Introduction
Gastrointestinal stromal tumors (GISTs) are the most
common mesenchymal tumors of the digestive tract (1); the annual incidence is between
11–14.5 per million people, and the prevalence is estimated to be
129 per million individuals (1).
Moreover, a National Cancer Database reported a cause-specific
mortality rate of 14.0% and a 5-year survival rate of ~82%
(2). GISTs arise predominantly in
the stomach but also occur in the small intestine, colon and rectum
(3). It is considered a spindle or
epithelioid cell neoplasm that expresses the mast/stem cell growth
factor receptor Kit (KIT) protein and that has a KIT or
platelet-derived growth factor receptor-α (PDGFRA) gene
mutation (4,5). KIT and PDGFRA are receptor tyrosine
kinases, and gain-of-function mutations of the KIT or
PDGFRA gene serve central roles in GIST pathogenesis by
activating KIT downstream signals such as the phosphoinositide
3-kinase/AKT/mTOR pathway (6).
Angiotensin II receptor blockers (ARBs) are widely
used to treat chronic kidney disease, heart failure and
hypertension (7). Previous studies
have shown that angiotensin II is associated with cancer
progression and that ARBs inhibit tumor growth by binding to the
angiotensin II type 1 receptor (8–10),
including in various common types of cancer cells such as breast
(11), endometrial (12) and stomach (13) in vitro and in vivo.
Furthermore, ARBs have been associated with reduced incidence and
mortality rates in several types of cancer in patients with
hypertension (14,15). Among these ARBs, telmisartan was
reported to inhibit proliferation of various types of cancer cells,
including urological (16,17) and colon (18) cancers, by inducing apoptosis.
AMP-activated protein kinase (AMPK) is an energy
sensor and a major regulator of cellular energy homeostasis
(19). It is hypothesized that
AMPK activation inhibits the mTOR signaling pathway. Since
ribosomal protein S6 kinase 1 (p70S6K) is downstream of mTOR,
inhibition of mTOR inhibits its activation, thus leading to reduced
protein synthesis, which is necessary for cancer cell growth
(20,21). Recent reports have demonstrated
that telmisartan contributes to AMPK activation and regulation of
the mTOR pathway in various types of cancers, and it induces a
reduction in p70S6K activation, thus decreasing protein synthesis
(22,23). However, the mechanisms underlying
the antiproliferative effects of telmisartan in GIST remains
unclear.
In the present study, the antiproliferative effect
of telmisartan and its mechanism of action were evaluated in
GIST-T1 cells. It has previously been reported that microRNA
(miRNA) signatures have a role in the antitumor effect of
telmisartan in esophageal adenocarcinoma and hepatocellular
carcinoma by modulating the cell cycle (22,23).
In the past 10 years, increasing evidence has indicated that miRNAs
directly control cell cycle progression by targeting cell cycle
regulators (24). Additionally,
miRNAs indirectly control cell cycle progression by targeting
signal transduction pathways in anticancer therapy (22,23).
In the present study it was hypothesized that unidentified miRNAs
have a role in the effect of telmisartan treatment on GIST and may
be targets for novel therapeutic options. The results revealed that
telmisartan inhibited the proliferation of GIT-T1 cells and induced
cell cycle arrest, as well as altering miRNA expression.
Materials and methods
Chemicals and antibodies
Telmisartan and valsartan were purchased from Tokyo
Chemical Industry Co., Ltd. Irbesartan was purchased from Wako Pure
Chemical Industries, Ltd. Candesartan was purchased from AdooQ
BioScience. Telmisartan was prepared as a 10 mM stock solution in
dimethyl sulfoxide (DMSO). Valsartan, irbesartan and candesartan
were prepared as 100 mM stock solutions in DMSO. The stock
solutions were stored at −20°C. The following materials were used:
Cell Cycle Phase Determination kit (Cayman Chemical Company),
Annexin V-FITC Early Apoptosis Detection kit (Cell Signaling
Technology, Inc.), protease inhibitor cocktail (Pro-Prep, complete
protease inhibitor mixture; Intron Biotechnology, Inc.) and
Angiogenesis Antibody Array kits (R&D Systems, Inc.).
Primary antibodies used for western blot analyses
were obtained from the following sources. β-actin antibody was
obtained from Sigma-Aldrich. Cyclin D1 and cyclin E antibodies were
obtained from Thermo Fisher Scientific, Inc. Cdk6, Cdk2, Cdk4 and
Rb antibodies were obtained from Santa Cruz Biotechnology, Inc.
Phosphorylated (p)-Rb was purchased from BD Pharmingen (BD
Bioscience). AMPKα, p-AMPKα Thr172, mTOR, p-mTOR, p70S6K and
p-p70S6K antibodies were purchased from Cell Signaling Technology,
Inc. Horseradish peroxidase (HRP)-conjugated anti-mouse and
anti-rabbit IgG secondary antibodies (Cell Signaling Technology,
Inc.) were used.
Cell culture
The human GIST-T1 cell line was obtained from Cosmo
Bio Co., Ltd. GIST-T1 cells were maintained at 37°C with 5%
CO2 in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (FBS; FUJIFILM Wako Pure
Chemical Corporation), 20 U/ml penicillin and 100 µg/ml
streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.).
Cell proliferation assay
Cell proliferation of GIST-T1 cells was assayed
using CCK-8 (Dojindo Molecular Technologies, Inc), according to the
manufacturer's instructions. Briefly, 5×103 cells of
each experiment group were equally seeded on 96-well plates and
cultured in 100 µl DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS for 24 h. Subsequently, 0, 1, 10 or 100
µm ARBs (telmisartan, candesartan, valsartan or irbesartan) or
vehicle were added to each well, and the cells were cultured for an
additional 48 h. CCK-8 reagent (10 µl) was added to each well, and
the plates were incubated at 37°C for 3 h, and the absorbance was
measured at 450 nm using a microplate reader (Thermo Fisher
Scientific, Inc.).
Cell cycle and apoptosis analyses
Cell cycle profiles were analyzed after telmisartan
treatment to assess growth inhibition. GIST-T1 cells
(1×106 cells in a 100-mm diameter dish) were treated
with or without 100 µm telmisartan for 24–48 h. Cell cycle
progression was analyzed by measuring the amount of propidium
iodide (PI)-labeled DNA in ethanol-fixed cells. The fixed cells
were washed with PBS and then stored at −20°C for flow cytometry
analysis. On the day of analysis, the cells were washed with cold
PBS, suspended in 100 µl of PBS with 10 µl of RNase A (250 µg/ml)
and incubated at 4°C for 30 min. A 110-µl aliquot of PI (100 µg/ml)
was added to each suspension, and the cells were incubated at 4°C
for at least 30 min prior to analysis. For apoptosis analysis,
cells were stained with FITC-conjugated Annexin V and PI (Annexin
V-FITC Early Apoptosis Detection kit; Cell Signaling Technology,
Inc.), then incubated for 10 min on ice in the dark. Tumor cells
were stained for 24 or 48 h, according to the manufacturer's
instructions. Flow cytometry was performed using a Cytomics FC500
flow cytometer (Beckman Coulter, Inc.). The percentages of cells
were determined using Kaluza Analysis v2.1 software (Beckman
Coulter, Inc.). All experiments were performed in triplicate.
Western blotting
Western blotting was performed according to a
previously described method (22).
The cells were lysed in a protease inhibitor cocktail (‘complete’
protease inhibitor mixture; iNtRON Biotechnology, Inc.) on ice for
20 min. Protein concentrations were measured using a NanoDrop 2000
fluorospectrometer (Thermo Fisher Scientific, Inc.). Protein
aliquots (1–10 µg) were resuspended in sample buffer and separated
on 10% Tris-glycine gradient gels via SDS-PAGE (25). After blocking in 5% dry skim milk
in TBS with 0.05% Tween-20 (TBST) for 1 h at room temperature, the
membranes were incubated with primary antibodies against cyclin D1
(cat. no AHF0082; 1:2,000), cyclin E (cat. no. MA5-14336; 1:1,000),
cdk6 (cat. no. sc-177; 1:5,000), cdk2 (cat. no. sc-163; 1:5,000),
cdk4 (cat. no. sc-749; 1:1,000), Rb (cat. no. sc-50; 1:4,000), p-Rb
(cat. no. 558385; 1:1,000), AMPKα (cat. no. 5832; 1:5,000), p-AMPKα
(cat. no. 2535; 1:1,000), mTOR (cat. no. 2983; 1:1,000), p-mTOR
(cat. no. 5536; 1:1,000), p70S6K (cat. no. 2708; 1:1,000), p-p70S6K
(cat. no. 9205; 1:1,000) and β-actin (cat. no. A5441; 1:5,000)
overnight at 4°C, followed by HRP-conjugated secondary antibodies
(cat. nos. 7074 and 7076; 1:2,000) in 5% dry skimmed milk in TBS
for 1 h at room temperature (26).
The proteins were visualized on X-ray film using an Enhanced
Chemiluminescence Detection system (PerkinElmer, Inc.). Band
intensities were semi-quantified using ImageJ software v1.52q
(National Institutes of Health) and normalized to β-actin.
Analysis of angiogenesis-related
protein profiles using an antibody array
A Human Angiogenesis Antibody Array (R&D
Systems, Inc.) was used according to the manufacturer's protocol.
This method is a dot-based assay that enables the detection and
comparison of 55 angiogenesis-specific cytokines. Each array
membrane was exposed to X-ray film using a chemiluminescence
detection system (PerkinElmer, Inc.).
miRNA array
miRNA array analysis was performed as described in a
previous study (17). Total RNA
was extracted from the cancer cell lines using a miRNeasy Mini kit
(Qiagen GmbH), according to the manufacturer's instructions. RNA
samples typically exhibited A260/280 ratios between 1.9
and 2.1, as determined using an Agilent 2100 Bioanalyzer (Agilent
Technologies, Inc.). After performing RNA measurements with an RNA
6000 Nano kit (Agilent Technologies, Inc.), the samples were
labeled using a miRCURY Hy3/Hy5 Power Labeling kit and subsequently
hybridized to a human miRNA Oligo chip (v.21.0; Toray Industries,
Inc.). The chips were scanned with a 3D-Gene® Scanner
3000 (Toray Industries, Inc.), and the results were analyzed using
3D-Gene Extraction software, v1.2 (Toray Industries, Inc.).
Differences in miRNA expression between the telmisartan-treated and
untreated control samples were assessed using GeneSpring GX v10.0
(Agilent Technologies, Inc.). Quantile normalization was performed
on the raw data that were above the background level.
Differentially expressed miRNAs were determined by the Mann-Whitney
U test. The false discovery rate was computed using the
Benjamini-Hochberg method for multiple testing (27). Hierarchical clustering was
performed using the furthest-neighbor method with the absolute
uncentered Pearson's correlation coefficient as a metric. A heat
map was produced with the relative expression intensity for each
miRNA, in which the log2 of the intensity was median-centered for
each row.
Statistical analyses
In vitro experiments were performed in
triplicate and results are expressed as the mean ± SD. All
statistical analyses were performed using GraphPad Prism 6 software
(GraphPad Software, Inc.). Non-parametric Wilcoxon/Man-Whitney U
test was utilized to examine statistical significance between the
two groups, and Kruskal-Wallis test followed by Dunns post-hoc test
was performed to analyze multiple comparisons. P<0.05 was
considered statistically significant.
Results
Telmisartan inhibits human GIST cell
proliferation and viability by inducing cell cycle arrest at G0/G1
phase
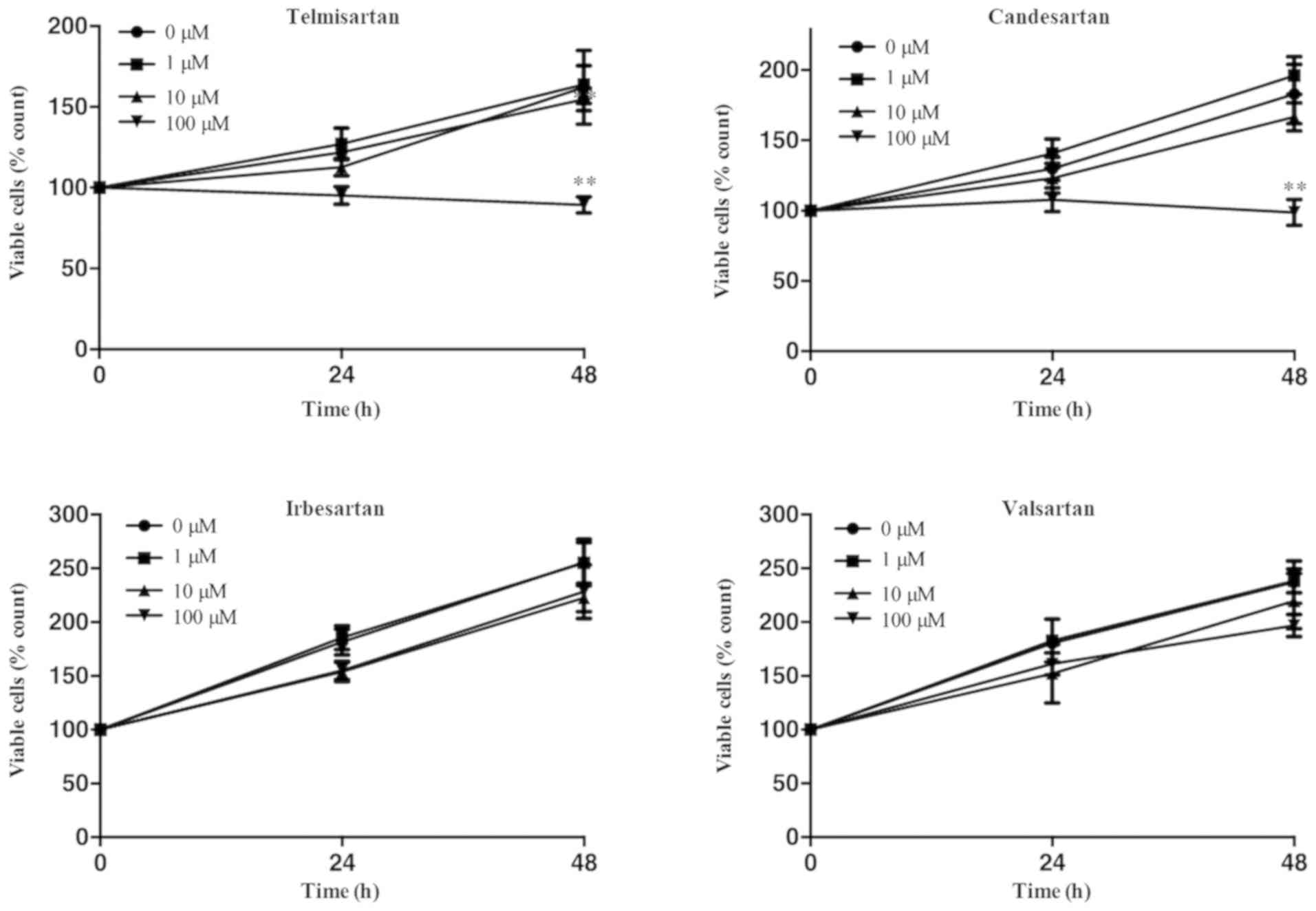
GIST-T1 cells were treated with 0, 1, 10 or 100 µm
of the ARBs (telmisartan, candesartan, irbesartan or valsartan) for
48 h. Telmisartan and candesartan inhibited the proliferation of
GIST-T1 cells (Fig. 1). No other
ARBs affected GIST-T1 cell viability. These results revealed that
telmisartan and candesartan inhibited cell proliferation dose- and
time-dependently in the GIST-1 cell lines (Fig. 1).
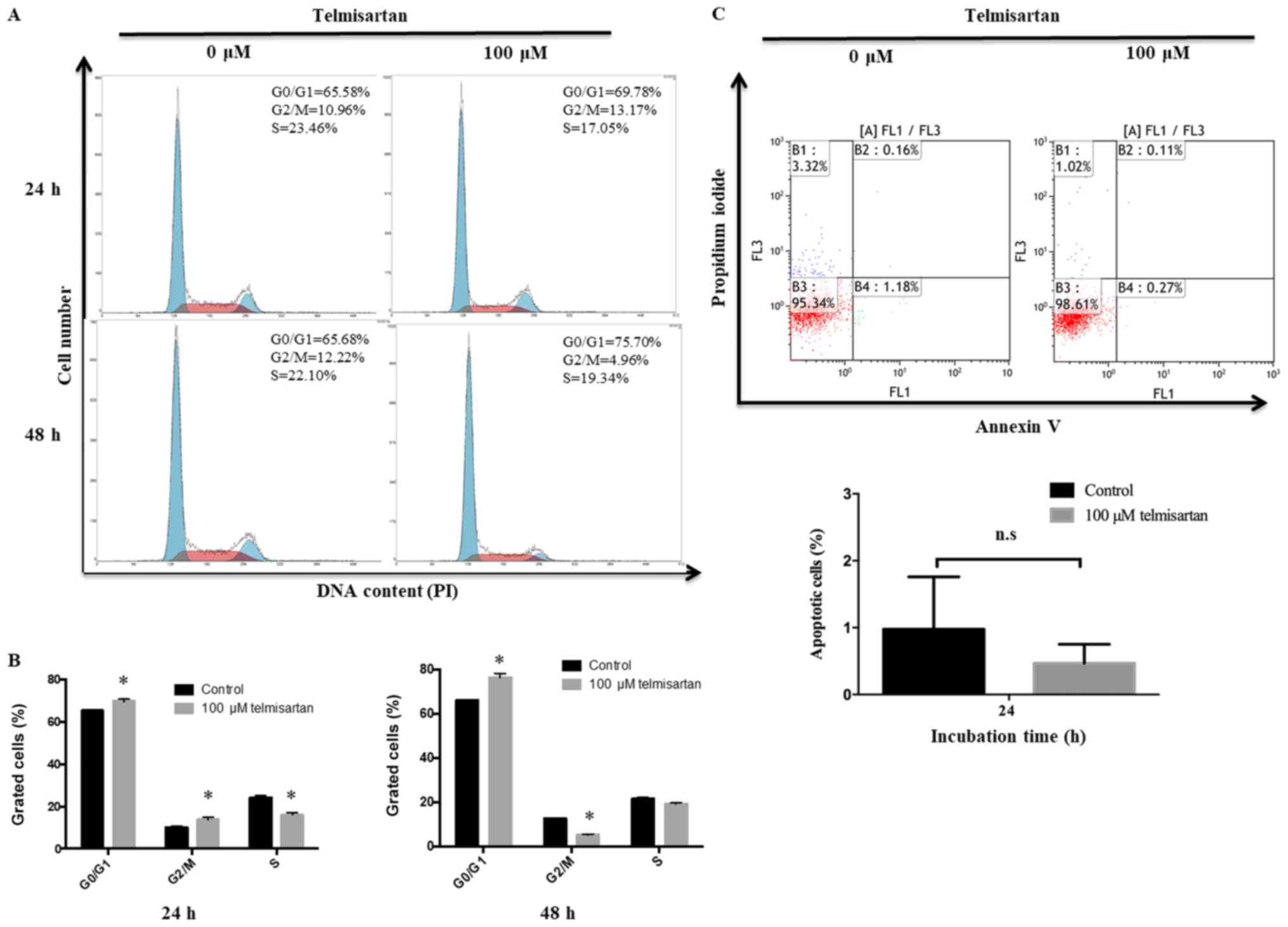
The effects of telmisartan on the cell cycle in
GIST-T1 cells was further investigated by flow cytometry. When
GIST-T1 cells were incubated with 100 µm of telmisartan, the number
of cells decreased in S and G2/M phases and increased in G0/G1
phases for 48 h compared with the control group (Fig. 2A and B). To investigate how
telmisartan influences GIST-T1 cell growth, apoptosis was analyzed.
The apoptotic effects of 0 and 100 µm telmisartan were measured by
flow cytometric analysis of annexin V-FITC/PI staining. According
to previous studies (18,28), 100 µm telmisartan induces apoptosis
at first 24 h, but not for 48 h at treatment. As shown in Fig. 2C, telmisartan did not induce a
significant change in the proportion of apoptotic GIST-T1 cells at
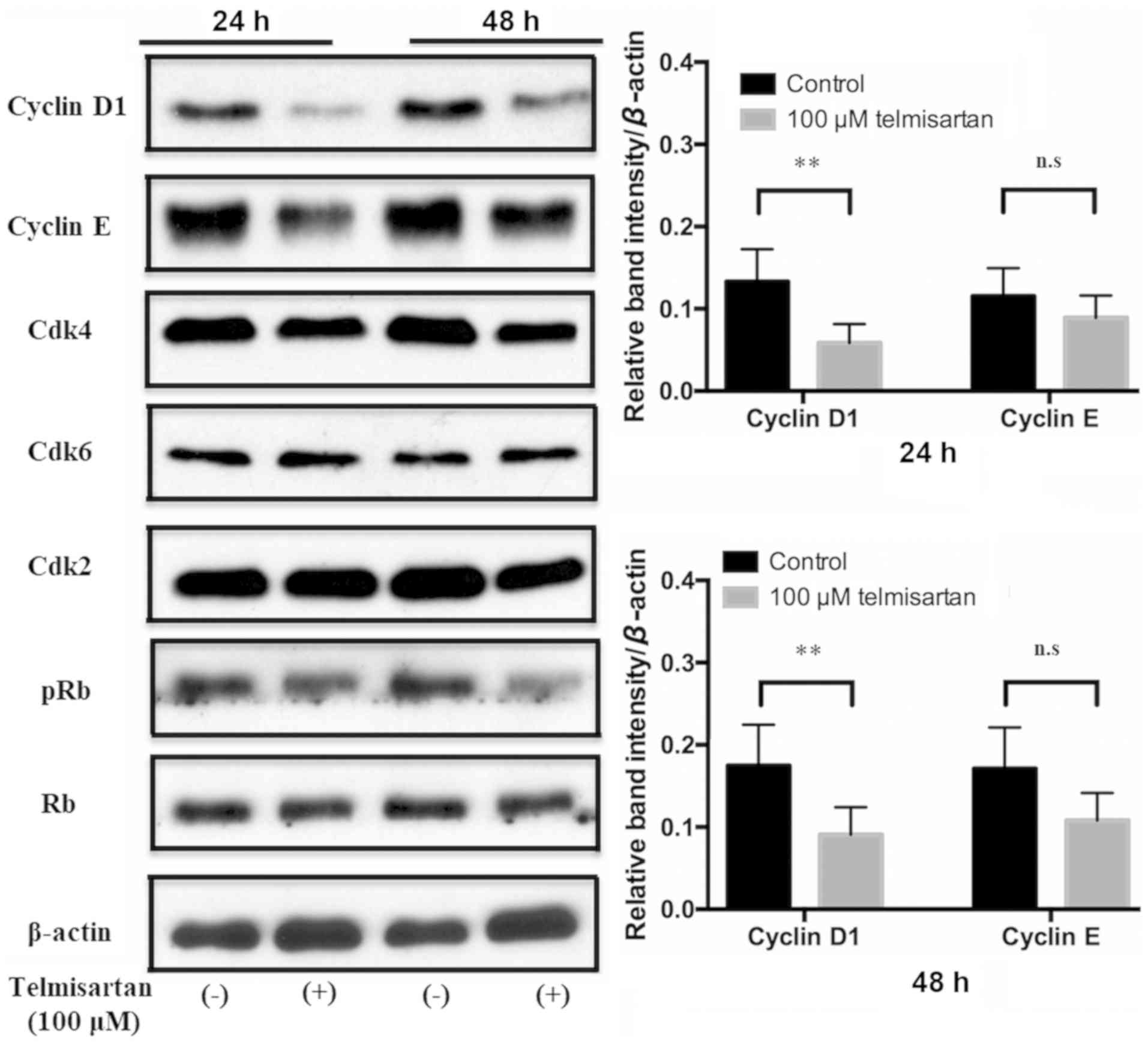
24 h post-treatment. Furthermore, the expression of cyclin D1
decreased in GIST-T1 cells after treatment with telmisartan
compared with the control group (Fig.
3). There were no notable changes in the protein expression
levels of cyclin E, Cdk2, Cdk4, Cdk6, p-Rb and Rb in
telmisartan-treated GIST-T1 cells. These results suggested that
telmisartan may inhibit cell cycle progression from G0/G1 to S
phase by decreasing the expression levels of cyclin D1.
Telmisartan induces AMPK
phosphorylation but does not suppress the AMPKα/mTOR pathway in
GIST-T1 cells
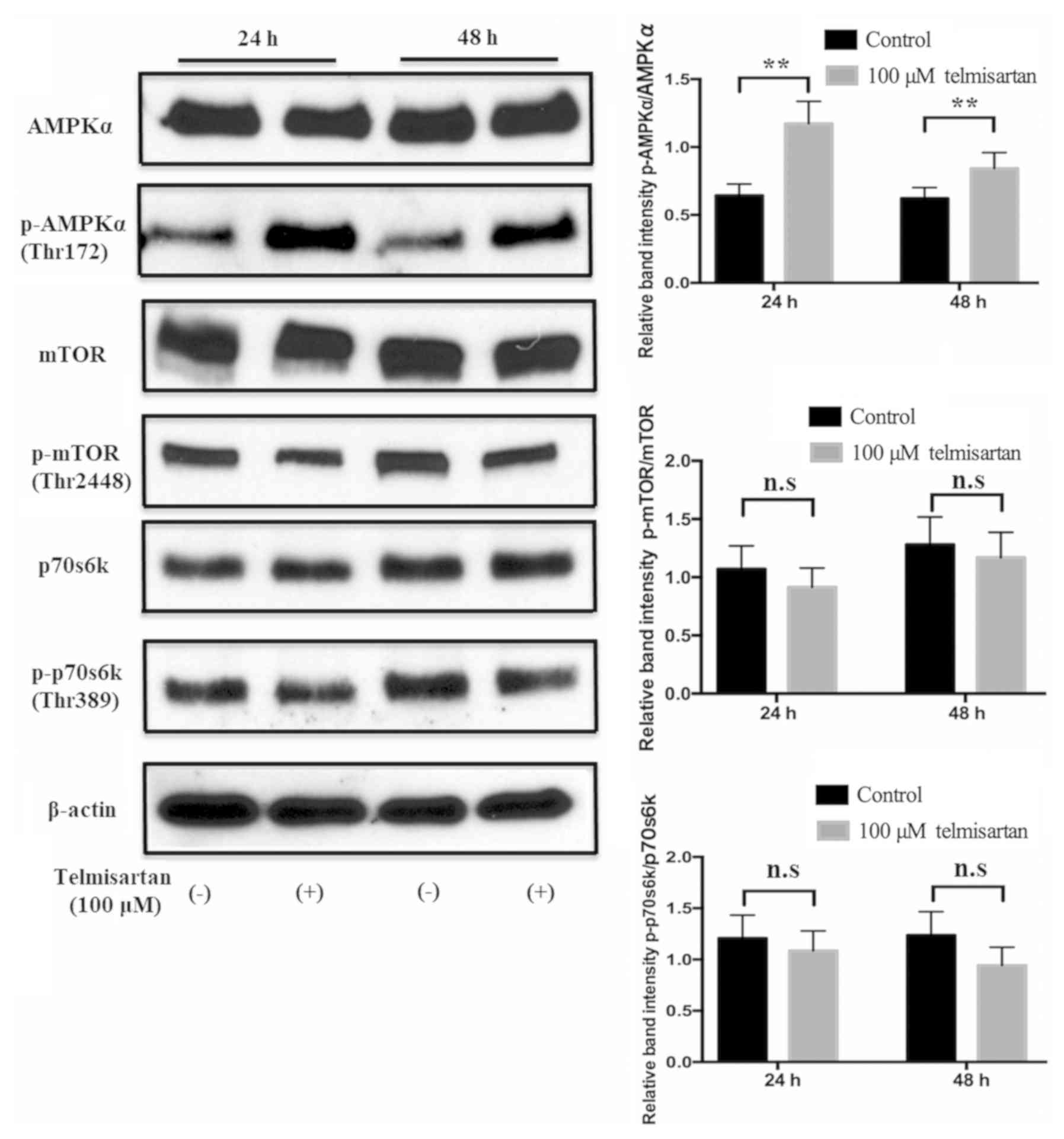
To examine the mechanism of telmisartan-induced cell
cycle arrest, AMPK/mTOR signaling was studied. Telmisartan
significantly induced phosphorylation of AMPKα in GIST-T1 cells
compared with the control group, and this activation lasted for at
least 48 h (Fig. 4). Conversely,
p-mTOR and p-p70S6K protein levels did not change in the GIST-T1
cells following treatment with telmisartan (Fig. 4). These results suggest that
telmisartan activated AMPKα, but did not suppress mTOR in GIST-T1
cells.
Association between telmisartan and
angiogenesis of GIST-T1 cells
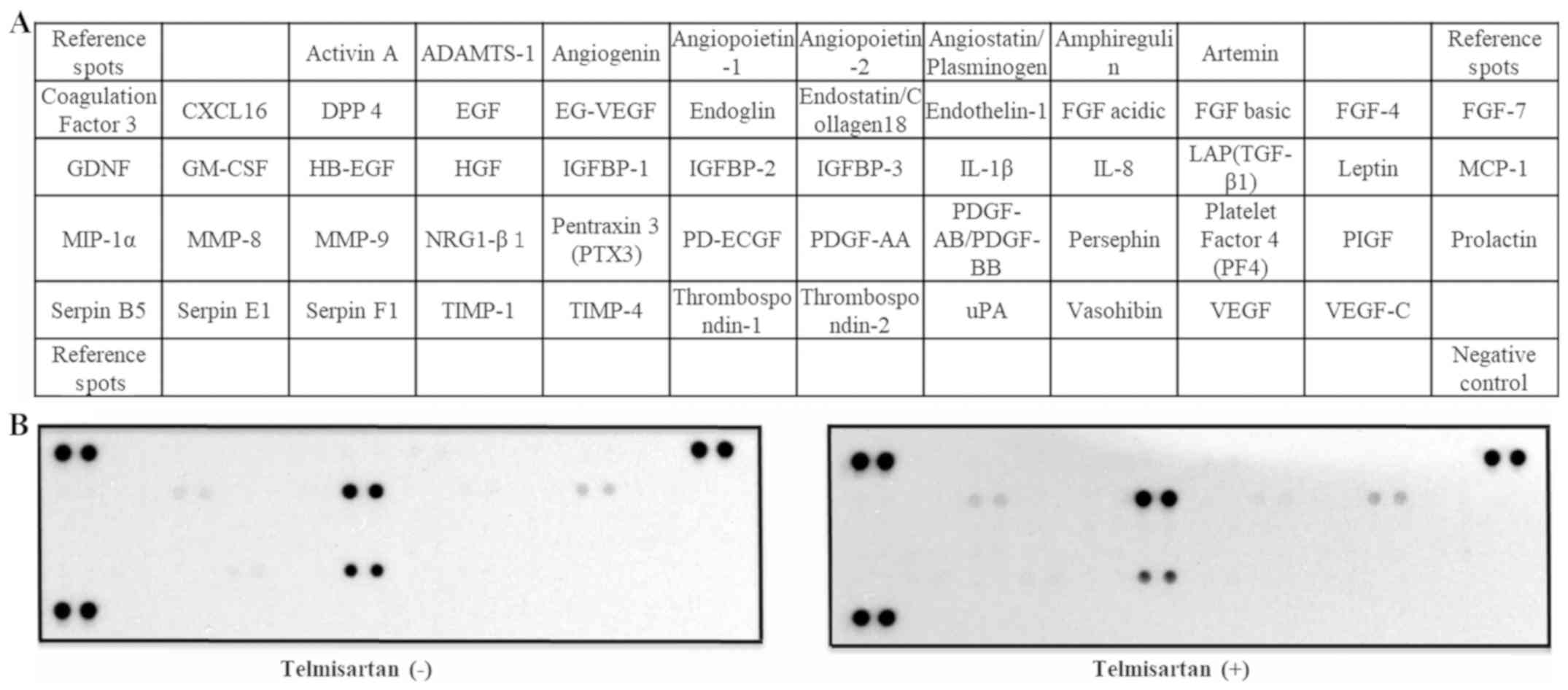
To identify angiogenic factors that may be affected
by telmisartan, a human angiogenesis array kit was applied to
GIST-T1 cells (Fig. 5A). The 55
angiogenesis-related proteins on the antibody array remained
unchanged after telmisartan treatment (Fig. 5B).
miRNA expression signatures are
different in telmisartan-treated and untreated GIST-1 cells
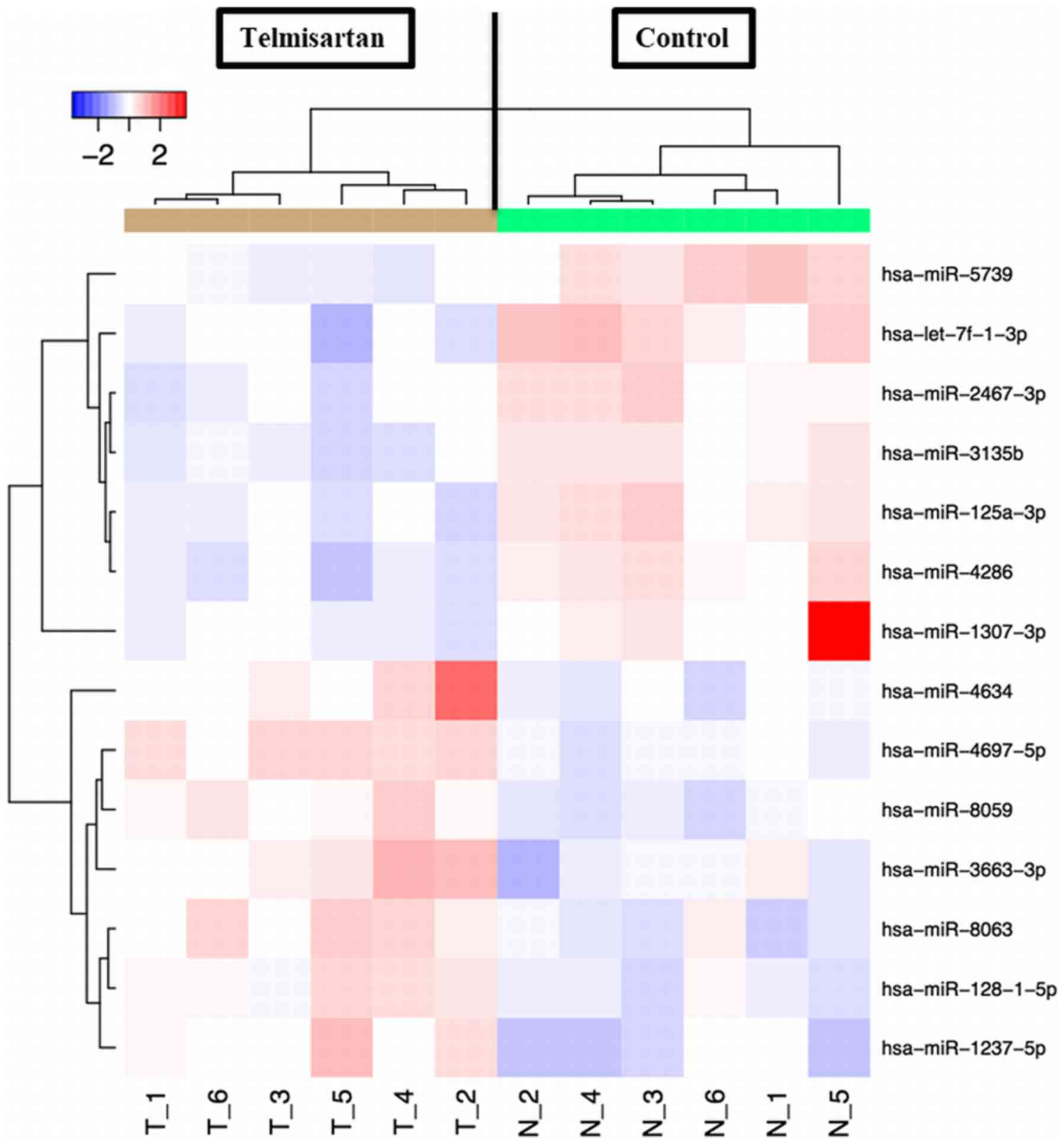
A miRNA microarray was used to examine the
expression levels of 2,555 miRNAs in GIST-T1 cells treated with or
without 100 µm telmisartan. After normalization and removing miRNAs
with missing values, 14 miRNAs were identified to be significantly
differentially expressed. Of them, 7 were found to be significantly
upregulated and the other 7 were downregulated (Fig. 6; Table
I).
 | Table I.Statistical results and chromosomal
locations of miRNAs in GIST-T1 cells treated with and without
telmisartan. |
Table I.
Statistical results and chromosomal
locations of miRNAs in GIST-T1 cells treated with and without
telmisartan.
| A, Upregulated
miRNAs | FC
(treated/untreated) | P-value | Chromosomal
location |
|---|
| hsa-miR-4634 | 2.03 | 0.0022 | 5q35.2 |
|
hsa-miR-1237-5p | 1.79 | 0.0087 | 11q13.1 |
|
hsa-miR-3663-3p | 1.70 | 0.0087 | 10q25.3 |
|
hsa-miR-4697-5p | 1.65 | 0.0022 | 11q25 |
| hsa-miR-8063 | 1.63 | 0.0087 | 15q14 |
| hsa-miR-8059 | 1.57 | 0.0050 | 21p12 |
|
hsa-miR-128-1-5p | 1.52 | 0.0087 | 2q21.3 |
|
| B, Downregulated
miRNAs | FC
(treated/untreated) | P-value | Chromosomal
location |
|
|
hsa-miR-1307-3p | 0.28 | 0.0087 | 10q24.33 |
|
hsa-let-7f-1-3p | 0.54 | 0.0043 | 9q22.32 |
| hsa-miR-5739 | 0.58 | 0.0022 | 22q12.1 |
| hsa-miR-4286 | 0.59 | 0.0050 | 8p23.1 |
| hsa-miR-3135b | 0.64 | 0.0050 | 6p21.32 |
|
hsa-miR-125a-3p | 0.64 | 0.0087 | 19q13.41 |
|
hsa-miR-2467-3p | 0.64 | 0.0050 | 2q37.3 |
Discussion
The present study focused on the antiproliferative
effects of telmisartan in GIST cells. According to previous
studies, telmisartan inhibits cell proliferation (6–8) and
tumor growth (9–11) in vitro and in vivo.
Moreover, epidemiologic studies have revealed that the use of ARBs
may increase the risk of cancer (29), whereas observational studies have
shown that ARBs may reduce cancer incidence and mortality (30,14).
However, the antiproliferative effects of telmisartan on GIST cells
remains unknown. To the best of our knowledge, the present study is
the first to demonstrate that telmisartan has antiproliferative
effects on human GIST-T1 cells in vitro.
Cyclin D1 and Cdks are important regulatory proteins
that promote the progression of the cell cycle during the crucial
restriction point at the G1/S phase transition (31). In the present study flow cytometric
analyses revealed that telmisartan induced cell cycle arrest in the
G0/G1 phase. Additionally, the level of the cell cycle regulatory
protein cyclin D1 was significantly reduced. Cyclin/Cdk complexes
are activated at different times during cell cycle progression
(32). Cdk4 and Cdk6 form
complexes with cyclin D1, which are required for G1 phase
progression, whereas Cdk2 forms a complex with cyclin E, which is
required for the G1-S transition (31,32).
The expression of various cell cycle-related molecules has been
associated with cancer cell metastasis and is related to cancer
prognosis (33,34). Previous studies have shown that
telmisartan induces cell cycle arrest in G0/G1 phase by decreasing
cyclin D1 and cyclin E (22,23).
Results from the present study indicated that telmisartan reduces
the expression of cyclin D1 and induces cell cycle arrest in
GIST-T1 cells.
Previous studies have shown that telmisartan
inhibits cell proliferation by inducing apoptosis in various types
of cancer, including gynecological (12) and urological (16,17)
cancer cell lines. Furthermore, it has been demonstrated that
telmisartan induces cell cycle progression, but not apoptosis in
esophageal adenocarcinoma (22).
In the present study, telmisartan did not increase the rates of
apoptosis of GIST-T1 cells, as shown by flow cytometry, which
suggested that telmisartan mainly inhibits GIST-T1 cell
proliferation by inducing cell cycle arrest but not apoptosis.
Telmisartan has been shown to activate AMPKα in
various cancer cells (22,23). Previous studies have shown that
telmisartan induces antiproliferative effects by phosphorylating
AMPKα in esophageal adenocarcinoma cells, suggesting that
AMPKα/mTOR pathway activation inhibits cell cycle regulatory
proteins (22,23). In the present study, proteins
involved in the AMPKα/mTOR signaling pathways, such as p-AMPKα,
p-mTOR and p-p70S6K, were evaluated. Telmisartan treatment led to
phosphorylation of AMPK, but it did not inhibit p70S6K or mTOR
phosphorylation in GIST-T1 cells, which suggested that telmisartan
induced AMPKα signaling in GIST-T1 cells but did not affect
downstream mTOR in GIST-T1 cells.
The tumor microenvironment is the product of
crosstalk between different cell types and serves a critical role
in promoting the initiation and progression of malignancy (35). Okazaki et al (13) reported that candesartan suppressed
tumor growth and angiogenesis in a xenograft mouse model. Although
telmisartan and candesartan are both known as ARBs, telmisartan is
also a partial agonist of PPAR-γ (7). However, in the present study, the 55
screened angiogenesis-related molecules had no changes after
treatment with telmisartan in GIST-T1 cells. Thus, it was
speculated that PPAR-γ does not play a major role in angiogenesis
of GIST-T1 cells.
miRNAs are endogenous mediators of gene expression
through site-specific binding at the 3′untranslated region of
target mRNAs leading to degradation or inactivate protein synthesis
(36). miRNAs regulate a number of
biological processes, such as cancer cell proliferation, tumor
growth, differentiation, apoptosis and energy metabolism (37). miR-1307-3p was downregulated in
GIST-T1 cells treated with telmisartan. miR-1307-3p originates from
the 3′end of pre-miR-1307, but its function remains largely
unknown. It has been reported that miR-1307 is overexpressed in
chemoresistant ovarian (38) and
prostate cancer tissues (39).
miR-1307 overexpression has been shown to inhibit levels of the
cell cycle inhibitors, p21 and p27 in prostate cancer cells
(39). Based on these studies and
the present data, it is suggested that the antiproliferative
effects of telmisartan may alter several miRNAs. However, there are
some limitations to the current study. Firstly, the inhibition
effects of telmisartan on GIST-T1 cells were optimized only for
in vitro growth and proliferation. Secondary, the in
vitro study was conducted using a higher dose of telmisartan
than that used in human treatments (1–10 µM) (7). Thus, it may be difficult to translate
these conditions in clinical setting, especially as the
pharmacokinetics of the drug may be different in a culture setting
and in the human body.
In conclusion, it was demonstrated that telmisartan
treatment inhibited human GIST cell proliferation, which may be by
inducing cell cycle arrest and decreasing the expression of cyclin
D1.
Acknowledgements
Not applicable.
Funding
The current study was supported by The Japan Society
for the Promotion of Science (JSPS) KAKENHI (grant no.
18K11023).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HK, SF and TsM conceived and deigned the study. TaM,
AF, TC, STT, NK, NN, TY, TT, KyO, JT, KF, TN, HY, AM, KeO, YS and
HM performed the experiments and were major contributors in writing
the manuscript. HI interpreted the data and drafted the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ARB
|
angiotensin II type 1 receptor
blocker
|
|
AMPK
|
AMP-activated protein kinase
|
|
GIST
|
gastrointestinal stromal tumor
|
|
mTOR
|
mammalian target of rapamycin
|
|
PDGFRA
|
platelet-derived growth factor
receptor-α
|
References
|
1
|
Rubin BP, Heinrich MC and Corless CL:
Gastrointestinal stromal tumor. Lancet. 369:1731–1741. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Giuliano K, Ejaz A, Reames BN, Choi W,
Sham J, Gage M, Johnston FM and Ahuja N: Comparing the long-term
outcomes among patients with stomach and small intestine
gastrointestinal stromal tumors: An analysis of the National Cancer
Database. J Surg Oncol. 118:486–492. 2018.PubMed/NCBI
|
|
3
|
Corless CL, Fletcher JA and Heinrich MC:
Biology of gastrointestinal stromal tumors. J Clin Oncol.
22:3813–3825. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fletcher CD, Berman JJ, Corless C,
Gorstein F, Lasota J, Longley BJ, Miettinen M, O'Leary TJ, Remotti
H, Rubin BP, et al: Diagnosis of gastrointestinal stromal tumors: A
consensus approach. Hum Pathol. 33:459–465. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miettinen M and Lasota J: Gastrointestinal
stromal tumors: Pathology and prognosis at different sites. Semin
Diagn Pathol. 23:70–83. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamamoto H and Oda Y: Gastrointestinal
stromal tumor: Recent advances in pathology and genetics. Pathol
Int. 65:9–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Benson SC, Pershadsingh HA, Ho CI,
Chittiboyina A, Desai P, Pravenec M, Qi N, Wang J, Avery MA and
Kurtz TW: Identification of telmisartan as a unique angiotensin II
receptor antagonist with selective PPAR-modulating activity.
Hypertension. 43:993–1002. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kinoshita J, Fushida S, Harada S, Yagi Y,
Fujita H, Kinami S, Ninomiya I, Fujimura T, Kayahara M, Yashiro M,
et al: Local angiotensin II-generation in human gastric cancer:
Correlation with tumor progression through the activation of
ERK1/2, NF-κB and survivin. Int J Oncol. 34:1573–1582. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Okamoto K, Tajima H, Ohta T, Nakanuma S,
Hayashi H, Nakagawara H, Onishi I, Takamura H, Ninomiya I, Kitagawa
H, et al: Angiotensin II induces tumor progression and fibrosis in
intrahepatic cholangiocarcinoma through an interaction with hepatic
stellate cells. Int J Oncol. 37:1251–1259. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Du N, Feng J, Hu LJ, Sun X, Sun HB, Zhao
Y, Yang YP and Ren H: Angiotensin II receptor type 1 blockers
suppress the cell proliferation effects of angiotensin II in breast
cancer cells by inhibiting AT1R signaling. Oncology Rep.
27:18932012.
|
|
11
|
Chen X, Meng Q, Zhao Y, Liu M, Li D, Yang
Y, Sun L, Sui G, Cai L and Dong X: Angiotensin II type 1 receptor
antagonists inhibit cell proliferation and angiogenesis in breast
cancer. Cancer Lett. 328:318–324. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koyama N, Nishida Y, Ishii T, Yoshida T,
Furukawa Y and Narahara H: Telmisartan induces growth inhibition,
DNA double-strand breaks and apoptosis in human endometrial cancer
cells. PLoS One. 9:e930502014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Okazaki M, Fushida S, Harada S, Tsukada T,
Kinoshita J, Oyama K, Tajima H, Ninomiya I, Fujimura T and Ohta T:
The angiotensin II type 1 receptor blocker candesartan suppresses
proliferation and fibrosis in gastric cancer. Cancer Lett.
355:46–53. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bhaskaran K, Douglas I, Evans S, van Staa
T and Smeeth L: Angiotensin receptor blockers and risk of cancer:
Cohort study among people receiving antihypertensive drugs in UK
general practice research database. BMJ. 344:e26972012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Makar GA, Holmes JH and Yang YX:
Angiotensin-converting enzyme inhibitor therapy and colorectal
cancer risk. J Natl Cancer Inst. 106:djt3742014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Funao K, Matsuyama M, Kawahito Y, Sano H,
Chargui J, Touraine JL, Nakatani T and Yoshimura R: Telmisartan is
a potent target for prevention and treatment in human prostate
cancer. Oncol Rep. 20:295–300. 2008.PubMed/NCBI
|
|
17
|
Funao K, Matsuyama M, Kawahito Y, Sano H,
Chargui J, Touraine JL, Nakatani T and Yoshimura R: Telmisartan as
a peroxisome proliferator-activated receptor-gamma ligand is a new
target in the treatment of human renal cell carcinoma. Mol Med Rep.
2:193–198. 2009.PubMed/NCBI
|
|
18
|
Lee L, Mafura B, Lauscher J, Seeliger H,
Kreis M and Gröne J: Antiproliferative and apoptotic effects of
telmisartan in human colon cancer cells. Oncol Lett. 8:2681–2866.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rehman G, Shehzad A, Khan AL and Hamayun
M: Role of AMP-activated protein kinase in cancer therapy. Arch
Pharm (Weinheim). 347:457–468. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dann SG and Thomas G: The amino acid
sensitive TOR pathway from yeast to mammals. FEBS Lett.
580:2821–2829. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fingar DC and Blenis J: Target of
rapamycin (TOR): An integrator of nutrient and growth factor
signals and coordinator of cell growth and cell cycle progression.
Oncogene. 23:3151–3171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fujihara S, Asahiro M, Ogawa K, Tadokoro
T, Chiyo T, Kato K, Kobara H, Mori H, Iwama H and Masaki T: The
angiotensin II type 1 receptor antagonist telmisartan inhibits cell
proliferation and tumor growth of esophageal adenocarcinoma via the
AMPKα/mTOR pathway in vitro and in vivo. Oncotarget. 8:8536–8549.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oura K, Tadokoro T, Fujihara S, Morishita
A, Chiyo T, Samukawa E, Yamana Y, Fujita K, Sakamoto T, Nomura T,
et al: Telmisartan inhibits hepatocellular carcinoma cell
proliferation in vitro by inducing cell cycle arrest. Oncol Rep.
38:2825–2835. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liang LH and He XH: Macro-management of
microRNAs in cell cycle progression of tumor cells and its
implications in anti-cancer therapy. Acta Pharmacol Sin.
32:1311–1320. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Towbin H, Staehelin T and Gordon J:
Electrophoretic transfer of proteins from polyacrylamide gels to
nitrocellulose sheets: Procedure and some applications. Proc Natl
Acad Sci USA. 76:4350–4354. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J Roy Stat Soc. 57:289–300. 1995.
|
|
28
|
de Araújo Júnior RF, Leitão Oliveira AL,
de Melo Silveira RF, de Oliveira Rocha HA, de França Cavalcanti P
and de Araújo AA: Telmisartan induces apoptosis and regulates Bcl-2
in human renal cancer cells. Exp Biol Med (Maywood). 240:34–44.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sipahi I, Debanne SM, Rowland DY, Simon DI
and Fang JC: Angiotensin-receptor blockade and risk of cancer:
Meta-analysis of randomised controlled trials. Lancet Oncol.
11:627–636. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shen J, Huang YM, Wang M, Hong XZ, Song
XN, Zou X, Pan YH, Ling W, Zhu MH, Zhang XX, et al:
Renin-angiotensin system blockade for the risk of cancer and death.
J Renin Angiotensin Aldosterone Syst. 17(pii):
14703203166566792016.PubMed/NCBI
|
|
31
|
Masaki T, Shiratori Y, Rengifo W, Igarashi
K, Yamagata M, Kurokohchi K, Uchida N, Miyauchi Y, Yoshiji H,
Watanabe S, et al: Cyclins and cyclin-dependent kinases:
Comparative study of hepatocellular carcinoma versus cirrhosis.
Hepatology. 37:534–543. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kato K, Gong J, Iwama H, Kitanaka A, Tani
J, Miyoshi H, Nomura K, Mimura S, Kobayashi M, Aritomo Y, et al:
The antidiabetic drug metformin inhibits gastric cancer cell
proliferation in vitro and in vivo. Mol Cancer Ther. 11:549–560.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Itami A, Shimada Y, Watanabe G and Imamura
M: Prognostic value of p27Kip1 and cyclin D1 expression
in esophageal cancer. Oncology. 57:311–317. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fukuchi M, Fukai Y, Kimura H, Sohda M,
Miyazaki T, Nakajima M, Masuda N, Tsukada K, Kato H and Kuwano H:
Prolyl isomerase Pin1 expression predicts prognosis in patients
with esophageal squamous cell carcinoma and correlates with
cyclinD1 expression. Int J Oncol. 29:329–334. 2006.PubMed/NCBI
|
|
35
|
Samples J, Willis M and Klauber-DeMore N:
Targeting angiogenesis and the tumor microenvironment. Surg Oncol
Clin N Am. 22:629–639. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Masaki T: MicroRNA and hepatocellular
carcinoma. Hepatol Res. 39:751–752. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Morishita A and Masaki T: miRNA in
hepatocellular carcinoma. Hepatology Res. 45:128–141. 2015.
View Article : Google Scholar
|
|
38
|
Zhou Y, Wang M, Wu J, Jie Z, Chang S and
Shuang T: The clinicopathological significance of miR-1307 in
chemotherapy resistant epithelial ovarian cancer. J Ovarian Res.
8:232015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qiu X and Dou Y: miR-1307 promotes the
proliferation of prostate cancer by targeting FOXO3A. Biomed
Pharmacother. 88:430–435. 2017. View Article : Google Scholar : PubMed/NCBI
|