Introduction
Carcinoma of the uterine cervix is a severe
malignant neoplasm that affects women globally (1). The mortality rate resulting from
cervical cancer remains high, and is second only to the mortality
rate of breast cancer (2).
Cervical cancer is a complex disease, and in addition to infection
with the well-studied human papillomavirus (HPV), many other
factors contribute to the tumorigenesis and progression of
neoplasms. Aberrant genes, multiparity, and smoking may exert a
significant role in the risk of cervical cancer (3). Therefore, novel therapeutic and
prognostic targets for cervical cancer are urgently needed.
There is increasing evidence that long non-coding
RNAs (lncRNAs) and mRNAs fulfill key roles in tumorigenesis and
tumor prognosis. Both lncRNAs and mRNAs may act as microRNA (miRNA)
sponges that bind to miRNA response elements (MREs) to regulate
gene expression (4). However, the
function of lncRNAs has yet to be adequately elucidated. Increasing
evidence has suggested that lncRNAs participate in various
biological processes, including cell differentiation, invasion,
migration, and the cell cycle (5).
The carcinogenic mechanisms of lncRNAs at the molecular level, as
well as their prognostic potential, are still largely unknown in
cervical cancer.
MiRNAs belong to the category of short non-coding
RNAs, which may be either upregulated or downregulated in malignant
tumors and may regulate the expression of protein-coding RNAs via
direct binding to these sequences. Similarly to lncRNAs and mRNAs,
miRNAs notably influence tumor growth, invasion, and migration by
acting as either oncogenes or tumor suppressor genes, and therefore
may influence disease outcome in patients with cancer (6).
Thus, a large-scale analysis of lncRNA-miRNA-mRNA
interactions as a competitive endogenous RNA (ceRNA) network is
essential for the comprehensive understanding of the oncogenesis
and prognosis of cervical cancer. To date, however, few studies
have been published that aimed to elucidate the association among
mRNAs, lncRNAs, and miRNAs through a ceRNA network in cervical
cancer.
The Cancer Genome Atlas (TCGA) is a public database
that provides access to genomic data from various types of cancer
(7). In the present study,
RNA-sequencing (RNA-seq) profiles were retrieved from TCGA to
analyze genes that are dysregulated in cervical cancer. The
relationship between aberrant RNA expression profiles and overall
survival was also analyzed. Moreover, Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) analyses were conducted to
annotate the DEmRNAs.
A total of 255 mRNAs, 15 miRNAs, and 12 lncRNAs were
identified as prognostic biomarkers for patients with cervical
cancer. Importantly, a miRNA-mediated lncRNA-miRNA-mRNA ceRNA
network was constructed and, to the best of the authors' knowledge
for the first time, the lncRNA EPB41L4A-AS1 was confirmed as a
pivotal regulator in cervical carcinoma through bioinformatics
analysis. Taken together, our results suggest this novel lncRNA is
a potential diagnostic biomarker, contributing to our knowledge of
the interactions between mRNAs and non-coding RNAs in cervical
cancer.
Materials and methods
Materials and data
A total of 307 cases were retrieved from TCGA
database, among which 304 were cervical squamous cell carcinoma and
endocervical adenocarcinoma (CESC) primary tumor tissues, and three
were solid normal tissues. RNA expression data, as well as clinical
information, were obtained from TCGA website (Date of access for
database: 27th August 2018). Note that medical ethics committee
approval was not required for the present study.
Differential gene expression
analysis
Linear Models for Microarray Data (Limma) (8) was used to identify DE genes (DEGs),
comparing between primary tumor tissues and solid normal tissues.
The ggplot2 (http://ggplot.yhathq.com/) package in R software was
employed to generate a volcano plot and to perform hierarchical
clustering analysis to distinguish statistically significant DEGs.
The selection criteria were P<0.05 and |fold change|>2 for
DEGs.
Functional enrichment analysis
R package clusterProfiler (9) was used to conduct functional
enrichment analysis, and significant enrichment in GO and KEGG
pathways (P<0.01 and q<0.01) were identified. P-values were
adjusted with the Benjamini-Hochberg method. A selection of DEGs
that were significantly enriched in cell cycle pathways were
displayed in a signal pathway diagram.
Univariate survival analysis
Kaplan-Meier (KM) analysis based on the survival
package (https://www.rdocumentation.org/packages/survival)
was used for univariate survival analysis. The KM analysis divides
patients into upregulated and downregulated groups according to
median expression levels.
Analysis of ceRNAs network
Three criteria were used to determine the competing
endogenous interactions between lncRNA-mRNA pairs: i) the lncRNA
and mRNA must share a significant number of miRNAs. A
hypergeometric test was used to determine whether a lncRNA and mRNA
shared a significant number of miRNAs. A newly developed algorithm
spongeScan (10) was employed to
predict MREs of lncRNAs. StarBase v.2.0 (11) was used to retrieve predicted and
experimentally confirmed miRNA-mRNA and/or miRNA-lncRNA
interactions. ii) Expression of lncRNA and mRNA must be positively
correlated. There have been a number of reports of miRNAs acting as
negative regulators of gene expression. If a greater number of
miRNAs are occupied by lncRNAs, fewer of these miRNAs will bind to
the target mRNA, thus increasing the expression level of mRNA.
Therefore, expression of lncRNAs and mRNAs in a ceRNA pair should
be positively correlated. iii) MiRNAs that share similar sequences
should serve similar roles in regulating the expression of lncRNAs
and mRNAs.
Two methods were used to determine the regulatory
role of miRNAs in the lncRNAs and mRNAs:
Regulation similarity
A similarity score was defined to analyze the
similarity between miRNA-lncRNA expression correlation and
miRNA-mRNA expression correlation. The following formula was
used:
Regulation similarity score=1-1m∑k=1M[|corr(mk,l)-corr(mk,g)||corr(mk,l)+corr(mk,g)|]M
Where M indicates the total number of shared
microRNAs, k stands for the kth shared miRNA, and
corr(mk, l) and corr(mk, g) represent the Pearson
correlation between the kth miRNA and lncRNA and the kth miRNA and
mRNA, respectively.
Sensitivity correlation
Sensitivity correlation, previously defined by Paci
et al (12), was used to
examine whether the interaction between mRNA and lncRNA is mediated
by miRNA in the lncRNA-miRNA-mRNA triplet. The average of all
triplets of a lncRNA-mRNA pair and their shared miRNAs was
considered as the sensitivity relevance between a selected mRNA and
lncRNA:
Sensitivity correlation=corr(l,g)=1M∑k=1Mcorr(l,g)-corr(mk,l)corr(mk,g)1-corr(mk,l)21-corr(mk,g)2
Where M indicates the total number of shared
miRNAs, k stands for the kth shared miRNA,
corr(l, g), corr(mk, l), and corr(mk, g) represent
the Pearson correlation among the lncRNA and the protein coding
gene, the kth miRNA and lncRNA, and the kth
miRNA and mRNA, respectively. The network was further filtered by
hyperP-value<0.05 and corP-value<0.05. The ceRNA network was
visualized with Cytoscape (v.3.6.1) (13). The Cytoscape plug-in called
‘cytoHubba’ (14) was used to
explore hub nodes. The maximal clique centrality (MCC) algorithm of
cytoHubba was used to identify hub nodes.
Results
Identification of DEmRNAs and
DEmiRNAs
RNA expression profiles of 304 CESC and three solid
normal tissues were obtained from TCGA database. DE analysis of RNA
expression levels in CESC tissues compared with those of normal
samples was performed. In total, 2,255 DEmRNAs and 133 DEmiRNAs
were confirmed with cut-off values of P<0.05 and |FC|>2.
There were 796 DEmRNAs that were significantly upregulated, and
1,459 DEmRNAs that were strongly downregulated. Moreover, 80
DEmiRNAs were upregulated, and 53 were downregulated based on the
differential expression profiles. FOXA1, PITX1, OTX1, SFN, and NMU
were the top five upregulated mRNAs, whereas DES, ACTG2, CNN1,
MYH11, and PTGIS were the top five downregulated mRNAs.
Additionally, the top five most highly expressed miRNAs were
miR-205-5p, miR-141-3p, miR-141-5p, miR-203b-3p, and miR-200c-5p,
whereas miR-204-5p, miR-133a-3p, miR-1-3p, miR-145-5p, and
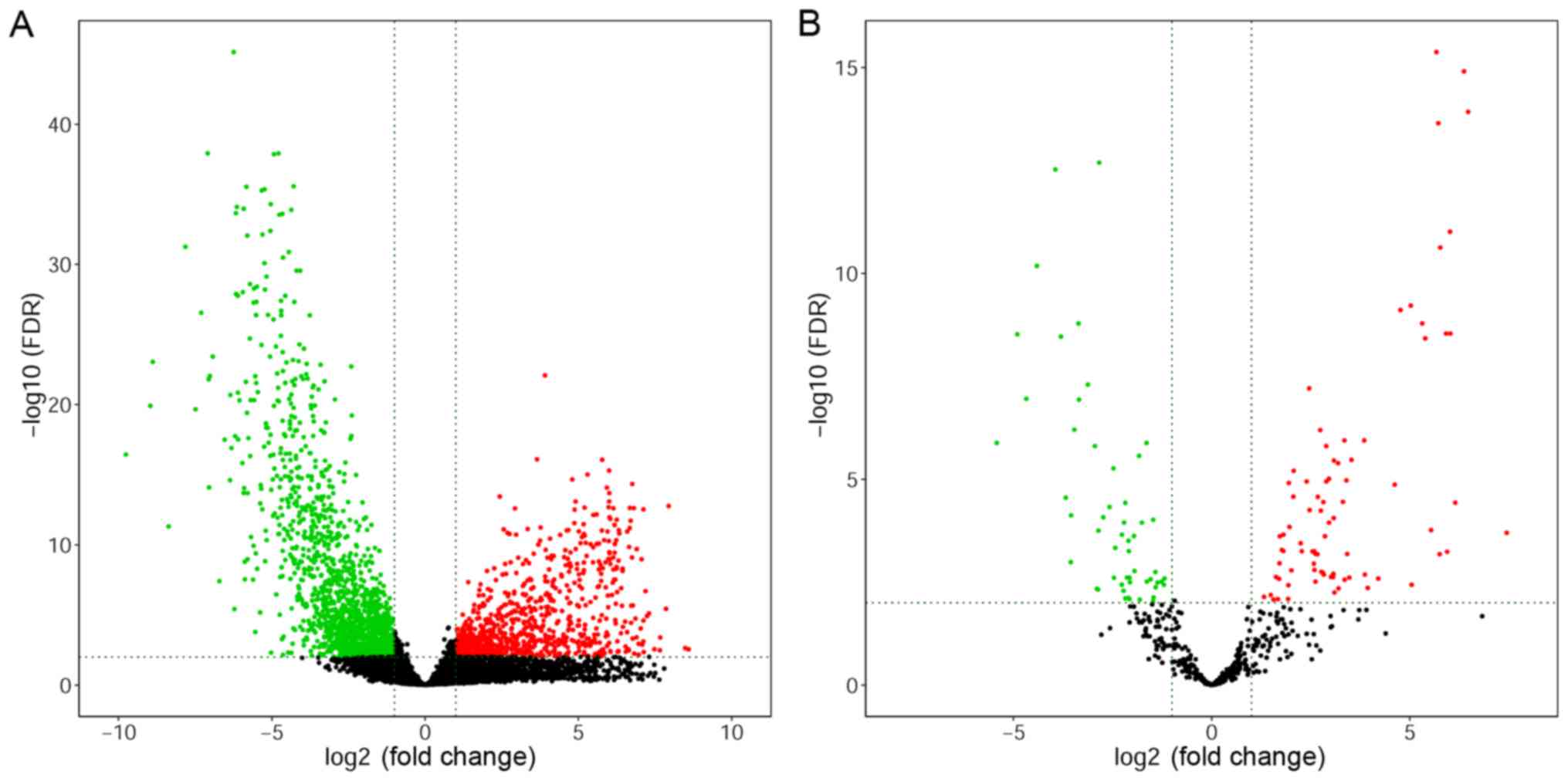
miR-10b-5p were the top five most downregulated miRNAs. Volcano
plots were subsequently generated to identify the DEGs with
statistically significant differences, comparing between primary
tumors and normal tissues (Fig.
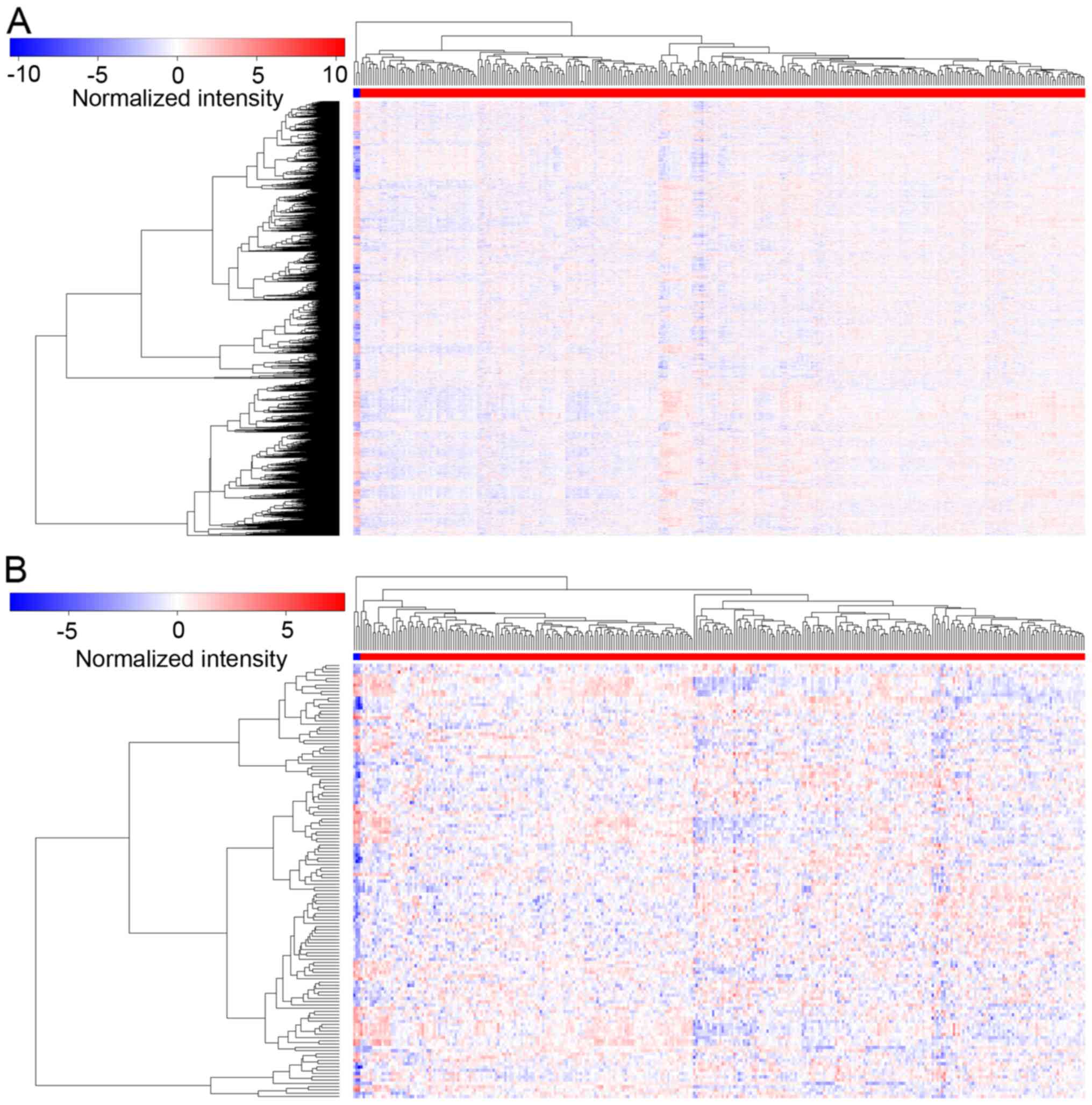
1). Heat maps of hierarchical clustering analysis were also
generated for the DEGs (Fig.
2).
Differentially expressed lncRNAs
(DElncRNAs) and survival analysis in CESC
A total of 150 DElncRNAs were identified to have
dysregulated expression profiles in CESC tissues compared with
those in normal samples. Among them, 60 DElncRNAs were upregulated
and 90 DElncRNAs were significantly downregulated. The top 10
DElncRNAs are presented in Table
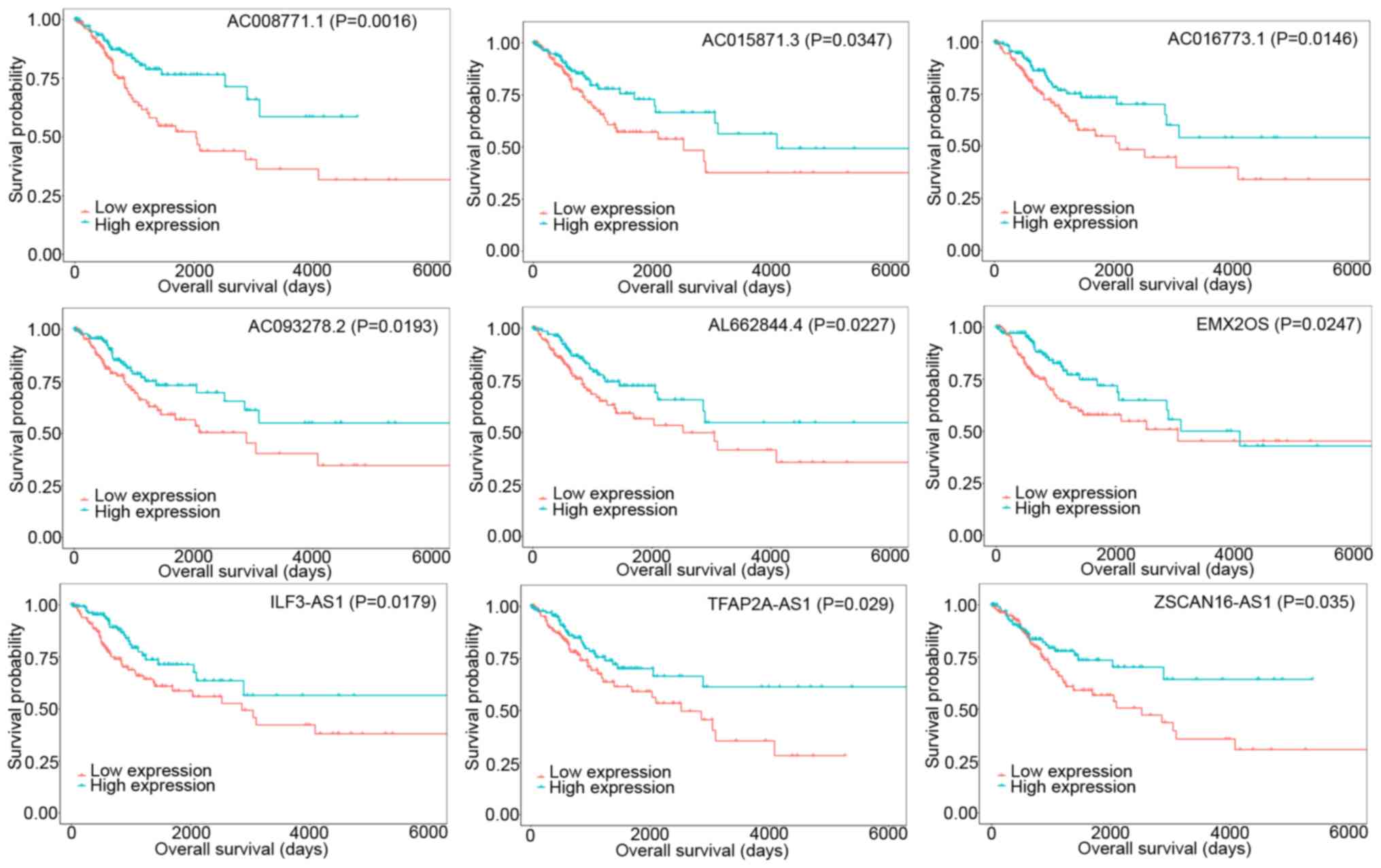
I. Subsequently, the association between DElncRNAs and the
prognosis of patients with CESC using KM analysis was studied. This
analysis revealed that 12 DElncRNAs were clearly related to overall
survival. The overall survival was positively associated with 9
DElncRNAs (AC008771.1, AC015871.3, AC016773.1, AC093278.2,
AL662844.4, EMX2OS, ILF3-AS1, TFAP2A-AS1, and ZSCAN16-AS1)
(Fig. 3), and negatively
associated with 3 DElncRNAs (AC019069.1, BAIAP2-AS1, and
CASC15).
 | Table I.Top 10 DElncRNAs in CESC tissues
compared with normal samples. |
Table I.
Top 10 DElncRNAs in CESC tissues
compared with normal samples.
| lncRNA | Expression
change | Log2 FC | P-value | FDR |
|---|
| EMX2OS | Downregulation | −6.54 |
2.93×10−20 |
3.19×10−18 |
| MAGI2-AS3 | Downregulation | −4.70 |
1.27×10−29 |
4.28×10−27 |
| MIR100HG | Downregulation | −4.36 |
6.34×10−16 |
4.17×10−14 |
| DIO3OS | Downregulation | −4.17 |
2.50×10−08 |
4.99×10−07 |
| DNM3OS | Downregulation | −3.91 |
3.65×10−15 |
2.16×10−13 |
| LINC01451 | Upregulation | 6.04 |
5.47×10−4 |
3.81×10−3 |
| FAM83A-AS1 | Upregulation | 6.28 |
6.38×10−4 |
4.32×10−3 |
| C5orf66-AS1 | Upregulation | 6.62 |
3.89×10−4 |
2.83×10−3 |
| AC245041.1 | Upregulation | 6.82 |
9.30×10−4 |
5.88×10−3 |
| AL049555.1 | Upregulation | 7.47 |
3.93×10−4 |
2.86×10−3 |
GO and KEGG pathway annotation of
DEmRNAs
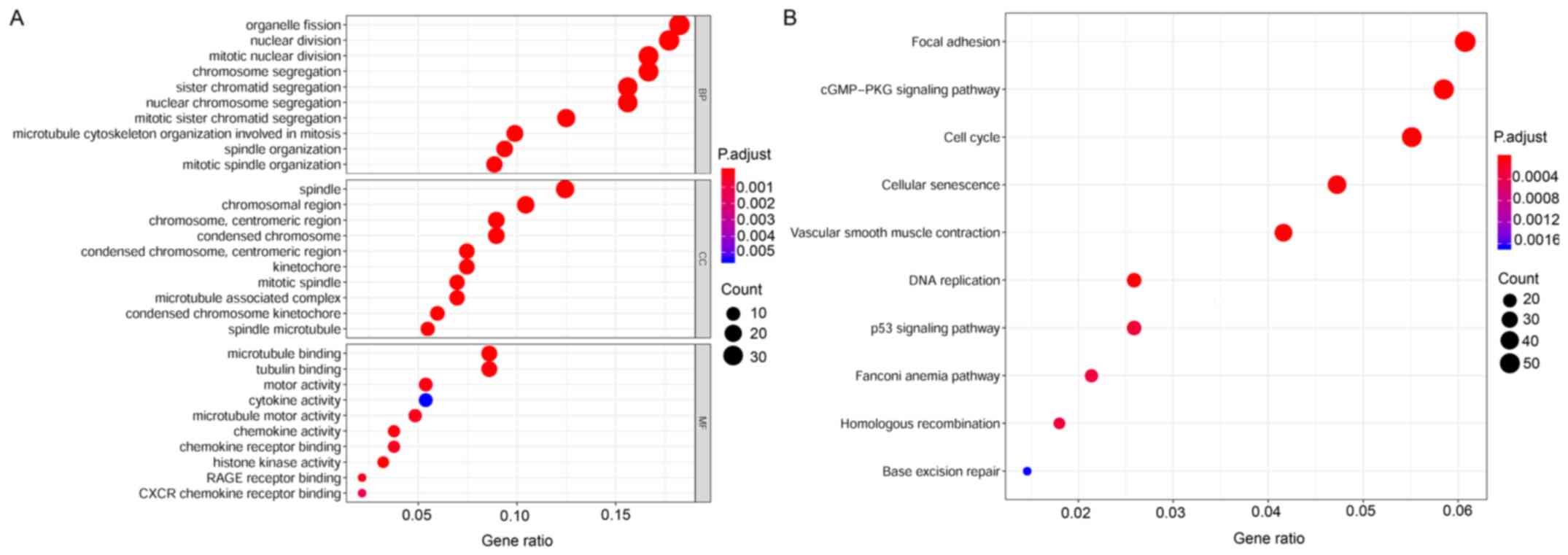
To explore the potential functions of aberrantly
expressed mRNAs that may be associated with the tumorigenesis and
development of CESC, GO and KEGG pathway analyses were conducted
using the R package clusterProfiler. The annotations of enriched GO
terms were related to cell division, cell proliferation, cellular
protein metabolic process, and cell differentiation. The top 30
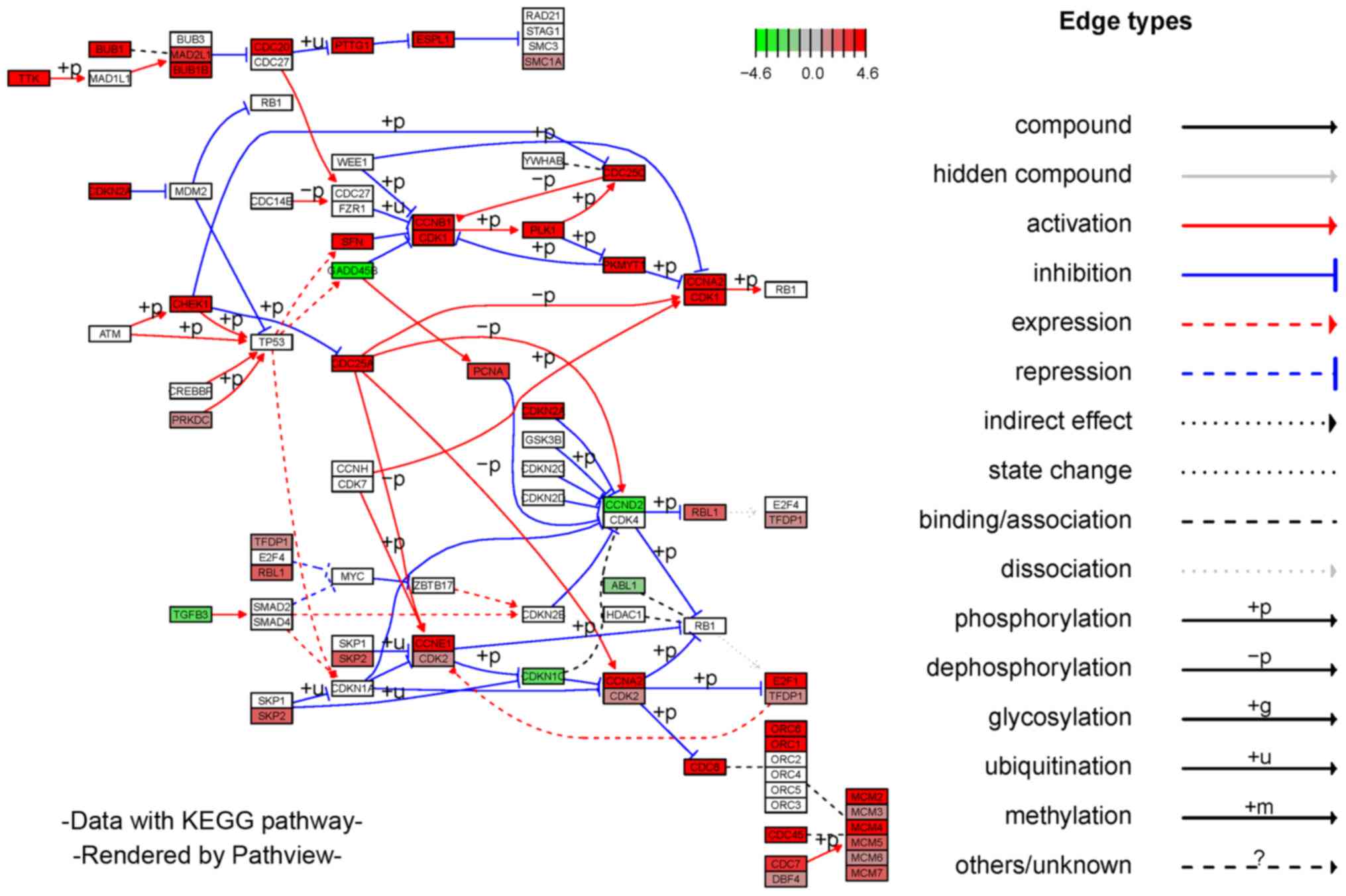
significantly enriched GO terms are shown in Fig. 4A. KEGG enrichment indicated that
the DEmRNAs were remarkably well-enriched in tumor-associated
pathways; including focal adhesion, cell cycle, cellular
senescence, DNA replication, p53 signaling pathway, Fanconi anemia
pathway, homologous recombination, and base excision repair. The
top 10 enriched pathways are shown in Fig. 4B. It was noted that DEmRNAs
enriched in the biological processes of the top 10 GO terms were
mainly associated with the cell cycle in KEGG pathways. Therefore,
the DEmRNAs of CESC are depicted in the signal pathway diagram of
the cell cycle. A plot describing the cell cycle and involvement of
DEmRNAs is shown in Fig. 5.
Gradient colors are used to indicate high- or low-expression genes.
The results of the functional enrichment analyses suggest that the
DEmRNAs may participate in oncogenicity and tumor progression in
CESC through regulating relevant biological processes and critical
pathways.
Assessment of DEmRNAs and DEmiRNAs
associated with overall survival
KM curves were generated to analyze the relationship
between RNA expression and overall survival in patients with CESC.
In the present study, 255 DEmRNAs were identified to be
significantly associated with overall survival and 15 DEmiRNAs were
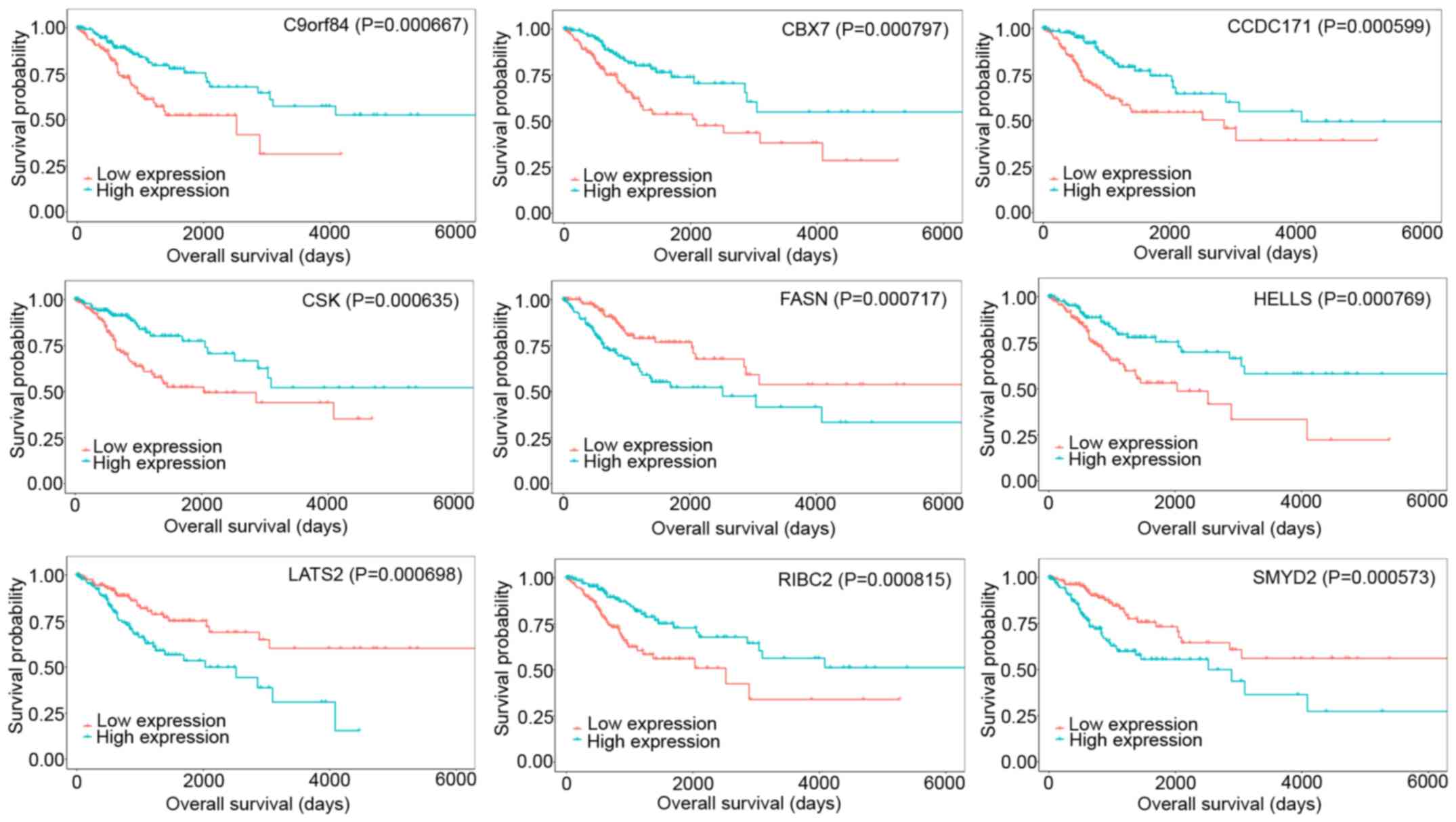
closely linked to prognosis in CESC. Among the top 9 mRNAs that
affected overall survival in CESC, C9orf84, CBX7, CCDC171, CSK,
HELLS, and RIBC2 were positively associated with overall survival,
whereas FASN, LATS2, and SMYD2 were negatively associated with
survival (Fig. 6). Overexpressed
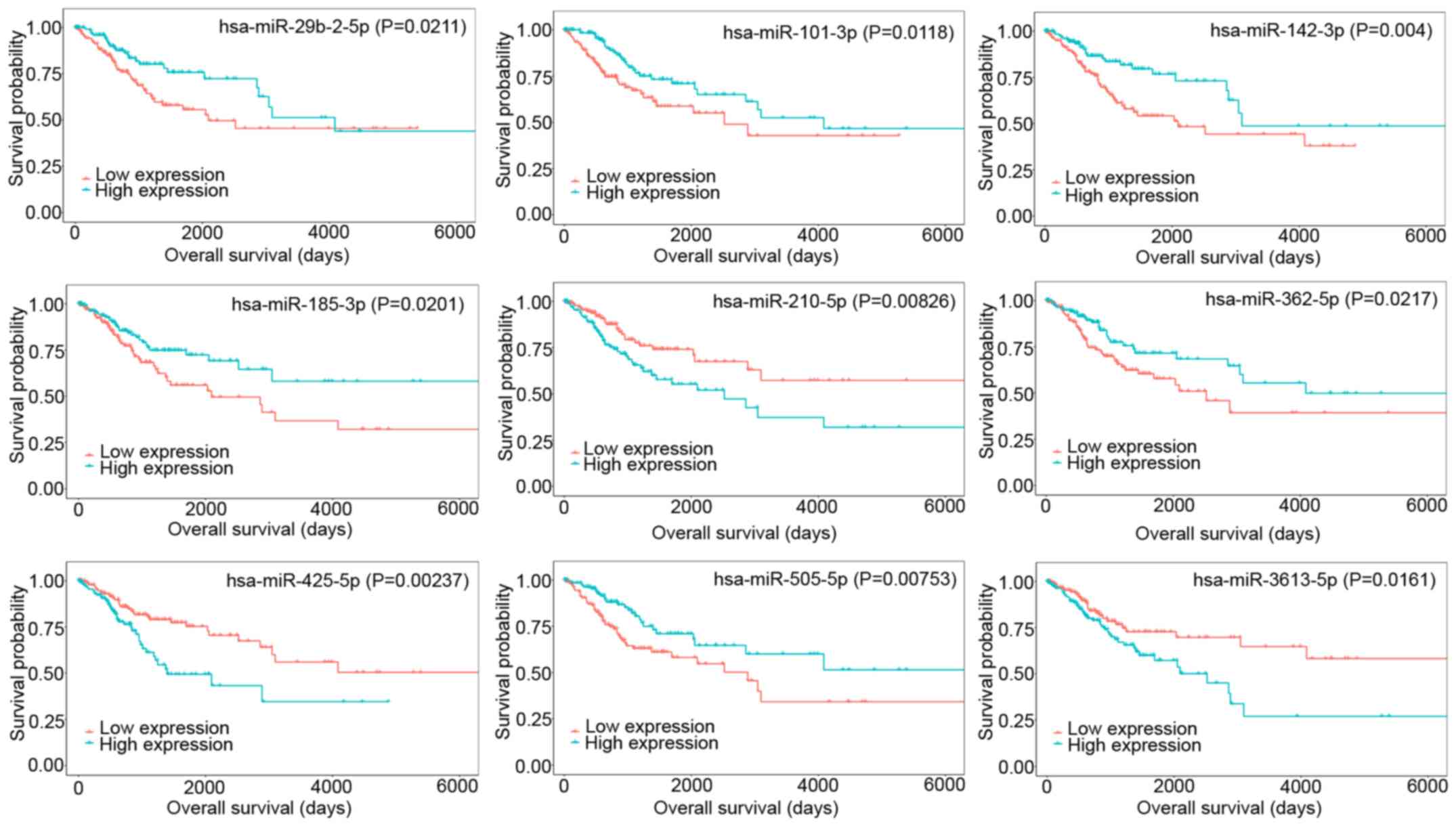
DEmiRNAs, including miR-99a-5p, miR-20b-3p, miR-101-3p, miR-145-5p,
miR-142-3p, miR-218-5p, miR-362-5p, miR-185-3p, miR-505-5p,
miR-642a-5p, and miR-29b-2-5p, were revealed to be strongly
associated with prolonged patient survival time, and upregulated
DEmiRNAs, including miR-210-5p, miR-425-5p, miR-3613-5p, and
miR-6892-5p, were associated with poor patient prognosis. The top 9
DEmiRNAs associated with overall survival are shown in Fig. 7.
Construction of ceRNA network in
CESC
Competing endogenous RNA network analysis was based
on three criteria (see the Materials and methods section) to
determine the competing endogenous interactions among mRNAs and
lncRNAs. A hypergeometric test of shared miRNAs, expression
correlation analysis of lncRNA-mRNA pairs, and regulation pattern
analysis of shared miRNAs were all implemented to build a ceRNA
regulatory network of lncRNAs, miRNAs, and mRNAs in CESC. StarBase
v2.0 was used to collect miRNA-lncRNA and/or miRNA-mRNA pairs for
prediction and experimental verification. A co-expression network
was constructed between the DElncRNAs and DEmRNAs and the network
was further filtered using hyper P-value <0.05 and cor P-value
<0.05. Cytoscape was used to visualize the ceRNA network.
Ultimately, 110 nodes and 190 edges were identified, including 5
lncRNAs, 15 miRNAs, and 90 mRNAs, for the ceRNA network. The
Cytoscape plug-in called ‘cytoHubba’ was used to detect hub nodes,
and the MCC algorithm of cytoHubba was used to identify hub nodes.
The top 10 genes were miR-374b-5p, miR-374a-5p, miR-590-3p,
miR-18a-5p, miR-18b-5p, miR-4735-3p, miR-376c-3p, EPB41L4A-AS1,
CCND2, and miR-153-3p, and their scores were 49, 48, 18, 17, 15,
10, 9, 7, 7, and 6, respectively. Among them, miR-18a-5p,
miR-376c-3p, and miR-590-3p were included with the DEmiRNAs,
whereas EPB41L4A-AS1 and CCND2 belonged to the DElncRNA and DEmRNA
groups, respectively. Based on the MCC scores, a sub-network was
established that centered around the lncRNA EPB41L4A-AS1, one of
the top 10 most highly expressed genes (Fig. 8).
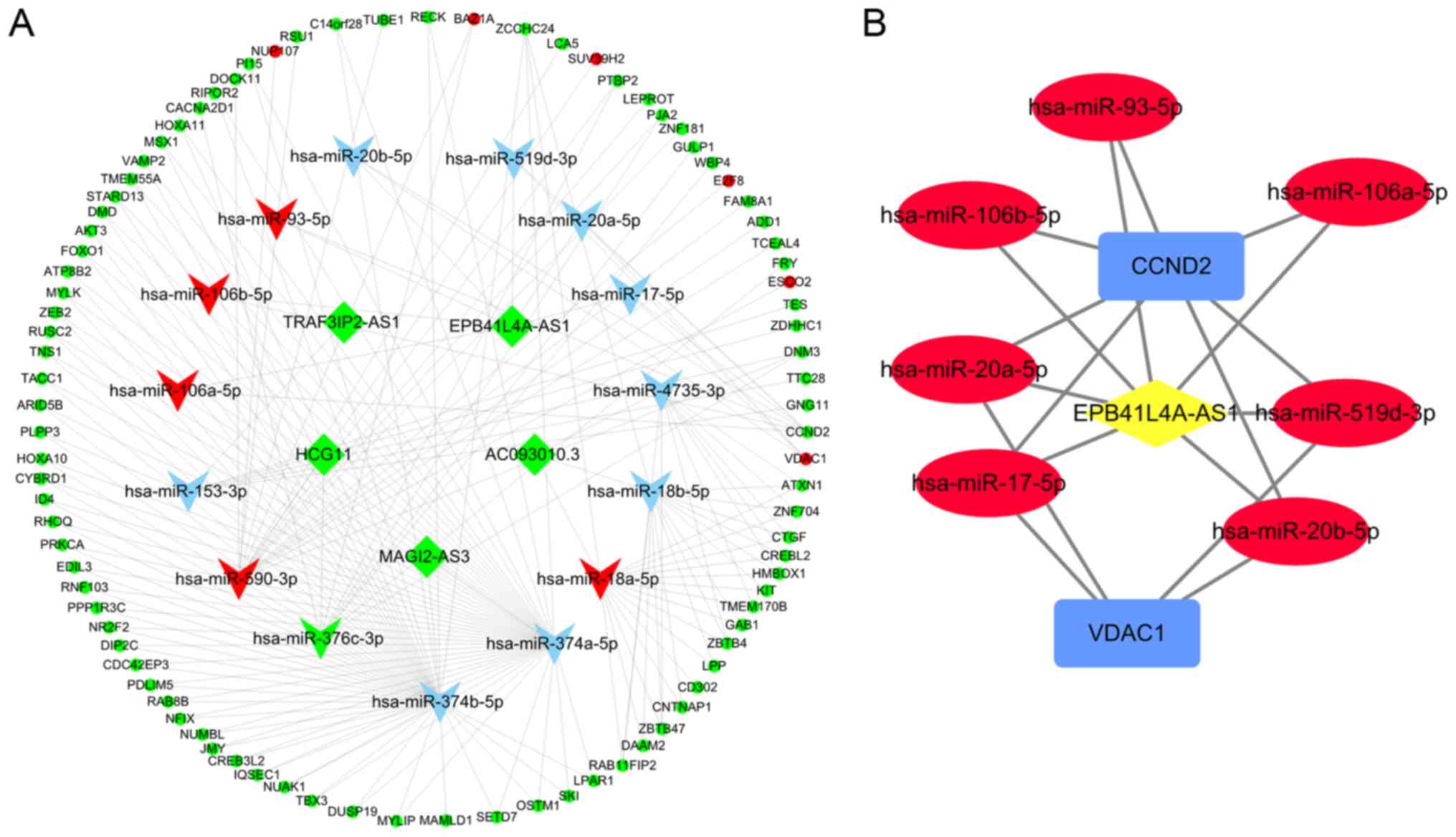 | Figure 8.ceRNA networks in CESC. (A) The ceRNA
network between DElncRNAs and DEmRNAs mediated by miRNAs. The
round, rhombus, and triangle nodes represent mRNAs, lncRNAs, and
miRNAs, respectively. Red indicates high expression, whereas green
represents low expression. Blue triangle nodes represent miRNAs
that do not belong to DEmiRNAs. (B) The subnetwork of
lncRNA-miRNA-mRNA. Red ellipse, blue rectangles, and yellow
diamonds represent miRNA, mRNA, and lncRNA, respectively. ceRNA,
competitive endogenous RNA; CESC, cervical squamous cell carcinoma
and endocervical adenocarcinoma; DE, differentially expressed. |
Discussion
LncRNAs are transcriptional products that are more
than 200 nucleotides in length and do not appear to possess any
significant coding protein function. LncRNAs are composed of
various heterogeneous non-coding RNAs (15). Although lncRNAs are abundant among
non-coding RNAs, their function has not been fully elucidated.
Notably, there has been increasing evidence that lncRNAs
significantly contribute to gene expression and regulation. In
recent years, multiple studies have revealed that lncRNAs
participate in various processes, including cell growth, cellular
death, and epigenetic regulation (16). Yang et al (17), confirmed Gm16551 as an lncRNA
regulator that suppresses lipogenesis and sterol regulatory
element-binding protein 1c activity in the mouse liver. Moreover,
the aberrant expression of lncRNAs is associated with pathogenesis
and development of multiple carcinomas via activation of
carcinogenic pathways and interaction with other RNAs (18).
The present study reported 60 upregulated and 90
downregulated lncRNAs among the lncRNAs that were differentially
expressed between tumor tissues and normal samples. Notably, lncRNA
MEG3 was found to be downregulated in our differential expression
profile. It was previously identified using experimental techniques
that the upregulation of MEG3 may inhibit cell growth through
reducing the expression of miR-21-5p in carcinoma of the uterine
cervix (19). The lncRNA
C5orf66-AS1 was overexpressed among the DE RNAs. Rui et al
(20) suggested that C5orf66-AS1
was markedly upregulated in uterine cervical neoplasms compared
with its expression in paracervical tissues. LncRNA C5orf66-AS1 may
regulate cancer proliferation and cell death in cervical cancer via
sponging miR-637 as a ceRNA. In addition, lncRNA LINC00511 was also
upregulated among aberrantly expressed lncRNAs. Lu et al
(21), indicated that the high
expression of LINC00511 was associated with poor disease outcome in
breast cancer. In addition, the study demonstrated that LINC00511
interacted with miR-185-3p as a competing ceRNA to facilitate
breast cancer tumorigenesis.
To explore the significance of DElncRNAs in
expression profiles further, KM analysis was performed to detect
the association between DElncRNAs and overall survival in patients
with uterine cervix carcinoma. It was noted that 12 DElncRNAs were
significantly associated with carcinoma outcome. The results of the
study by He et al (22),
were consistent with our findings, as the present study reported
that high expression of lncRNA CASC15 was associated with poor
disease outcome in hepatocellular carcinoma. Yin et al
(23), demonstrated that
AC016773.1 is an independent factor influencing survival in clear
cell renal carcinoma via multivariate Cox regression analysis. The
present study noted that some lncRNAs (AC008771.1, AC015871.3,
AC019069.1, AC093278.2, and AL662844.4) that have been studied
poorly to date are significantly associated with survival time,
although further experimental verification is required for
validation.
MiRNAs form one group of small non-coding RNA
molecules that exist in animals, plants, and some viruses (24), and function in RNA silencing and
post-transcription to regulate gene expression. Guay et al
(25), suggested that transfer of
miRNAs to rodent and human pancreatic β-cells results in chemokine
expression and apoptosis of β-cells. Previous studies have
demonstrated multiple roles for miRNAs in many biological
processes. For example, Wüst et al (26), verified that loss of miR-1/133a
hinders mitochondrial activity in skeletal muscle. Lee et al
(27), demonstrated that
epithelial microvesicle-containing cav-1/hnRNPA2B1 complex bound to
miR-17/93-activated tissue macrophages and initiated an immune
response. Furthermore, aberrant miRNA expression is implicated in
disease states. Mehta et al (28), suggested that miR-132 may modulate
B lymphopoiesis via Sox4 and inhibit development of cancerous
B-cells. Singh et al (29),
showed that miR-17~92-deficient type 2 innate lymphoid cells
relieved lung inflammation after exposure to allergens in mice.
MiRNAs may be upregulated or downregulated, and aberrantly
expressed miRNAs are involved in tumorigenesis and the development
of multiple types of cancer, including cervical cancer (30,31).
In the present study, 133 miRNAs were differentially
expressed between cervical tumors and normal solid samples. Among
the DEmiRNAs, 80 miRNAs showed increased expression, and 53 miRNAs
showed decreased expression, respectively. The data revealed that
miRNA-205-5p, miRNA-210-5p, miRNA-425-5p, miRNA-21-5p, and
miRNA-141-3p were highly expressed. Several studies have detected
the relationship between these miRNAs and various types of cancer.
Chen et al (32), showed
that miRNA-205-5p may restrict the reproductive and invasive
capabilities of colorectal carcinoma by inhibiting PTK7.
Interestingly, miRNA-205-5p was the most highly expressed miRNA in
our expression profiles, a finding which is consistent with those
of Vilming Elgaaen et al (33), who reported that miRNA-205-5p was
the most highly expressed (FC=74) miRNA in high-grade ovarian
carcinoma. Qu et al (34),
reported that miRNA-21-5p was highly expressed in pancreatic
cancer, and further suggested a favorable sensitivity and
specificity (0.77 and 0.80, respectively) for the diagnostic value
of miRNA-21-5p. Their study highlighted that miRNA-21-5p may be a
prospective indicator in pancreatic cancer. It has also been
observed that the expression of miR-141-3p is inversely associated
with p53 expression in glioblastoma and normal brain tissues.
MiR-141-3p facilitated glioma progression and temozolomide
resistance by regulating p53 expression, and therefore may act as a
new diagnostic and therapeutic indicator in glioblastoma (35). Hang et al (36), confirmed that miR-145-5p is
strongly and negatively associated with connective tissue growth
factor (CTGF) (r=0.1126, P=0.02188), and downregulation of
miR-145-5p was found to be associated with poor disease outcome and
higher stages in ovarian cancer. The present study also found that
miR-145-5p was one of the downregulated miRNAs. By comprehensively
analyzing the relationship between DEmiRNAs and overall survival of
patients with cervical cancer, miR-210-5p and miR-425-5p were found
to be upregulated and had a significant negative association with
overall survival. Ying et al (37), suggested that upregulation of
miR-210-5p exerted an impact on carcinogenic signaling pathways,
and may participate in the progression of kidney carcinoma. Low
expression of miR-425-5p was indicated to inhibit cell growth,
infiltration, and migration in gastric carcinoma. Moreover,
downregulated miR-425-5p may suppress tumor metastasis to lung
tissues in nude mice (38).
The importance of mRNAs in malignant neoplasms has
been well studied. Therefore, the differences in mRNA expression
between cervical cancer samples and normal tissues were analyzed. A
total of 2,255 DEmRNAs were confirmed in our profile. KM analysis
was simultaneously performed to explore the relationship between
DEmRNAs and patient outcome. Fatty acid synthase gene (FASN) was
determined to be highly expressed in cervical tumor samples, and an
inverse association between FASN and overall survival was detected
in patients with cervical cancer. Xia et al (39) demonstrated that FASN may serve as a
potential prognostic biomarker in patients with cervical cancer
through Cox regression analysis. Notably, the mRNAs CXCL12 and
CCL21 were both downregulated in our data, and are known to be
involved in invasion, migration, and progression of cervical cancer
(40,41). Moreover, a low expression of mRNA
FOXO1 was observed, and it has been previously reported that
downregulation of FOXO1 may induce cellular proliferation and
activate tumor cell viability in carcinoma of the uterine cervix
(42). Zhao et al (43) indicated that dysfunction of the
mRNAs CHEK1 and CDKN2A, which were upregulated within our group of
DEmRNAs, could facilitate cellular proliferation in cervical
cancer.
The ceRNA network was originally proposed by Salmena
et al (44) to highlight
the notion that mRNAs, lncRNAs, and other RNAs may interact with
miRNAs by sponging the miRNAs through MREs. Several studies have
revealed that lncRNAs are able to indirectly regulate mRNAs through
miRNAs (45,46). Therefore, GO and KEGG analyses of
DEmRNAs were conducted to provide insights into the impact of
dysregulated lncRNAs. The enriched GO terms were mainly involved in
cell division, cellular proliferation, cellular protein metabolic
processes, and cell differentiation. The KEGG pathways of DEmRNAs
were markedly enriched in tumor-associated pathways, including
focal adhesion, cell cycle, cellular senescence, DNA replication,
p53 signaling, Fanconi anemia, homologous recombination, and base
excision repair. Moreover, the top 10 GO terms enriched in DEmRNAs
were primarily associated with the cell cycle in KEGG pathways.
Subsequently, the effects of DEmRNAs on overall
survival were assessed, and 255 DEmRNAs with prognostic value in
cervical cancer were identified. The DEmRNAs CBX7, SMYD2, and LATS2
were strongly associated with the clinical outcome of cervical
cancer patients by KM analysis. Previous studies have shown that
CBX7 is a vital tumor suppressor, and the absence of CBX7
contributes to tumorigenesis, whereas high expression of CBX7 is
correlated with longer survival time (47,48).
High expression of SMYD2 was found in papillary thyroid carcinoma
and hepatocellular carcinoma tissues, and patients with papillary
thyroid cancer and hepatocellular cancer with upregulated SMYD2
were observed to have poor disease outcomes (49,50).
Consistent with our findings, Luo et al (51) suggested that the expression of the
mRNA LATS2 was a significant predictive factor of overall survival
in lung adenocarcinoma.
A hypothesis of ceRNA network provided a new avenue
for studies that could improve our understanding of the interaction
between lncRNAs and mRNAs mediated by miRNAs. The construction of a
lncRNA-miRNA-mRNA network offered new clues for revealing the key
RNAs involved in tumorigenesis and progression of cervical cancer.
Here, a miRNA-mediated lncRNA-miRNA-mRNA cross-talk network from
TCGA database was generated. In the ceRNA network, there were 110
nodes and 190 edges, including 5 lncRNAs, 15 miRNAs, and 90 mRNAs.
The scores of RNAs that were identified in the network were
calculated, and a subnetwork was constructed that centered on the
lncRNA EPB41L4A-AS1 in the top 10 genes based on MCC scores. The
figure created for the ceRNA sub-network indicated that the lncRNA
EPB41L4A-AS1 could indirectly interact with the mRNAs CCND2 and
VDAC1 via miR-106a-5p, miR-93-5p, miR-106b-5p, miR-519d-3p,
miR-17-5p, miR-20a-5p, and miR-20b-5p. This suggested that the
lncRNA EPB41L4A-AS1 may function as a vital regulator in carcinoma
of the uterine cervix. In line with this, Zhan et al
(52) also provided evidence in
support of EPB41L4A-AS1 acting as a potential candidate for stage
progression and poor prognosis in ovarian cancer. It was shown in
the present study how lncRNAs affect the expression of coding genes
mediated by miRNAs, highlighting the importance of a
lncRNA-miRNA-mRNA network in cervical cancer.
It is important to verify the function of the RNAs
implicated in cervical cancer as well as the interaction among
mRNAs, miRNAs, and lncRNAs, both in vitro and in
vivo, using mice, for example, as an animal model. During in
vivo research, the homology of mRNA, miRNA, and lncRNA must be
considered. To answer this, it is necessary to obtain the mouse (or
others) RNA-seq data and human RNA-seq data to check the homology
via tools such as BLAST (Basic Local Alignment Search Tool). In the
present study, RNA-seq profiles from mice or other animal models
were not used, and would be useful to analyze in future
studies.
In the literature, some mRNAs and miRNAs in mouse
have functions that are similar to those of humans. For example, Qi
et al (53) showed mRNA
paired like homeodomain 1 (PITX1), which was overexpressed in the
present study, can suppress both human telomerase reverse
transcriptase (hTERT) and mouse TERT (mtert) promoter activity.
Sastre-Perona et al (54)
revealed that PITX1 is specifically expressed in basal tumor
propagating cells, where it co-localizes with SOX2 and TRP63 and
determines cell fate in mouse and human squamous cell carcinoma.
Osei-Amponsa et al (55)
confirmed a critical role for hypermethylation of FOXA1 in
heterogeneity of bladder cancer, and epigenetic silencing of FOXA1
drives squamous differentiation in mice. In the present study,
miRNA-205 was upregulated; Son et al (56) suggested that miRNA-205 of humans
shares similar sequences to that of miRNA-712 in mice, and is
highly conserved in most vertebrates. miRNA-205 and miRNA-712 also
share >50% of the cell signaling targets, including TIMP3. For
the lncRNAs, this appears not to be the case. Further
investigations into vertebrate lncRNAs revealed that, although
lncRNAs are conserved in sequence, they are not conserved in terms
of their transcription (57,58).
This means that, even when the sequence of a human lncRNA is
conserved in another vertebrate species, there is often no
transcription of a lncRNA in the orthologous genomic region.
Our results have uncovered the potential mechanisms
of tumorigenesis and prognosis in cervical cancer, and suggest that
the lncRNA EPB41L4A-AS1 is both a novel and vital biomarker for
carcinogenesis and progression, and also a candidate therapeutic
target in cervical cancer that needs to be studied further. Drugs
targeting lncRNA EPB41L4A-AS1, which is implicated in the
overexpression of miR-106a/106b-5p, will have usefulness in the
treatment of cervical cancer. However, to achieve this, further
in vitro and in vivo experiments are needed to verify
the mechanisms of mutual interaction between the lncRNA
EPB41L4A-AS1 and related target genes in the ceRNA sub-network
mentioned above.
In conclusion, in the present study, the expression
profiles of lncRNA, miRNA, and mRNA in cervical cancer were
analyzed from TCGA database. Cancer-specific RNAs that may be
relevant for tumorigenesis, development, and prognosis were
screened via differential gene expression analysis, KM survival
analysis, and a comprehensive ceRNA network. Our study has
significantly contributed to the current understand¬ing of cervical
cancer pathogenesis, and has identified novel RNAs as potential
markers for diagnosis, prognosis, and therapy. Due to the
limitations of bioinformatics analysis, however, large-scale
prospective research will be necessary for further validation.
Acknowledgements
We are grateful to Mr Jiajia Xu and Ms Wenjing Xu
(Shanghai Jieyi Biotechnology Co., Ltd, China) for their technical
computer support.
Funding
No funding was received.
Availability of data and materials
The results in the present study are based upon data
generated by the TCGA Research Network: https://www.cancer.gov/tcga. The datasets used and/or
analyzed during the present study are available from the
corresponding author on reasonable request.
Authors' contributions
PC and WZ designed the study and drafted the
manuscript. YC, XZ, and DY performed the data analysis. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Waggoner SE: Cervical cancer. Lancet.
361:2217–2225. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu S, Zhang P, Chen Z, Liu M, Li X and
Tang H: MicroRNA-7 downregulates XIAP expression to suppress cell
growth and promote apoptosis in cervical cancer cells. FEBS Lett.
587:2247–2253. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang H, Zhao Y, Chen M and Cui J:
Identification of novel long non-coding and circular RNAs in human
papillomavirus-mediated cervical cancer. Front Microbiol.
8:17202017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li LJ, Zhao W, Tao SS, Leng RX, Fan YG,
Pan HF and Ye DQ: Competitive endogenous RNA network: Potential
implication for systemic lupus erythematosus. Expert Opin Ther
Targets. 21:639–648. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huarte M: The emerging role of lncRNAs in
cancer. Nat Med. 21:1253–1261. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang R, Xing L, Wang M, Chi H, Zhang L and
Chen J: Comprehensive analysis of differentially expressed profiles
of lncRNAs/mRNAs and miRNAs with associated ceRNA networks in
triple-negative breast cancer. Cell Physiol Biochem. 50:473–488.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tomczak K, Czerwińska P and Wiznerowicz M:
The Cancer Genome Atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19((1A)): A68–A77. 2015.PubMed/NCBI
|
|
8
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Furió-Tarí P, Tarazona S, Gabaldón T,
Enright AJ and Conesa A: spongeScan: A web for detecting microRNA
binding elements in lncRNA sequences. Nucleic Acids Res. 44((W1)):
W176–80. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA
interaction networks from large-scale CLIP-Seq data. Nucleic Acids
Res. 42(D1): D92–D97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Paci P, Colombo T and Farina L:
Computational analysis identifies a sponge interaction network
between long non-coding RNAs and messenger RNAs in human breast
cancer. BMC Syst Biol. 8:832014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT and
Lin CY: cytoHubba: Identifying hub objects and sub-networks from
complex interactome. BMC Syst Biol. 8 (Suppl 4):S112014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang H, Chen Z, Wang X, Huang Z, He Z and
Chen Y: Long non-coding RNA: A new player in cancer. J Hematol
Oncol. 6:372013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu W, Alvarez-Dominguez JR and Lodish HF:
Regulation of mammalian cell differentiation by long non-coding
RNAs. EMBO Rep. 13:971–983. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang L, Li P, Yang W, Ruan X, Kiesewetter
K, Zhu J and Cao H: Integrative transcriptome analyses of metabolic
responses in mice define pivotal LncRNA metabolic regulators. Cell
Metab. 24:627–639. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bolha L, Ravnik-Glavač M and Glavač D:
Long noncoding RNAs as biomarkers in cancer. Dis Markers.
2017:72439682017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Yao T, Wang Y, Yu J, Liu Y and
Lin Z: Long noncoding RNA MEG3 is downregulated in cervical cancer
and affects cell proliferation and apoptosis by regulating miR-21.
Cancer Biol Ther. 17:104–113. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rui X, Xu Y, Jiang X, Ye W, Huang Y and
Jiang J: Long non-coding RNA C5orf66-AS1 promotes cell
proliferation in cervical cancer by targeting miR-637/RING1 axis.
Cell Death Dis. 9:11752018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu G, Li Y, Ma Y, Lu J, Chen Y, Jiang Q,
Qin Q, Zhao L, Huang Q, Luo Z, et al: Long noncoding RNA LINC00511
contributes to breast cancer tumourigenesis and stemness by
inducing the miR-185-3p/E2F1/Nanog axis. J Exp Clin Cancer Res.
37:2892018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
He T, Zhang L, Kong Y, Huang Y, Zhang Y,
Zhou D, Zhou X, Yan Y, Zhang L, Lu S, et al: Long non-coding RNA
CASC15 is upregulated in hepatocellular carcinoma and facilitates
hepatocarcinogenesis. Int J Oncol. 51:1722–1730. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yin H, Wang X, Zhang X, Wang Y, Zeng Y,
Xiong Y, Li T, Lin R, Zhou Q, Ling H, et al: Integrated analysis of
long noncoding RNA associated-competing endogenous RNA as
prognostic biomarkers in clear cell renal carcinoma. Cancer Sci.
109:3336–3349. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guay C, Kruit JK, Rome S, Menoud V, Mulder
NL, Jurdzinski A, Mancarella F, Sebastiani G, Donda A, Gonzalez BJ,
et al: Lymphocyte-derived exosomal microRNAs promote pancreatic β
cell death and may contribute to type 1 diabetes development. Cell
Metab. 29:348–361.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wüst S, Dröse S, Heidler J, Wittig I,
Klockner I, Franko A, Bonke E, Günther S, Gärtner U, Boettger T, et
al: Metabolic maturation during muscle stem cell differentiation is
achieved by miR-1/133a-mediated inhibition of the Dlk1-Dio3 mega
gene cluster. Cell Metab. 27:1026–1039.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee H, Li C, Zhang Y, Zhang D, Otterbein
LE and Jin Y: Caveolin-1 selectively regulates microRNA sorting
into microvesicles after noxious stimuli. J Exp Med. 216:2202–2220.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mehta A, Mann M, Zhao JL, Marinov GK,
Majumdar D, Garcia-Flores Y, Du X, Erikci E, Chowdhury K and
Baltimore D: The microRNA-212/132 cluster regulates B cell
development by targeting Sox4. J Exp Med. 212:1679–1692. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Singh PB, Pua HH, Happ HC, Schneider C,
von Moltke J, Locksley RM, Baumjohann D and Ansel KM: MicroRNA
regulation of type 2 innate lymphoid cell homeostasis and function
in allergic inflammation. J Exp Med. 214:3627–3643. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Giza DE, Vasilescu C and Calin GA:
MicroRNAs and ceRNAs: Therapeutic implications of RNA networks.
Expert Opin Biol Ther. 14:1285–1293. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pardini B, De Maria D, Francavilla A, Di
Gaetano C, Ronco G and Naccarati A: MicroRNAs as markers of
progression in cervical cancer: A systematic review. BMC Cancer.
18:6962018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen S, Wang Y, Su Y, Zhang L, Zhang M, Li
X, Wang J and Zhang X: miR-205-5p/PTK7 axis is involved in the
proliferation, migration and invasion of colorectal cancer cells.
Mol Med Rep. 17:6253–6260. 2018.PubMed/NCBI
|
|
33
|
Vilming Elgaaen B, Olstad OK, Haug KB,
Brusletto B, Sandvik L, Staff AC, Gautvik KM and Davidson B: Global
miRNA expression analysis of serous and clear cell ovarian
carcinomas identifies differentially expressed miRNAs including
miR-200c-3p as a prognostic marker. BMC Cancer. 14:802014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qu K, Zhang X, Lin T, Liu T, Wang Z, Liu
S, Zhou L, Wei J, Chang H, Li K, et al: Circulating miRNA-21-5p as
a diagnostic biomarker for pancreatic cancer: Evidence from
comprehensive miRNA expression profiling analysis and clinical
validation. Sci Rep. 7:16922017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou X, Wu W, Zeng A, Nie E, Jin X, Yu T,
Zhi T, Jiang K, Wang Y, Zhang J, et al: MicroRNA-141-3p promotes
glioma cell growth and temozolomide resistance by directly
targeting p53. Oncotarget. 8:71080–71094. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hang W, Feng Y, Sang Z, Yang Y, Zhu Y,
Huang Q and Xi X: Downregulation of miR-145-5p in cancer cells and
their derived exosomes may contribute to the development of ovarian
cancer by targeting CT. Int J Mol Med. 43:256–266. 2019.PubMed/NCBI
|
|
37
|
Ying G, Wu R, Xia M, Fei X, He QE, Zha C
and Wu F: Identification of eight key miRNAs associated with renal
cell carcinoma: A meta-analysis. Oncol Lett. 16:5847–5855.
2018.PubMed/NCBI
|
|
38
|
Zhang Z, Li Y, Fan L, Zhao Q, Tan B, Li Z
and Zang A: microRNA-425-5p is upregulated in human gastric cancer
and contributes to invasion and metastasis in vitro and
in vivo. Exp Ther Med. 9:1617–1622. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xia L, Wang H, Cai S, Su X, Shen J, Meng
Q, Chen Y, Li L, Yan J, Zhang C, et al: Integrated analysis of a
competing endogenous RNA network revealing a prognostic signature
for cervical cancer. Front Oncol. 8:3682018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Teicher BA and Fricker SP: CXCL12
(SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 16:2927–2931.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xiong Y, Huang F, Li X, Chen Z, Feng D,
Jiang H, Chen W and Zhang X: CCL21/CCR7 interaction promotes
cellular migration and invasion via modulation of the MEK/ERK1/2
signaling pathway and correlates with lymphatic metastatic spread
and poor prognosis in urinary bladder cancer. Int J Oncol.
51:75–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Prasad SB, Yadav SS, Das M, Govardhan HB,
Pandey LK, Singh S, Pradhan S and Narayan G: Down regulation of
FOXO1 promotes cell proliferation in cervical cancer. J Cancer.
5:655–662. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao L, Zhang Z, Lou H, Liang J, Yan X, Li
W, Xu Y and Ou R: Exploration of the molecular mechanisms of
cervical cancer based on mRNA expression profiles and predicted
microRNA interactions. Oncol Lett. 15:8965–8972. 2018.PubMed/NCBI
|
|
44
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li F, Huang C, Li Q and Wu X: Construction
and comprehensive analysis for dysregulated long non-coding RNA
(lncRNA)-associated competing endogenous RNA (ceRNA) network in
gastric cancer. Med Sci Monit. 24:37–49. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Song J, Ye A, Jiang E, Yin X, Chen Z, Bai
G, Zhou Y and Liu J: Reconstruction and analysis of the aberrant
lncRNA-miRNA-mRNA network based on competitive endogenous RNA in
CESC. J Cell Biochem. 119:6665–6673. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ni S, Wang H, Zhu X, Wan C, Xu J, Lu C,
Xiao L, He J, Jiang C, Wang W, et al: CBX7 suppresses cell
proliferation, migration, and invasion through the inhibition of
PTEN/Akt signaling in pancreatic cancer. Oncotarget. 8:8010–8021.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Forzati F, Federico A, Pallante P, Abbate
A, Esposito F, Malapelle U, Sepe R, Palma G, Troncone G, Scarfò M,
et al: CBX7 is a tumor suppressor in mice and humans. J Clin
Invest. 122:612–623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xu W, Chen F, Fei X, Yang X and Lu X:
Overexpression of SET and MYND domain-containing protein 2 (SMYD2)
is associated with tumor progression and poor prognosis in patients
with papillary thyroid carcinoma. Med Sci Monit. 24:7357–7365.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zuo SR, Zuo XC, He Y, Fang WJ, Wang CJ,
Zou H, Chen P, Huang LF, Huang LH, Xiang H, et al: Positive
expression of SMYD2 is associated with poor prognosis in patients
with primary hepatocellular carcinoma. J Cancer. 9:321–330. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Luo SY, Sit KY, Sihoe AD, Suen WS, Au WK,
Tang X, Ma ES, Chan WK, Wistuba II, Minna JD, et al: Aberrant large
tumor suppressor 2 (LATS2) gene expression correlates with EGFR
mutation and survival in lung adenocarcinomas. Lung Cancer.
85:282–292. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhan L, Li J and Wei B: Long non-coding
RNAs in ovarian cancer. J Exp Clin Cancer Res. 37:1202018.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Qi DL, Ohhira T, Fujisaki C, Inoue T, Ohta
T, Osaki M, Ohshiro E, Seko T, Aoki S, Oshimura M, et al:
Identification of PITX1 as a TERT suppressor gene located on human
chromosome 5. Mol Cell Biol. 31:1624–1636. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sastre-Perona A, Hoang-Phou S, Leitner MC,
Okuniewska M, Meehan S and Schober M: De novo PITX1 expression
controls bi-stable transcriptional circuits to govern self-renewal
and differentiation in squamous cell carcinoma. Cell Stem Cell.
24:390–404.e8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Osei-Amponsa V, Buckwalter JM, Shuman L,
Zheng Z, Yamashita H, Walter V, Wildermuth T, Ellis-Mohl J, Liu C,
Warrick JI, et al: Hypermethylation of FOXA1 and allelic loss of
PTEN drive squamous differentiation and promote heterogeneity in
bladder cancer. Oncogene. 39:1302–1317. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Son DJ, Kumar S, Takabe W, Kim CW, Ni CW,
Alberts-Grill N, Jang IH, Kim S, Kim W, Won Kang S, et al: The
atypical mechanosensitive microRNA-712 derived from pre-ribosomal
RNA induces endothelial inflammation and atherosclerosis. Nat
Commun. 4:30002013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Washietl S, Kellis M and Garber M:
Evolutionary dynamics and tissue specificity of human long
noncoding RNAs in six mammals. Genome Res. 24:616–628. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kutter C, Watt S, Stefflova K, Wilson MD,
Goncalves A, Ponting CP, Odom DT and Marques AC: Rapid turnover of
long noncoding RNAs and the evolution of gene expression. PLoS
Genet. 8:e10028412012. View Article : Google Scholar : PubMed/NCBI
|