary-cilia defects.
Am J Hum Genet. 99:770–776. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zeitz C, Jacobson SG, Hamel CP, Bujakowska
K, Neuillé M, Orhan E, Zanlonghi X, Lancelot ME, Michiels C,
Schwartz SB, et al: Whole-exome sequencing identifies LRIT3
mutations as a cause of autosomal-recessive complete congenital
stationary night blindness. Am J Hum Genet. 92:67–75. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang X, Wang H, Sun V, Tuan HF, Keser V,
Wang K, Ren H, Lopez I, Zaneveld JE, Siddiqui S, et al:
Comprehensive molecular diagnosis of 179 Leber congenital amaurosis
and juvenile retinitis pigmentosa patients by targeted next
generation sequencing. J Med Genet. 50:674–688. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li S, Yang M, Liu W, Liu Y, Zhang L, Yang
Y, Sundaresan P, Yang Z and Zhu X: Targeted Next-generation
sequencing reveals Novel RP1 mutations in autosomal recessive
retinitis pigmentosa. Genet Test Mol Biomarkers. 22:109–114. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang M, Li S, Liu W, Yang Y, Zhang L,
Zhang S, Jiang Z, Yang Z and Zhu X: Targeted Next-generation
sequencing reveals a novel Frameshift mutation in the MERTK gene in
a chinese family with retinitis pigmentosa. Genet Test Mol
Biomarkers. 22:165–169. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rosenberg T, Haim M, Hauch AM and Parving
A: The prevalence of Usher syndrome and other retinal
dystrophy-hearing impairment associations. Clin Genet. 51:314–321.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kimberling WJ, Weston MD, Möller C,
Davenport SL, Shugart YY, Priluck IA, Martini A, Milani M and Smith
RJ: Localization of Usher syndrome type II to chromosome 1q.
Genomics. 7:245–249. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lewis RA, Otterud B, Stauffer D, Lalouel
JM and Leppert M: Mapping recessive ophthalmic diseases: linkage of
the locus for Usher syndrome type II to a DNA marker on chromosome
1q. Genomics. 7:250–256. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bhattacharya G, Miller C, Kimberling WJ,
Jablonski MM and Cosgrove D: Localization and expression of
usherin: A novel basement membrane protein defective in people with
Usher's syndrome type IIa. Hear Res. 163:1–11. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eudy JD, Weston MD, Yao S, Hoover DM, Rehm
HL, Ma-Edmonds M, Yan D, Ahmad I, Cheng JJ, Ayuso C, et al:
Mutation of a gene encoding a protein with extracellular matrix
motifs in Usher syndrome type IIa. Science. 280:1753–1757. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hartong DT, Berson EL and Dryja TP:
Retinitis pigmentosa. Lancet. 368:1795–809. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McGee TL, Seyedahmadi BJ, Sweeney MO,
Dryja TP and Berson EL: Novel mutations in the long isoform of the
USH2A gene in patients with Usher syndrome type II or non-syndromic
retinitis pigmentosa. J Med Genet. 47:499–506. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rivolta C, Sweklo EA, Berson EL and Dryja
TP: Missense mutation in the USH2A gene: Association with recessive
retinitis pigmentosa without hearing loss. Am J Hum Genet.
66:1975–1978. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seyedahmadi BJ, Rivolta C, Keene JA,
Berson EL and Dryja TP: Comprehensive screening of the USH2A gene
in Usher syndrome type II and non-syndromic recessive retinitis
pigmentosa. Exp Eye Res. 79:167–173. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jiang L, Liang X, Li Y, Wang J, Zaneveld
JE, Wang H, Xu S, Wang K, Wang B, Chen R and Sui R: Comprehensive
molecular diagnosis of 67 Chinese Usher syndrome probands: High
rate of ethnicity specific mutations in Chinese USH patients.
Orphanet J Rare Dis. 10:1102015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang L, Yang Y, Li S, Tai Z, Huang L, Liu
Y, Zhu X, Di Y, Qu C, Jiang Z, et al: Whole Exome sequencing
analysis identifies mutations in LRP5 in Indian families with
familial exudative vitreoretinopathy. Genet Test Mol Biomarkers.
20:346–351. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Di Y, Huang L, Sundaresan P, Li S, Kim R,
Ballav Saikia B, Qu C, Zhu X, Zhou Y, Jiang Z, et al: Whole-exome
sequencing analysis identifies mutations in the EYS gene in
retinitis pigmentosa in the Indian population. Sci Rep.
6:194322016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dreyer B, Brox V, Tranebjaerg L, Rosenberg
T, Sadeghi AM, Möller C and Nilssen O: Spectrum of USH2A mutations
in Scandinavian patients with Usher syndrome type II. Hum Mutat.
29:4512008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu W, Dai H, Lu T, Zhang X, Dong B and Li
Y: Seven novel mutations in the long isoform of the USH2A gene in
Chinese families with nonsyndromic retinitis pigmentosa and Usher
syndrome Type II. Mol Vis. 17:1537–1552. 2011.PubMed/NCBI
|
|
37
|
Huang L, Mao Y, Yang J, Li Y, Li Y and
Yang Z: Mutation screening of the USH2A gene in retinitis
pigmentosa and USHER patients in a Han Chinese population. Eye
(Lond). 32:1608–1614. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu X, Bulgakov OV, Darrow KN, Pawlyk B,
Adamian M, Liberman MC and Li T: Usherin is required for
maintenance of retinal photoreceptors and normal development of
cochlear hair cells. Proc Natl Acad Sci USA. 104:4413–4418. 2007.
View Article : Google Scholar : PubMed/NCBI
|