Introduction
Pulmonary epithelial cells are key regulators of
lung homeostasis, providing a large surface area for gas exchange
(1). Moreover, the physical
barrier formed by the alveolar epithelium protects the body from
damage caused by inhaled external materials, such as pathogenic
microorganisms, and it also regulates the transport of water and
ions (2). Pulmonary epithelial
cells are particularly vulnerable to acute lung injury (ALI)/acute
respiratory distress syndrome (ARDS) (3). Pulmonary epithelial barrier function
is critically dependent on barrier integrity; the cells must engage
in self-repair and self-renewal, and interact with various other
cells in the context of pulmonary inflammation (4). Previous studies have revealed the
potential relationships between dynamic cytoskeletal damage and
epithelial barrier dysfunction, suggesting that cytoskeletal
re-arrangement plays an important role in the development of
lipopolysaccharide (LPS)-induced epithelial hyper permeability
(5–7). Our previous study of the pulmonary
microvascular endothelial barrier revealed that LPS modulated
endothelial barrier function by regulating the dynamics of the
cytoskeleton and intercellular junctions via the PI3K/Akt signaling
pathway, which is a skeletal system-related pathway (8).
Effective pulmonary epithelial barrier repair is
essential for preventing pathogen invasion and ALI/ARDS (1). In an ARDS animal model, inflammatory
mediators such as interleukin (IL)-1β, which triggers alveolar
edema, mediate alveolar epithelial repair via a cytokine-dependent
pathway involving epidermal growth factor and transforming growth
factor-α (9). Furthermore,
keratinocyte and hepatocyte growth factors in the pulmonary edema
fluid of patients with ALI/ARDS stimulate alveolar epithelial
repair in vitro (9).
Several cells in the pulmonary inflammatory microenvironment,
including endothelial cells and innate immune cells, also exert
specific effects on the pulmonary epithelial barrier (10). Alveolar macrophages resident in the
bronchi and alveoli respond immediately to harmful materials such
as pathogens that reach the lower respiratory tract, as do alveolar
epithelial cells (11).
Macrophages can become polarized to one of two subtypes, M1 and M2,
in several situations. M1 macrophages usually promote host defenses
against bacteria or viruses and cause tissue damage, while M2
macrophages exhibit anti-inflammatory functions and promote tissue
repair (10,12). Moreover, M2 macrophages contribute
to lung epithelial repair in patients with bacterial pneumonia,
ventilator-associated lung injury and ALI/ARDS, but the specific
mechanisms remain unknown (12).
Our previous studies revealed that T-cell immunoglobulin mucin 3
(Tim-3), an immunomodulatory molecule, plays an important role in
the mononuclear cell/macrophage system of patients with severe
sepsis (13), and that Tim-3 may
be involved in macrophage polarization (14). Therefore, the present study
investigated whether there was a correlation between Tim-3
expression by macrophages and the regulation of alveolar pulmonary
barrier function, and also whether barrier protection was induced
by activation of a signaling cascade in epithelial cells.
Materials and methods
Cell isolation and culture
Murine bone marrow-derived macrophages (BMDMs) were
generated from the femurs of male adult C57BL/6J mice [age, 6–7
weeks; wild-type; weight, 20–25 g; housed in a pathogen-free area
at a controlled temperature (20-26°C) with humidity (40-60%) and
free to access to food and water; Shanghai Model Organisms Biotech
Co., Ltd.] as described previously (14). In brief, ten mice were anesthetized
by the intraperitoneal injection of pentobarbital sodium (1%; 50
mg/kg) and sacrificed by cervical dislocation; mortality was
verified by observing the indications of breathing, heartbeat,
pupil and nerve reflex. Bone marrow was flushed from the femurs,
then bone marrow cell pellets were collected and treated with 10%
(v/v) red blood cell lysis buffer (v/v) on ice for 10 min. Cell
pellets were centrifuged at 1,500 × g at 4°C for 10 min and
resuspended in complete medium [DMEM (Hyclone; Cytiva) containing
10% FBS (cat. no. 0025; ScienCell Research Laboratories, Inc.) and
10 ng/ml macrophage colony-stimulating factor (cat. no. 315–02;
PeproTech, Inc.)]. Cells were incubated at 37°C and 5%
CO2 for 3 days, and on day 4 half of the medium was
replaced with new complete DMEM. After 7 days in culture, adherent
cells were used in subsequent experiments. All animal studies were
approved by the Animal Care and Use Committee of Zhejiang
University and conformed to the Guide for the Care and Use of
Laboratory Animals published by the US National Institutes of
Health (NIH Publication no. 85–23; revised 1996).
The mouse peritoneal macrophage cell (RAW 264.7) and
mouse lung epithelial cell (MLE-12) lines were obtained from the
American Type Culture Collection; mycoplasma testing was performed
for the cell lines. Cells were incubated in complete medium (DMEM
with 10% FBS) at 37°C and 5% CO2.
Co-culture of macrophages and
epithelial cells
Macrophages (RAW 264.7 and BMDMs) were
differentiated to the M1 or M2 subtype using methods previously
described (15). RAW 264.7 and
BMDMs were differentiated into M1-polarized or M2-polarized
macrophages by the addition of LPS (100 ng/ml; cat. no. L2880;
Sigma-Aldrich; Merck KGaA) or IL-4 and IL-13 (10 ng/ml each;
PeproTech, Inc.) at 37°C with 5% CO2 for 48 h.
Transwell inserts (pore size, 0.4 µm; cat. no. 3450;
Costar; Corning, Inc.) were used for macrophages and MLE-12
co-culture. In total, 1×106 M1 or M2 macrophages (RAW
264.7 and BMDMs) were seeded into the upper chamber with DMEM. In
the lower chamber, 2х106 MLE-12 cells were seeded and
treated with or without LPS (10 µg/ml) in complete DMEM at 37°C for
24 h. For PI3K inhibition, 25 µmol/l LY294002 (cat. no. 9901; Cell
Signaling Technology, Inc.), which had no significant effect on
untreated MLE-12 cells (Fig.
S1A-E), was added to LPS (10 µg/ml)-treated MLE-12 cells and
incubated in serum-free and growth factor-free medium at 37°C and
5% CO2 for 24 h.
Flow cytometry
Adherent BMDMs were extracted as previously
described (14). After cells were
detached and washed using PBS, 0.5 µg phycoerythrin-conjugated
anti-mouse F4/80 (cat. no. 70-AM048004; MultiSciences Biotech Co.,
Ltd.) and 0.4 µg allophycocyanin (APC)-conjugated anti-Mouse CD86
(cat. no. 70-AM08605; MultiSciences Biotech Co., Ltd.) were added
to each tube. After incubation at room temperature for 30 min,
washing with PBS, and fixing and permeabilizing for 15 min with the
FIX&PERM kit (cat. no. 70-GAS003; MultiSciences Biotech Co.,
Ltd.), BMDMs were incubated with 0.5 µg APC-conjugated anti-Mouse
CD206 (cat. no. 141707; BioLegend, Inc.) for 1 h at room
temperature. BMDMs were then washed and analyzed by flow cytometry
(BD FACS Calibur; BD Biosciences).
Immunofluorescence
RAW 264.7 and BMDMs were treated as previously
described (8). After washing with
PBS and fixing with 4% (w/v) paraformaldehyde solution at room
temperature for 20 min, cells were incubated with Tim-3 antibody
(1:500; cat. no. 60355-1-Ig; ProteinTech Group, Inc.) overnight at
4°C. After washing three times with PBS, the cells were incubated
with CoraLite594-conjugated antibody (1:200; cat. no. SA00013-3;
ProteinTech Group, Inc.) at room temperature for 1 h. Cells were
washed with PBS and incubated with DAPI (1 µg/ml; cat. no. D9564;
Sigma-Aldrich; Merck KGaA) at room temperature for 10 min. Then,
the cells were washed in PBS and observed under a fluorescent
microscope (magnification, ×400; Leica Microsystems).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells and RT to cDNA
was performed by using the RNA Extraction kit (cat. no. 9767;
Takara Bio, Inc.) and the PrimeScript RT Master Mix (cat. no.
RR036A; Takara Bio, Inc.) as previously described (14). Subsequently, qPCR was performed
using SYBR RT-qPCR Master mix (cat. no. Q311-02-AA; Vazyme Biotech
Co., Ltd.). The following thermocycling conditions were used for
qPCR: Initial denaturation at 95°C for 30 sec, followed by 40
cycles at 95°C for 5 sec and 60°C for 30 sec. mRNA expression
levels were quantified using the 2−ΔΔCq method (16). Sequences of the primers used in
this study are presented in Table
SI.
Small interfering RNA (siRNA)
transfections and Tim-3 blockade
Negative control siRNA and Tim-3-specific siRNA
(forward, CCUAACCACGGAGAGAAAUTT and reverse, AUUUCUCUCCGUGGUUAGGGT;
Gene Chemical Technology Co., Ltd.) were transfected (30 pmol) into
RAW 264.7 cells (1×105 cells) using INTERFERin (cat. no.
409-10; Polyplus-transfection SA) for 24 h before subsequent
experimentation. BMDMs (1х106) were treated with 10
µg/ml anti-mouse Tim-3-blocking monoclonal Ab (mAb; cat. no.
134002; BioLegend, Inc.) at 37°C for 48 h for function
blocking.
Transwell-Evans blue monolayer
permeability assay
Transwell inserts (pore size, 3.0-µm; cat. no. 3472;
Costar; Corning, Inc.) were used to evaluate permeability as
previously described (8). In
brief, 1×105 MLE-12 cells were seeded in inserts and
treated with LPS (10 µg/ml) at 37°C for 24 h, then co-cultured with
RAW 264.7 or BMDMs (1×106). Subsequently, 100 µl Evans
Blue (EB)-conjugated albumin (0.67 mg/ml; Sigma-Aldrich; Merck
KGaA) was added to the upper chamber as previously described
(17). To the lower chamber, 500
µl 4% BSA (Sigma-Aldrich; Merck KGaA) was added. After incubation
for 1 h at 37°C, the medium in the lower chambers was collected.
The absorbance was determined at a wavelength of 620 nm using a
micro-plate reader. The trans-epithelial cell EB-albumin leak was
calculated according to standard curve.
Wound healing assay
Based on a previous description (8), 2х106 MLE-12 cells were
seeded in 6-well plates. Confluent monolayer cells were scraped by
using a 1,000 µl pipette tip and then washed with PBS. MLE-12 cells
were treated with LPS (10 µg/ml) at 37°C for 24 h, then co-cultured
in DMEM (with 2% FBS) with RAW 264.7 or BMDMs. The images were
captured using a light microscope at 0 and 24 h at the same
position of the wound. The migratory ability was assessed by the
rate of scratch wound confluence using Adobe Photoshop 2017
software (Adobe Systems, Inc.) and the following formula: % wound
confluence=(a-b) × 100%/a, where a is the initial scratch wound
area at 0 h and b is the scratch wound area at 24 h.
Western blotting
Total protein was extracted from MLE-12 cells using
RIPA buffer (Beyotime Institute of Biotechnology) and quantified
using the BCA kit (Beyotime Institute of Biotechnology), as
previously described (14).
Protein samples (10 µg per lane) were separated via 10% SDS-PAGE
and transferred to PVDF membranes. The membranes were then blocked
with 5% (w/v) skimmed milk at room temperature for 1 h. Then, the
membranes were incubated with primary antibodies at 4°C overnight,
including phosphorylated (p)-Akt (1:2,000; cat. no. 4060; Cell
Signaling Technology, Inc.) and Akt (1:1,000; cat. no. 4685; Cell
Signaling Technology, Inc.). The membranes were incubated with a
horseradish peroxidase-conjugated secondary antibody (1:2,000; cat.
no. A0208; Beyotime Institute of Biotechnology) and developed with
an enhanced chemiluminescence kit (cat. no. 70-P1421; Multi
Sciences Biotech) and exposed to X-ray film. Protein expression was
semi-quantified using Image J software (version 1.8.0, National
Institutes of Health).
Statistical analysis
All experiments were repeated at least three times.
Data are presented as the mean ± SD. A two-tailed Student's t-test
was used for two-group comparisons and one-way ANOVA followed by
Tukey's multiple comparisons test was used for multiple-group
comparisons. GraphPad Prism 7.0 (GraphPad Software, Inc.) was used
for analysis. P<0.05 was considered to indicate a statistically
significant difference.
Results
Co-culture with macrophages affects
the migration and monolayer permeability of LPS-treated MLE-12
cells
To identify and quantify the number of M1/M2 cells
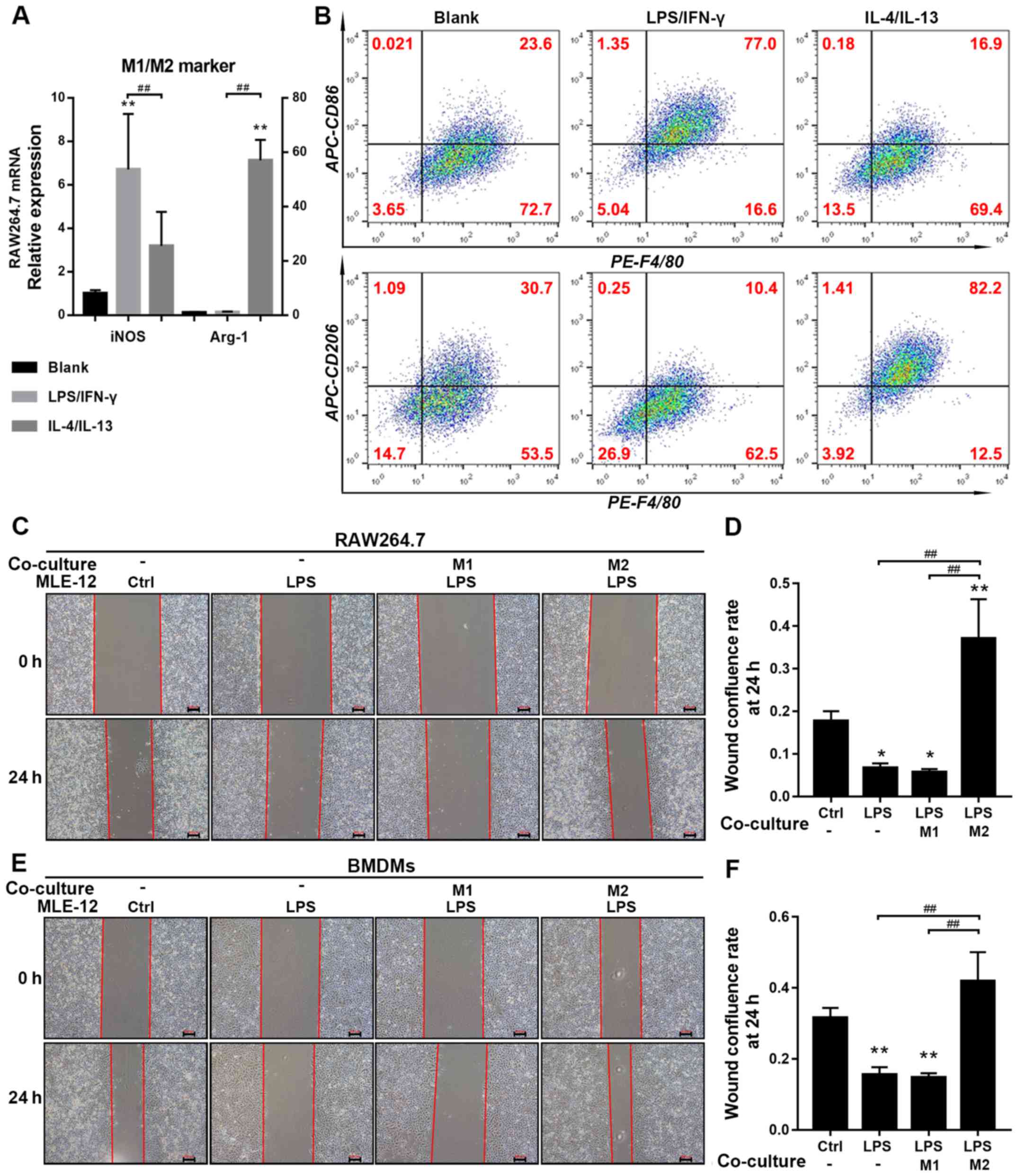
present after various standard treatments, the expression levels of
M1/M2 markers were assessed in RAW 264.7 cells and BMDMs using both
RT-qPCR and flow cytometry. The mRNA expression of the M1 marker
inducible nitric oxide synthase (iNOS) in RAW 264.7 cells and the
number of F4/80+CD86+ BMDMs were
significantly upregulated by LPS stimulation, thus indicating
successful M1 polarization. Moreover, it was revealed that mRNA
expression of the M2 marker Arginase 1 (Arg-1) in RAW 264.7 cells
and the number of F4/80+CD206+ BMDMs were
significantly upregulated by IL-4 and IL-13 stimulation, indicating
successful M2 polarization (Fig. 1A
and B). In addition, LPS-pretreated MLE-12 cells upregulated
the expression levels of M1 markers, but did not downregulate the
expression levels of M2 markers in M2 macrophages compared with
untreated M2 macrophages (Fig.
S1G).
To determine the effects of different macrophage
subtypes (RAW 264.7 and BMDMs) on MLE-12 cell migration after
treatment with LPS, migratory rates were measured using a wound
healing assay. The extent of scratch wound confluence at 24 h was
significantly decreased in the LPS group compared with the control
and M2 macrophage co-culture groups, and also significantly reduced
in the M1 macrophage co-culture group compared with the control
group (Fig. 1C-F). However, no
significant difference was demonstrated between the LPS and M1
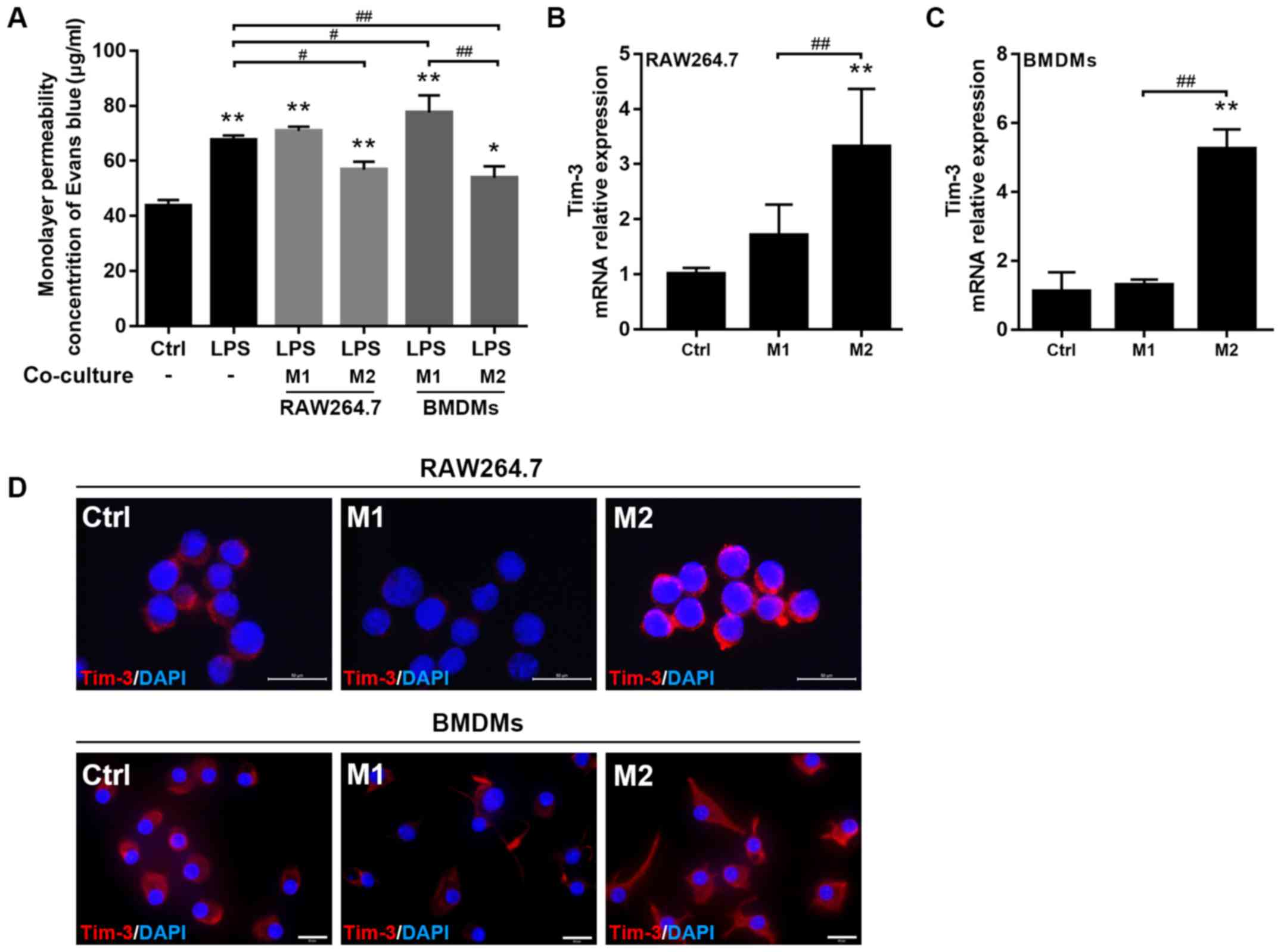
macrophage co-culture groups. The results revealed that EB leakage
from the upper chamber, reflecting monolayer permeability, was
significantly increased in the LPS group compared with the control
and M2 macrophage co-culture groups, and was also significantly
greater in the M1 macrophage (BMDMs) co-culture group compared with
the LPS group (Fig. 2A).
Tim-3 expression in different subtypes
of macrophages
To assess Tim-3 expression in different subtypes of
macrophages, RT-qPCR and immunofluorescence labeling were used to
quantify Tim-3 expression in M1/M2 macrophages. RT-qPCR results
indicated that the mRNA expression of Tim-3 was significantly
increased in M2 macrophages (RAW 264.7 and BMDMs); however, the
expression in the control and M1 macrophage groups was not
significantly different (Fig. 2B and
C). Furthermore, it was revealed that Tim-3 fluorescence
intensity exhibited a similar trend, as it was increased in M2 but
decreased in M1 macrophages (Fig.
2D).
Tim-3 blockade or knockdown in M2
macrophages abolishes the restoration of the migratory ability and
monolayer permeability in LPS-treated MLE-12 cells
To investigate whether co-culture with M2
macrophages modulated the lung epithelial barrier function mediated
by Tim-3, RAW 264.7 cells were transfected with Tim-3 siRNA, and
knockdown efficiency was ~70% (Fig.
S1F; Fig. 3A). It was
demonstrated that knockdown of endogenous Tim-3 significantly
compromised the barrier protection induced by co-culture of
M2-polarized RAW 264.7 cells (Fig.
3F). Moreover, Tim-3 siRNA significantly inhibited the
promoting effect of M2-polarized RAW 264.7 cells on MLE-12 cell
migratory ability, which was inhibited by LPS (Fig. 3B and C). It was also determined
that the extent of EB leakage from the upper chamber was
significantly increased in the M2 + Tim-3 siRNA co-culture group
compared with the M2 and M2 + negative control co-culture groups
(Fig. 3F). Similar effects were
observed when M2-polarized BMDMs were incubated with a
Tim-3-blocking mAb; the extent of MLE-12 cell confluence at 24 h
was significantly reduced in the M2 + Tim-3 mAb co-culture group
compared with the M2 co-culture group (Fig. 3D and E). The results indicated that
EB leakage from the upper chamber was significantly increased in
the M2 + Tim-3 mAb co-culture group compared with the M2 co-culture
group (Fig. 3G).
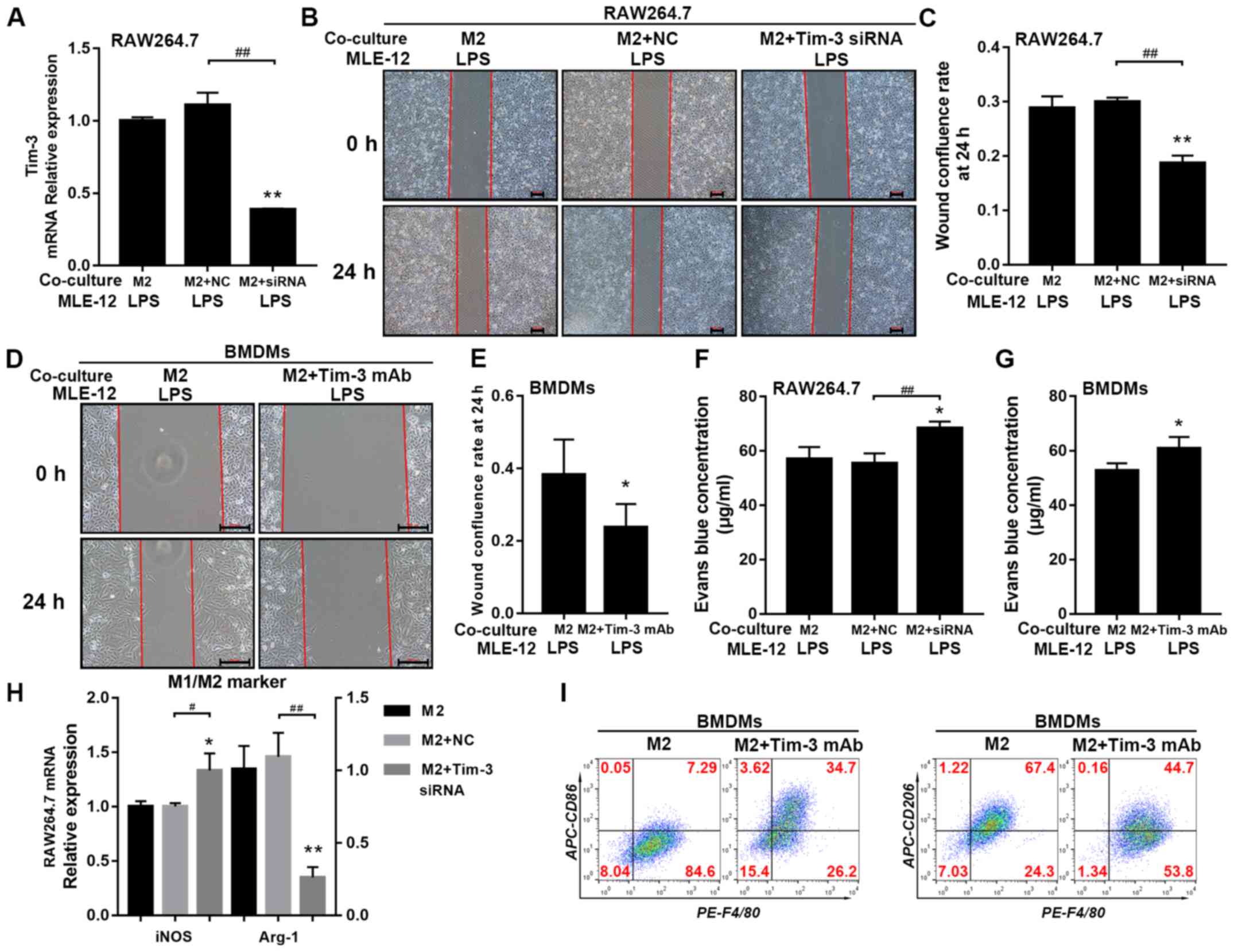 | Figure 3.Effects of Tim-3 blockade on
monolayer permeability and migration of LPS-injured MLE-12 cells
co-cultured with M2 macrophages. (A) Tim-3-siRNA knockdown
efficiency (~70%) was measured using reverse
transcription-quantitative PCR. (B) Wound healing assay was used to
assess the (C) migration of MLE-12 cells co-cultured with M2-like
RAW 264.7 cells treated with Tim-3 specific siRNA. (D) Wound
healing assay was used to assess the (E) migration of MLE-12 cells
co-cultured with M2-like BMDMs treated with Tim-3 mAb. EB leakage
from MLE-12 monolayers co-cultured with (F) Tim-3-knockdown M2-like
RAW 264.7 cells and (G) Tim-3 mAb-treated M2-like BMDMs. (H) mRNA
expression levels of the M1 marker iNOS and the M2 marker Arg-1 in
Tim-3-knockdown M2-like RAW 264.7 cells. (I) Proportions of M1
subtype (F4/80+CD86+) and M2 subtype
macrophages (F4/80+CD206+) in Tim-3
mAb-treated M2-like BMDMs. Scale bar, 200 µm. *P<0.05,
**P<0.01 vs. the M2 co-culture group; #P<0.05,
##P<0.01. EB, Evans Blue; LPS, lipopolysaccharide;
Tim-3, T-cell immunoglobulin mucin 3; BMDM, bone marrow-derived
macrophages; mAb, monoclonal antibody; iNOS, inducible nitric oxide
synthase; Arg-1, Arginase 1; siRNa, small interfering RNA; NC,
negative control. |
Next, the present study assessed M1/M2 marker
expression levels in M2 macrophages after Tim-3 signaling was
reduced or blocked. mRNA expression of the M1 marker iNOS in RAW
264.7 cells and the number of F4/80+CD86+
BMDMs were significantly upregulated in the presence of Tim-3 siRNA
or Tim-3 mAb. However, the mRNA expression of the M2 marker Arg-1
in RAW 264.7 cells and the number of
F4/80+CD206+ BMDMs were significantly
downregulated by Tim-3 siRNA or Tim-3 mAb (Fig. 3H and I).
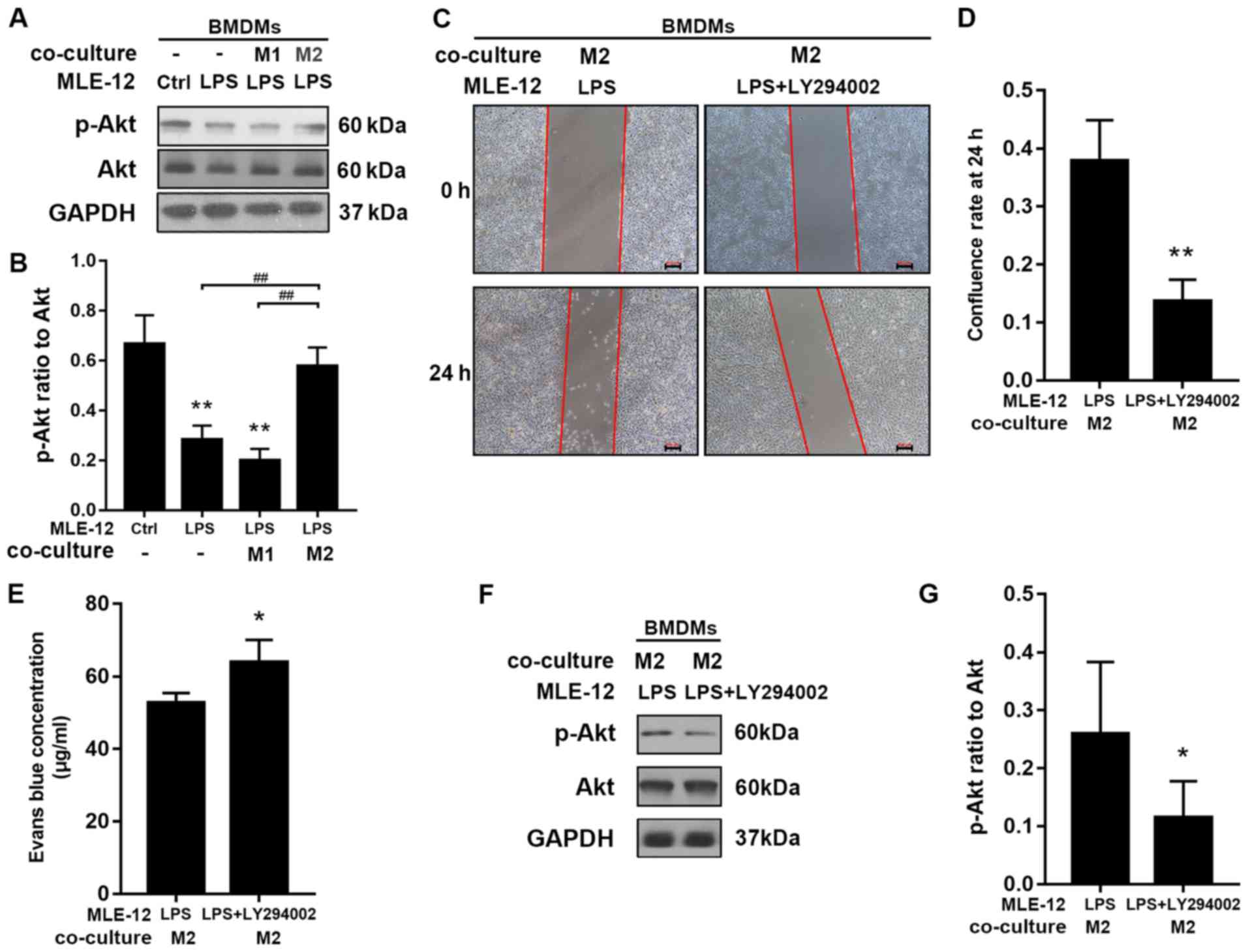
M2 macrophages restore epithelial
barrier via the PI3k/Akt signaling pathway in epithelial cells
Western blotting was used to assess the ratio of
p-Akt to total Akt expression. The ratio of p-Akt to total Akt was
significantly higher in MLE-12 cells co-cultured with M2
macrophages (BMDMs) compared with cells exposed to LPS alone and
cells co-cultured with M1 macrophages (Fig. 4A and B). The present study also
examined whether the PI3K/Akt pathway in MLE-12 cells was involved
in the barrier protection provided by co-culture with M2
macrophages. LY294002 (25 µM), a PI3K inhibitor, was used to assess
whether inhibition of the PI3K/Akt pathway compromised the
protective effect. It was determined that LY294002 inhibited the
restoration of MLE-12 cell migration and monolayer permeability
after co-culture with M2 macrophages (Fig. 4C-E). Furthermore, LY294002 reduced
p-Akt protein expression (Fig. 4F and
G).
Discussion
The respiratory system is often exposed to harmful
substances from the airway or bloodstream (10). Moreover, a variety of lung cells
are involved in the maintenance of pulmonary homeostasis; alveolar
macrophages, epithelial cells and other cells play important roles
(4,12). Host-pathogen interactions can
injure pulmonary epithelial cells and trigger barrier dysfunction;
thus, the alveolar space is no longer protected against fluid entry
from adjacent vascular compartments (10). The resulting alveolar edema is
termed ALI/ARDS, which is a syndrome characterized by intractable
hypoxemia, respiratory distress, progressive respiratory failure
and high mortality and morbidity rates (18). LPS, also known as endotoxins, is a
pro-inflammatory component of the outer membrane of Gram-negative
bacteria and plays an important role in infection (10). LPS is the principal pathogenic
component of airway microorganisms that can cross the epithelial
barrier to trigger sepsis-related barrier dysfunction, where it
activates innate immune cells, triggers local and systemic
inflammatory responses and disrupts barrier function; LPS is
typically used to induce ALI/ARDS in animal models (8,16).
The present study used LPS to stimulate mouse lung epithelial cells
to simulate the damage induced in the pulmonary epithelial barrier
under inflammatory conditions. It was revealed that epithelial
barrier permeability increased and cell migration decreased after
LPS treatment.
Previous studies have revealed that pulmonary
epithelial cells are involved in the maintenance of lung
homeostasis, as epithelial cell secretions contain enzymes such as
lysozyme that damage bacteria (19,20).
Furthermore, it has been revealed that pulmonary epithelial cells
maintain humoral and ionic homeostasis by regulating the
cytoskeleton, cell junctions and ion channels (4,21).
Destruction of the epithelial barrier can also trigger recurrent
infections and ALI (1). It has
been revealed that pulmonary epithelial barrier repair is useful
for treating lung injury (22) and
that certain cytokines may play important roles in injury repair,
and thus are attracting increasing attention (9,23).
Numerous cellular interactions are involved in the alveolar
inflammatory microenvironment; alveolar macrophages play important
roles in pathogen recognition and recruitment of immune cells to
the alveoli (4). Different
macrophage subtypes inhibit inflammation via multiple pathways,
including switching of M1 macrophages to the M2 subtype, induction
of efferocytosis to clear neutrophils and secretion of
anti-inflammatory mediators (4,12),
thus enhancing damage repair. A previous study revealed that
adaptive transfer of M2 macrophages promotes lung injury repair and
reduces pulmonary inflammation in mice; in contrast, M2 macrophages
aggravate lung injury (24). In
line with these previous results, the present results indicated
that co-culture of epithelial cells with M1 macrophages aggravated
the epithelial barrier permeability induced by LPS, whereas
co-culture with M2 macrophages significantly attenuated LPS-induced
epithelial leakage and upregulated the migration of epithelial
cells. Therefore, the present study identified the important roles
played by different macrophage subtypes in the induction and repair
of pulmonary epithelial damage.
Tim-3 is an immunomodulatory molecule that is highly
expressed in cytotoxic T and helper T1 (Th1) cells, which engages
in inhibitory signaling that triggers apoptosis of cytotoxic T and
helper T1 cells (25). Tim-3 is
also expressed in monocytes, macrophages and dendritic cells, where
it plays an important role in immune regulation (26). Furthermore, Tim-3 has been revealed
to regulate the functions of monocytes and macrophages in both
humans and mice; a previous study based on a sepsis model revealed
that Tim-3 signaling reduced the release of inflammatory factors,
inhibited macrophage function and provided pulmonary protection
(27). Furthermore, it has been
revealed that blockade of Tim-3 signaling increases sepsis severity
and significantly reduces mouse survival (28). Our previous study using clinical
samples revealed that Tim-3 expression was increased in T
lymphocytes, but decreased in mononuclear cells and macrophages in
patients with sepsis (13). In
addition, upregulation of Tim-3 expression in T cells may be
associated with T-cell apoptosis and functional depletion, but the
function of Tim-3 in monocytes is not fully understood (13). Therefore, the present study
investigated whether Tim-3 expression in M2 macrophages affected
epithelial barrier function in a macrophage-epithelial cell
co-culture model. It was demonstrated that Tim-3 was weakly
expressed in M1 macrophages, but highly expressed in M2
macrophages; it was also determined that interference or blockade
of Tim-3 in M2 macrophages compromised repair of the lung
epithelial barrier, which was associated with macrophage
polarization. Therefore, it was speculated that Tim-3 may be
involved in macrophage M1/M2 switching.
It is important to identify regulatory molecules
expressed in epithelial cells that are associated with macrophages.
The PI3K/Akt pathway not only regulates various cellular functions,
but also has roles in the signaling underlying epithelial barrier
function during sepsis and ALI/ARDS (29,30).
The present study identified activated PI3K/Akt signaling in MLE-12
cells co-cultured with M2 macrophages; however, the pathway was
inhibited by LPS. The PI3K inhibitor LY294002 was used in the
present study to examine whether the PI3K/Akt pathway mediates M2
macrophage-induced barrier protection. It was demonstrated that
inhibition of PI3K/Akt signaling suppressed cell migration and
barrier restoration induced by M2 macrophage co-culture, suggesting
that Tim-3 regulates the function of macrophages, which indirectly
regulates PI3K/Akt activity in epithelial cells rather than
macrophages, thus affecting the barrier function of epithelial
cells. Based on the Transwell assay results, cell-cell direct
contact may not be required for barrier protection by M2
macrophage; however, some M1/M2-produced cytokines or chemokines
may be involved in the process. It has been reported that
epithelial and innate immune cell interactions mediate wound
healing and tissue restoration (31). Moreover, cytokines such as IL-10
(32) and extracellular vesicles
(33) produced by macrophages
participate in epithelial wound repair, and thus future studies
will investigate whether Tim-3 mediates these processes in
macrophages. In addition, MLE-12 in wound healing assay should be
serum-starved, however, due to its intolerance to serum-free
medium, 2% FBS was used, which was a limitation of this study.
In conclusion, it was revealed that macrophage Tim-3
affects macrophage polarization. Furthermore, the present results
indicated that M1 macrophages promoted inflammation and aggravated
pulmonary epithelial barrier dysfunction. However, M2 macrophages
attenuated inflammation, thus promoting repair of the lung
epithelial barrier after injury, and this function was speculated
to be mediated by PI3K/Akt signaling in epithelial cells. Given the
protective role of M2 macrophages, control of macrophage phenotype
rather than complete macrophage depletion may be a potential
therapeutic approach to treat injury repair. Furthermore, the
present results suggested that Tim-3 may be a valuable therapeutic
target.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by Zhejiang
Provincial Natural Science Foundation (grant no. LY16H150003).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ and WZ wrote the main manuscript text and
performed the experiments. YZ also analyzed the data.
Ethics approval and consent to
participate
All animal studies were approved by the Animal Care
and Use Committee of Zhejiang University and conformed to the Guide
for the Care and Use of Laboratory Animals published by the US
National Institutes of Health (NIH Publication no. 85-23; revised
1996).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Brune K, Frank J, Schwingshackl A, Finigan
J and Sidhaye V: Pulmonary epithelial barrier function: Some new
players and mechanisms. Am J Physiol Lung Cell Mol Physiol.
308:L731–L745. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gunther J and Seyfert HM: The first line
of defence: Insights into mechanisms and relevance of phagocytosis
in epithelial cells. Semin Immunopathol. 40:555–565. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guillot L, Nathan N, Tabary O, Thouvenin
G, Le Rouzic P, Corvol H, Amselem S and Clement A: Alveolar
epithelial cells: Master regulators of lung homeostasis. Int J
Biochem Cell Biol. 45:2568–2573. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bhattacharya JK: Westphalen,
Macrophage-epithelial interactions in pulmonary alveoli. Semin
Immunopathol. 38:461–469. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rodgers LS and Fanning AS: Regulation of
epithelial permeability by the actin cytoskeleton. Cytoskeleton
(Hoboken, N.J.). 68:653–660. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Herold S, Gabrielli NM and Vadasz I: Novel
concepts of acute lung injury and alveolar-capillary barrier
dysfunction. Am J Physiol Lung Cell Mol Physiol. 305:L665–L681.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang W, Weng J, Yu L, Huang Q, Jiang Y and
Guo X: Role of TLR4-p38 MAPK-Hsp27 signal pathway in LPS-induced
pulmonary epithelial hyperpermeability. BMC Pulm Med. 18:1782018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zheng X, Zhang W and Hu X: Different
concentrations of lipopolysaccharide regulate barrier function
through the PI3K/Akt signalling pathway in human pulmonary
microvascular endothelial cells. Sci Rep. 8:99632018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Geiser T: Mechanisms of alveolar
epithelial repair in acute lung injury-a translational approach.
Swiss Med Wkly. 133:586–590. 2003.PubMed/NCBI
|
|
10
|
Nova Z, Skovierova H and Calkovska A:
Alveolar-capillary membrane-related pulmonary cells as a target in
endotoxin-induced acute lung injury. Int J Mol Sci. 20(pii):
E8312019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Reynier F, de Vos AF, Hoogerwerf JJ,
Bresser P, van der Zee JS, Paye M, Pachot A, Mougin B and van der
Poll T: Gene expression profiles in alveolar macrophages induced by
lipopolysaccharide in humans. Mol Med. 18:1303–1311. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aggarwal NR, King LS and D'Alessio FR:
Diverse macrophage populations mediate acute lung inflammation and
resolution. Am J Physiol Lung Cell Mol Physiol. 306:L709–L275.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xia Q, Wei L and Zhang Y, Sheng J, Wu W
and Zhang Y: Immune checkpoint receptors Tim-3 and PD-1 regulate
monocyte and T Lymphocyte function in septic patients. Mediators
Inflamm. 2018:16329022018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang W, Zhang Y, He Y, Wang X and Fang Q:
Lipopolysaccharide mediates time-dependent macrophage M1/M2
polarization through the Tim-3/Galectin-9 signalling pathway. Exp
Cell Res. 376:124–132. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hunter MM, Wang A, Parhar KS, Johnston MJ,
Van Rooijen N, Beck PL and McKay DM: In vitro-derived alternatively
activated macrophages reduce colonic inflammation in mice.
Gastroenterology. 138:1395–1405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Patterson CE, Rhoades RA and Garcia JG:
Evans blue dye as a marker of albumin clearance in cultured
endothelial monolayer and isolated lung. J Appl Physiol (1985).
72:865–873. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ellison RT III and Giehl TJ: Killing of
gram-negative bacteria by lactoferrin and lysozyme. J Clin Invest.
88:1080–1091. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ibrahim HR, Aoki T and Pellegrini A:
Strategies for new antimicrobial proteins and peptides: Lysozyme
and aprotinin as model molecules. Curr Pharm Des. 8:671–693. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hamacher J, Hadizamani Y, Borgmann M,
Mohaupt M, Männel DN, Moehrlen U, Lucas R and Stammberger U:
Cytokine-Ion channel interactions in pulmonary inflammation. Front
Immunol. 8:16442017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ito Y, Correll K, Schiel JA, Finigan JH,
Prekeris R and Mason RJ: Lung fibroblasts accelerate wound closure
in human alveolar epithelial cells through hepatocyte growth
factor/c-Met signaling. Am J Physiol Lung Cell Mol Physiol.
307:L94–L105. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lindsay CD: Novel therapeutic strategies
for acute lung injury induced by lung damaging agents: The
potential role of growth factors as treatment options. Hum Exp
Toxicol. 30:701–724. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Du Z, Zhang S, Lin Y, Zhou L, Wang Y, Yan
G, Zhang M, Wang M, Li J, Tong Q, et al: Momordicoside G Regulates
macrophage phenotypes to stimulate efficient repair of lung injury
and prevent urethane-induced lung carcinoma lesions. Front
Pharmacol. 10:3212019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Monney L, Sabatos CA, Gaglia JL, Ryu A,
Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel
RA, et al: Th1-specific cell surface protein Tim-3 regulates
macrophage activation and severity of an autoimmune disease.
Nature. 415:536–541. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sabatos CA, Chakravarti S, Cha E, Schubart
A, Sánchez-Fueyo A, Zheng XX, Coyle AJ, Strom TB, Freeman GJ and
Kuchroo VK: Interaction of Tim-3 and Tim-3 ligand regulates T
helper type 1 responses and induction of peripheral tolerance. Nat
Immunol. 4:1102–1110. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu X, Jiang S, Zhang Q, Xu S, Bao X, Cao
W, Bai J and Tang L: Tim-3 regulates Tregs' ability to resolve the
inflammation and proliferation of acute lung injury by modulating
macrophages polarization. Shock. 50:455–464. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang X, Jiang X, Chen G, Xiao Y, Geng S,
Kang C, Zhou T, Li Y, Guo X, Xiao H, et al: T cell Ig mucin-3
promotes homeostasis of sepsis by negatively regulating the TLR
response. J Immunol. 190:2068–2079. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu X, Li H, Gong Y, Zheng H and Zhao D:
Hydrogen sulfide ameliorated lipopolysaccharide-induced acute lung
injury by inhibiting autophagy through PI3K/Akt/mTOR pathway in
mice. Biochem Biophys Res Commun. 507:514–518. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu XT, Ansari AR, Pang XX, Li HZ, Zhang
ZW, Luo Y, Arshad M and Song H: Visfatin plays a significant role
in alleviating lipopolysaccharide-induced apoptosis and autophagy
through PI3K/AKT signaling pathway during acute lung injury in
mice. Arch Immunol Ther Exp (Warsz). 67:249–261. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brazil JC, Quiros M, Nusrat A and Parkos
CA: Innate immune cell-epithelial crosstalk during wound repair. J
Clin Invest. 129:2983–2993. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saraiva M, Vieira P and O'Garra A: Biology
and therapeutic potential of interleukin-10. J Exp Med. 217(pii):
e201904182019.
|
|
33
|
Kim SY, Joglekar MV, Hardikar AA, Phan TH,
Khanal D, Tharkar P, Limantoro C, Johnson J, Kalionis B and
Chrzanowski W: Placenta Stem/Stromal cell-derived extracellular
vesicles for potential use in lung repair. Proteomics.
19:e18001662019. View Article : Google Scholar : PubMed/NCBI
|