Introduction
Chronic kidney disease (CKD) is an increasing public
health problem with substantial health care costs and morbidity
(1). Tubulointerstitial fibrosis
(TIF) is a common result of chronic kidney disease; it leads to
destruction of the normal kidney structures and irreversible loss
of kidney function (2,3). The induction of TIF causes the
disease to progress to renal failure (4), and several hypotheses have been
developed to elucidate the progression of TIF, including mechanisms
such as inflammation, epithelial-mesenchymal transition,
senescence, chronic hypoxia and the contribution of reactive oxygen
species (5,6). The progression of TIF has been found
to be strongly correlated with renal tubular lesions (7). Renal tubular epithelial cells (RTECs)
are the most abundant cell type in the kidney and produce various
active factors, such as growth factors, interleukins, inflammatory
factors, chemokines and cell adhesion molecules (8,9).
Thus, RTECs are involved in numerous processes, such as epithelial
cell transdifferentiation, inflammatory cell activation and cell
proliferation, and notably, they have also been observed to serve
an important role in TIF progression (9,10).
RTECs were originally considered as terminally
differentiated cells; however, they have since been found to exert
significant proliferative ability (11). Kidney injury has been discovered to
stimulate RTECs to release cytokines and rapidly enter the cell
cycle, which has been demonstrated to be important for the rapid
recovery of renal function following acute injury (12,13).
Proliferation is accompanied by the presence of damage factors
(12); RTECs are highly sensitive
to damage factors upon entering the cell cycle, and when the damage
factors persist, they are also found to have an adverse effect on
the subsequent proliferation response (14–16).
In a TIF model, RTEC injury was found to be positively correlated
with compensatory proliferation, in which the proliferation of
RTECs subsequently promoted cell death (17,18).
The sensitivity of cells to kidney injury is different, that is,
differentiated complete cells are more sensitive compared with stem
cells, which are more sensitive compared with proliferating cells
(14,19). Upon injury, the stimulation of
surviving RTECs to enter the division cycle has been found to
increase their sensitivity to the external environment, such as
hypoxia and drug toxicity, which promotes the death of RTECs;
therefore, a vicious circle of ‘proliferation and death’ is formed
(10). In this cycle, the
signaling pathways in RTECs are contradictory; however, the
specific mechanisms involved remain unclear.
The DNA damage response (DDR) is a multicomplex
network of signaling pathways that are involved in DNA damage
repair, cell cycle checkpoints and apoptosis (20). A previous tumor cell study showed
that drugs or radiation will not cause cell death due to damage to
DNA, which may be associated with proliferative and
death-associated mechanisms (21).
Ataxia telangiectasia mutated (ATM) serves a central role in
phosphorylating several important proteins that activate the DDR
and mobilize this intricate DDR network (22).
Aristolochic acids (AA) are nephrotoxic and
carcinogenic phytochemicals found in many plant species (23). AA-dependent human nephropathy
occurs as the result of the environmental exposure to
Aristolochia subspecies or its use as a traditional
botanical therapy and is characterized by severe renal fibrosis and
upper urothelial carcinoma (24).
In the present study, an AA renal fibrosis model was established
in vitro to investigate whether the vicious
proliferation-death cycle is a pathophysiological process of TIF
following chronic injury to the kidneys. In addition, the
underlying molecular mechanisms of the proliferation-death cycle
were investigated in the TIF model. It was hypothesized that this
malignant cycle of RTECs serves as the main driver of TIF
progression following the persistent release of injury factors, and
that DDR-induced cell death serves an important role in its
molecular mechanism.
Materials and methods
Reagents
FBS, trypsin and RPMI-1640 medium were obtained from
Gibco (Thermo Fisher Scientific, Inc.), and streptomycin and
penicillin were purchased from Sigma-Aldrich (Merck KGaA). AA was
obtained from Chengdu Manst Biotech Co., Ltd. (http://www.cdmust.com/).
Cell culture and transfection. The human proximal
tubular epithelial cell line, human kidney (HK)-2, was purchased
from The Cell Bank of Type Culture Collection of the Chinese
Academy of Sciences. Cells were cultured in RPMI-1640 medium,
supplemented with 10% FBS and 1% antibiotics (0.1 mg/ml
streptomycin and 100 U/ml penicillin), and maintained in a
humidified with 5% CO2 at 37°C.
Cells were subsequently transfected with lentiviral
vectors containing short hairpin (sh)RNA (Gima Gene) targeting p21
(shp21), ATM (shATM) or shRNA-negative control (shCon). The
lentiviral vector system consists of three plasmids: GV lentiviral
vector series, pHelper 1.0 vector and pHelper 2.0 vector.
http://www.genechem.com.cn/Zaiti.aspx?zt=GV115.
Briefly, HK-2 cells were plated and cultured for 12 h at 37°C and
subsequently, lentiviral vectors encoding shp21, shATM or shCon (45
µg/ml) were mixed with the culture medium. Polybrene, at a final
concentration of 8 µg/ml, was added to the culture medium to
facilitate the transfection. Following incubation for 6 h at 37°C,
fresh RPMI-1640 medium was added to the cells and cultured for a
further 48 h at 37°C. When cells reached 70–90% confluence, the
medium of the transfected cells was replaced with fresh RPMI-1640
medium containing 10% FBS and 20 µg/ml AA, whereas the control
groups (untreated group) were replaced with normal RPMI-1640 medium
containing 10% FBS. The cells were then cultured for 24 h before
the cells were collected for subsequent analysis.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and total RNA was reverse transcribed into cDNA (Takara Bio,
Inc.) according to the manufacturers' protocol. RT-qPCR was
subsequently performed using the SYBR® Green Master Mix
kit (Takara Bio, Inc.) and a CFX96 Touch Real-Time PCR Detection
system (Bio-Rad Laboratories, Inc.) according to the manufacturers'
protocol (holding Stage, 95°C for 3 min; cycling stage, 95°C for 5
sec, 60°C for 30 sec, 40 cycles; melt curve stage, 95°C for 15 sec
then 60°C for 1 min). The following primer pairs were used for the
qPCR: p21, forward 5′-TCTCAGGGTCGAAAACGG-3′, reverse
5′-TGGGCGGATTAGGGCTTC-3′; ATM, forward
5′-ATAGATTGTGTAGGTTCCGATGG-3′, reverse
5′-CATCTTGTCTCAGGTCATCACG-3′; GAP DH, forward
5′-GTGAACCATGAGAAGTATGACAAC-3′, reverse
5′-CATGAGTCCTTCCACGATACC-3′. Expression levels normalized to the
internal reference gene GAPDH and were quantified using the
2−ΔΔCq method (25).
Western blotting
The cells were homogenized in ice-cold RIPA lysate
(Beyotime Institute of Biotechnology) for cleavage, and phosphatase
inhibitor (Beyotime Institute of Biotechnology) and PMSF (Swiss
Roche, Inc.) were added at the same time (RIPA lysate:phosphatase
inhibitor:PMSF=100:10:1). Western blot analysis was performed as
previously described (26). Total
protein was extracted from transfected HK-2 cells and quantified
using a BCA assay kit (Beyotime Institute of Biotechnology). A
total of 60 µg of protein were separated by 10% SDS-PAGE and
subsequently transferred onto polyvinylidene fluoride membranes
(0.2/0.45 µm; EMD Millipore) at 300 mA and blocked with 5% nonfat
milk for 1 h at room temperature. The membranes were incubated at
4°C overnight with primary antibodies against the following target
proteins: Phosphorylated (p)-ATM (rabbit; 1:1,000; cat. no.
ab81292; Abcam); ATM (mouse; 1:2,000; cat. no. ab78; Abcam);
p-checkpoint kinase-2 (p-Chk2; rabbit; 1:1,000; cat. no. 64o0492;
Affinity Biosciences); checkpoint kinase 2 (Chk2; rabbit; 1:5,000;
cat. no. ab109413; Abcam); rabbit histone H2 A.X (r-H2AX (rabbit;
1:1,000; cat. no. ab11175; Abcam); p-p53 (rabbit; 1:1,000; cat. no.
ab1431; Abcam); p53 (mouse; 1:1,000; cat. no. ab26; Abcam) p21
(rabbit; 1:1,000; cat. no. BS6561; Bioworld Technology, Inc.); CDK2
(rabbit; 1:1,000; cat. no. ab32147; Abcam); cyclin D1 (rabbit;
1:1,000; cat. no. ab16663; Abcam); proliferating cell nuclear
antigen (PCNA; mouse; 1:1,000; ab29; Abcam); Bax (rabbit; 1:1,000;
cat. no. 2772S; Cell Signaling Technology, Inc.); Bcl-2 (mouse;
1:1,000; cat. no. 15071; Cell Signaling Technology, Inc.) and
anti-GAPDH (rabbit; 1:10,000; cat. no. AP0063; Bioworld Technology,
Inc.). Following the primary antibody incubation, the membranes
were washed with TBST (0.1% Tween-20) and subsequently incubated
with a horseradish peroxidase-conjugated goat anti-rabbit (rabbit;
1:5,000; cat. no. BL003A; BioSharp Technology, Inc.) or anti-mouse
IgG secondary antibody (mouse; 1:5,000; cat. no. BL001A; BioSharp
Technology, Inc.) for 1 h at room temperature. Protein expression
levels were normalized to GAPDH. Protein bands were visualized
using an enhanced chemiluminescence kit and protein expressions
were semi-quantified using Image-Pro Plus software 6.0 (Media
Cybernetics, Inc.).
Flow cytometric analysis of
apoptosis
Transfected HK-2 cells were harvested, routinely
digested with trypsin and washed twice with PBS. The cells were
subsequently resuspended in 500 µl binding buffer and stained with
5 µl Annexin V-FITC and 5 µl propidium iodide for 10 min at room
temperature in the dark (Apoptosis Detection kit, C1062M, Beyotime
Institute of Biotechnology). Apoptotic cells were subsequently
analyzed using a CytoFLEX flow cytometer (Beckman Coulter, Inc.),
according to the manufacturer's protocol. The results were analyzed
with FlowJo 7.6 software (FlowJo LLC).
Caspase-3 activity measurement
Caspase-3 activity was analyzed using a Caspase-3
Activity Assay kit (Beyotime Institute of Biotechnology) according
to the manufacturer's protocol. Briefly, attached and floating
cells were lysed for 15 min at 4°C after centrifugation at 4°C
(2×106 cells added to 100 µl lysate) and then incubated
with Ac-DEVD-p-nitroaniline (p-NA) for 1 h at 37°C. The levels of
p-NA, which reflects the caspase-3 activity, were then determined
at a wavelength of 405 nm with a microplate reader.
Statistical analysis
Statistical analysis was performed using SPSS 20.0
software (IBM Corp.) and data were presented as the mean ± standard
error of the mean. Statistical differences were determined using a
one-way ANOVA, followed by Tukey's post hoc test for multiple
comparisons, whereas an ANOVA and Tamhane T2 post hoc test was
performed with data demonstrating a heterogeneous variance. All
data were obtained from >3 independent experiments. P<0.05
was considered to indicate a statistically significant
difference.
Results
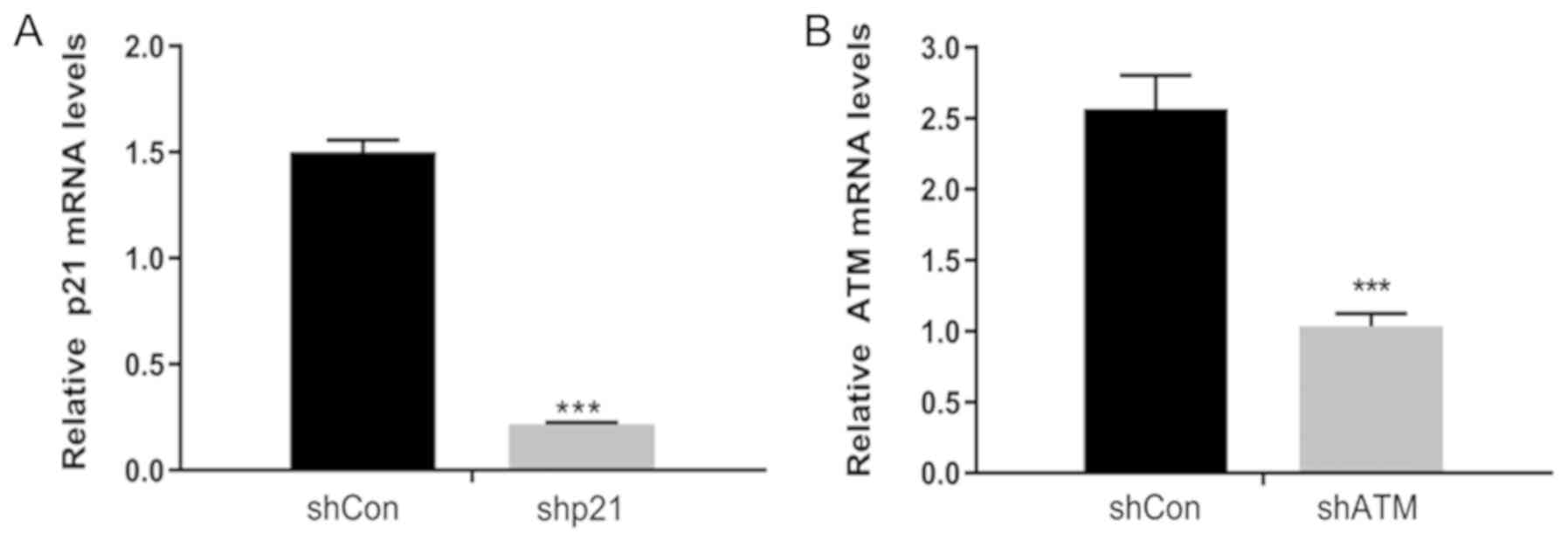
Successful transfection of
lentiviral-vectors encoding shRNAs into HK-2 cells
To confirm that the shRNA vectors have been
successfully transfected into the HK-2 cells, the mRNA expression
levels of p21 and ATM following transfection were analyzed using
RT-qPCR. shp21 and shATM were successfully transfected into HK-2
cells, as demonstrated by the significantly reduced expression
levels of p21 and ATM, respectively, compared with the shCon groups
(Fig. 1A and B).
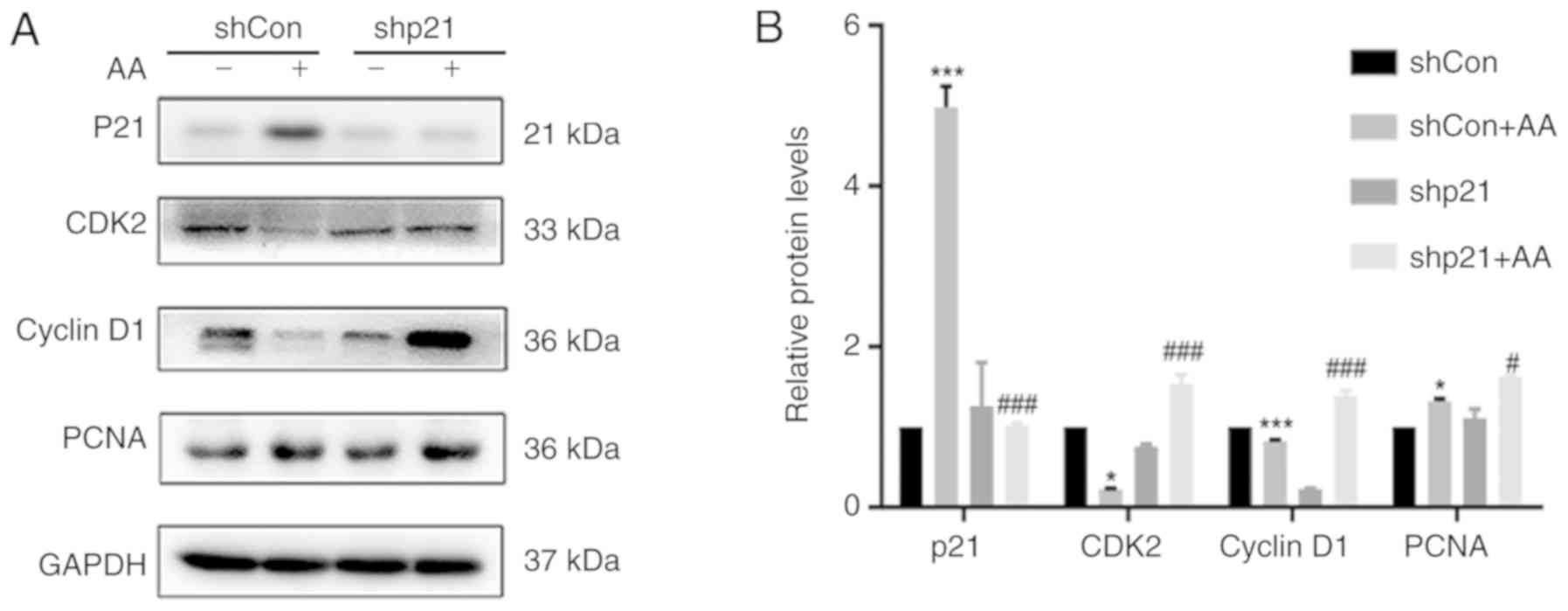
p21 knockdown promotes cell cycle
progression and cell proliferation in HK-2 cells
To investigate the effects of p21 on the cell cycle
and cell proliferation, the protein expression levels of CDK2,
cyclin D1 and PCNA following AA-induced injury were analyzed using
western blotting (Fig. 2A and B).
The shCon + AA group demonstrated significantly increased p21
protein expression levels and significantly decreased CDK2 and
cyclin D1 protein expression levels compared with non-AA treated
shCon group; however, the shp21 + AA group significantly inhibited
the expression levels of p21 and significantly increased the
expression levels of CDK2 and cyclin D1 proteins compared with the
shCon + Aa group. In addition, following AA-induced injury, cell
proliferation was increased, as demonstrated by increased PCNA
expression levels, whereas following p21 knockdown with shRNA
(shp21 + AA group), PCNA expression levels were significantly
increased and cell proliferation was promoted compared with shCon +
AA.
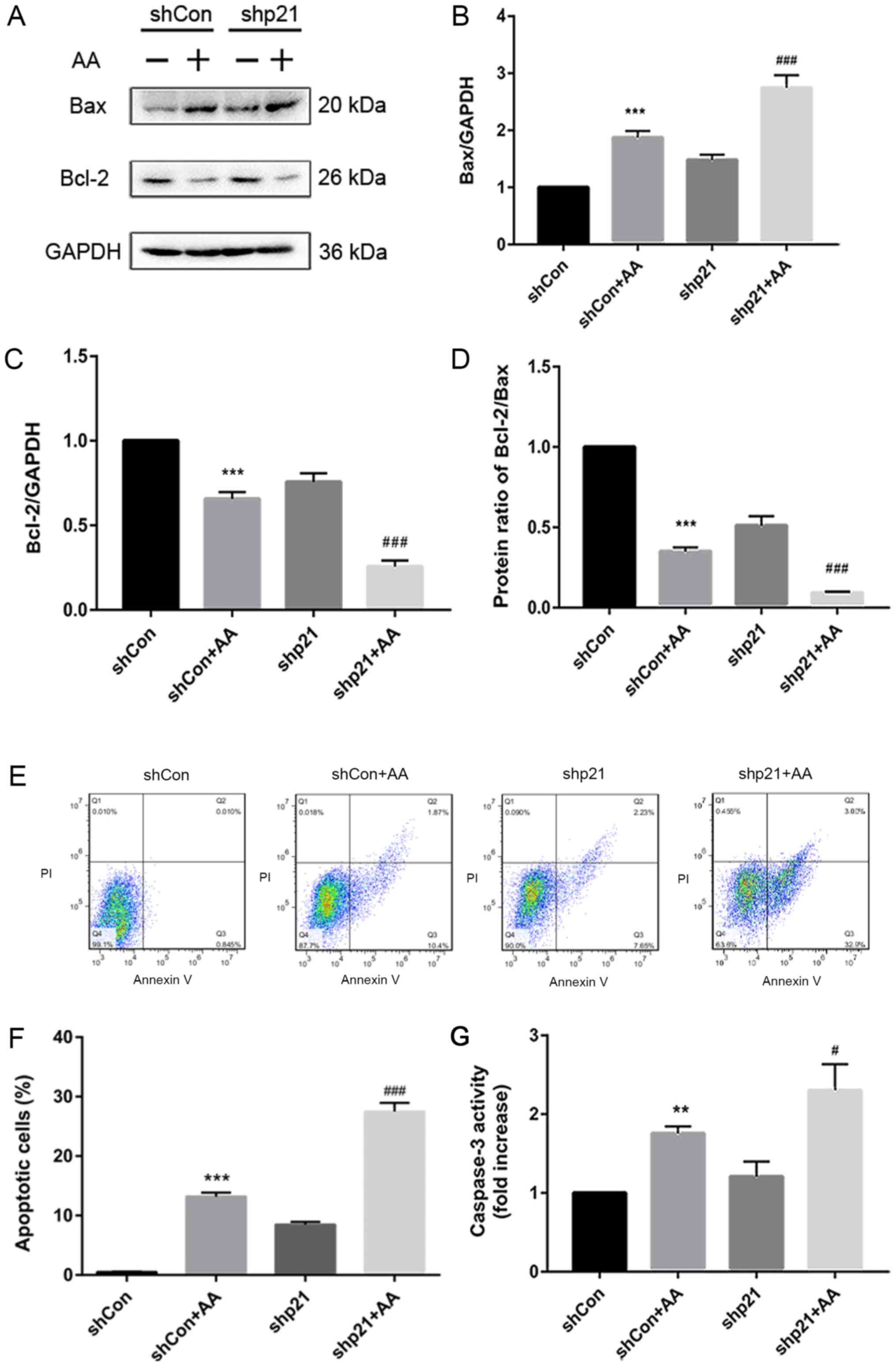
p21 knockdown increases AA-induced
apoptosis in HK-2 cells
The apoptotic role of p21 in AA-treated HK-2 cells
was subsequently investigated; AA treatment significantly increased
the protein expression levels of Bax and decreased the protein
expression levels of Bcl-2 in the shCon + AA group compared with
the non-AA treated shCon group, whereas p21 knockdown (shCon or
shCon + AA) significantly increased the protein expression levels
(Fig. 3A-C). The protein ratio of
Bcl-2/Bax also increased or decreased correspondingly (Fig. 3D). In addition, AA-induced injury
significantly increased the apoptotic rate in the shCon + AA group
compared with the shCon group (Fig. 3E
and F). Under the same conditions (shCon or shCon + AA), shp21
transfection significantly increased the number of AA-induced
apoptotic cells compared with the non-shp21 transfection group
(Fig. 3E and F). Caspase-3
activity was also analyzed using a caspase-3 activity kit; it was
observed that p21 knockdown significantly increased caspase-3
activity in the AA-induced HK-2 cells compared with the shCon + AA
group (Fig. 3G). These results
suggested that p21 deficiency may accelerate AA-induced RTEC
apoptosis.
p21 knockdown decreases AA-induced DDR
activity in HK-2 cells
The ability to induce cell death during the cell
cycle through the DDR signaling pathway was investigated using a
p21 gene knockdown cell model. The protein expression levels of
p-ATM, ATM, p-Chk2, Chk2, rH2AX, p-p53 and p53 were analyzed using
western blotting (Fig. 4). In
AA-treated cells following p21 knockdown, the expression levels of
these phosphorylated proteins were decreased compared with the
shCon + AA group. Shp21 treatment significantly decreased the
protein ratio of p-p53/p53, p-ATM/ATM and p-Chk2/Chk2 in the shCon
or shCon + AA group compared with the non-p21 knockdown group
(Fig. 3C-E). These finding
indicated that p21 may exert a protective effect over cell survival
and can increase the DNA repair ability of cells.
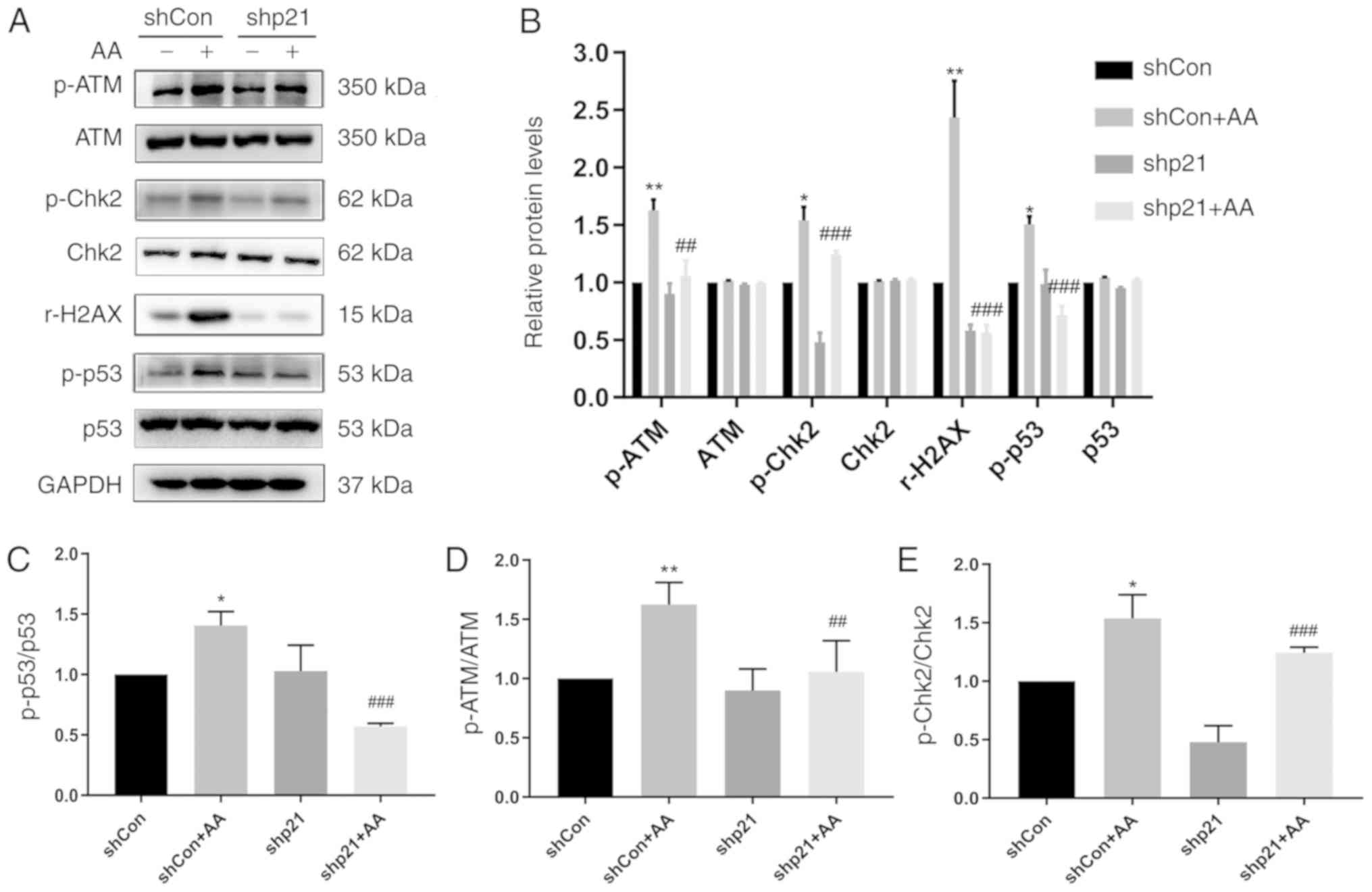 | Figure 4.p21 knockdown reduces the DNA damage
response in HK-2 cells. (A) Protein expression levels of p-ATM,
ATM, p-Chk2, Chk2, rH2AX, p-p53 and p53 were analyzed using western
blotting in cells transfected with shp21 or shCon with or without
AA-induced injury. (B) Semi-quantification of data from (A). (C-E)
Phosphorylated/total protein expression ratios of (C) p-p53/p53,
(D) p-ATM/ATM and (E) p-Chk2/Chk2 from data from. (A) Data are
presented as the mean ± standard error of the mean; n=3;
*P<0.05, **P<0.01 vs. shCon; ##P<0.01,
###P<0.001 vs. shCon + AA. AA, aristolochic acid;
ATM, ataxia telangiectasia mutated; Chk2, checkpoint kinase 2; Con,
control; p, phosphorylated; r-H2AX, H2AX phosphorylation at Serine
139; sh, short hairpin RNA. |
Knockdown of ATM expression levels
using shRNA reduces the DDR and decreases p21 expression to induce
cell cycle arrest
The protein expression levels of p-ATM, ATM, p-Chk2,
Chk2, r-H2AX, p-p53, p53, p21, CDK2, and cyclin D1 in shCon- or
shATM-transfected cells following 24 h of AA treatment were
analyzed using western blotting (Figs.
5 and 6). Cells in the shATM +
AA group demonstrated a decreased DNA repair ability through the
observed significantly reduced the protein ratio of p-Chk2/Chk2,
p-p53/p53 and the protein expression level of r-H2AX compared with
the shCON +AA group (Fig. 5). In
addition, AA-induced shCon group significantly increased the
protein expression levels of p21 compared with non-AA treated shCon
group, which would result in cell cycle arrest (Fig. 6); however, following ATM knockdown,
the expression levels of p21, CDK2 and cyclin D1 were significantly
decreased, thereby promoting the cell cycle process.
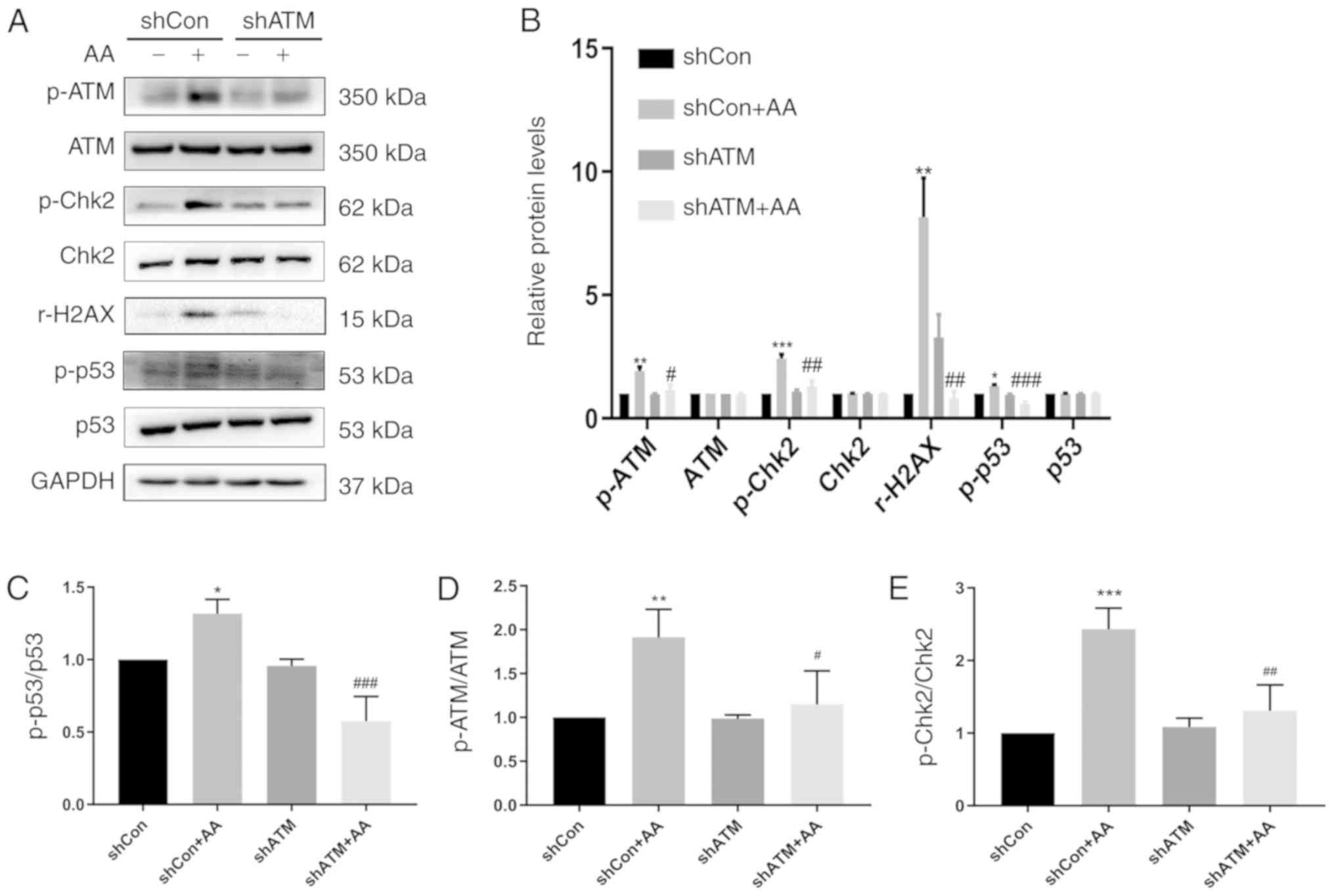 | Figure 5.Genetic knockdown of ATM reduces the
DNA damage response and p-p53 expression levels. (A) Protein
expression levels of p-ATM, ATM, p-Chk2, Chk2, r-H2AX, p-p53 and
p53 were detected using western blotting in cells transfected with
shATM or shCon with or without AA-induced injury. (B)
Semi-quantification of data from (A). (C-E) Phosphorylated/total
protein expression ratio of (C) p-p53/p53, (D) p-ATM/ATM and (E)
p-Chk2/Chk2 from data from. (A) Data are presented as the mean ±
standard error of the mean; n=3; *P<0.05, **P<0.01,
***P<0.001 vs. shCon; #P<0.05,
##P<0.01, ###P<0.001 vs. shCon + AA.
AA, aristolochic acid; ATM, ataxia telangiectasia mutated; Con,
control; Chk2, checkpoint kinase 2; p, phosphorylated; r-H2AX, H2AX
phosphorylation at Serine 139; sh, short hairpin RNA. |
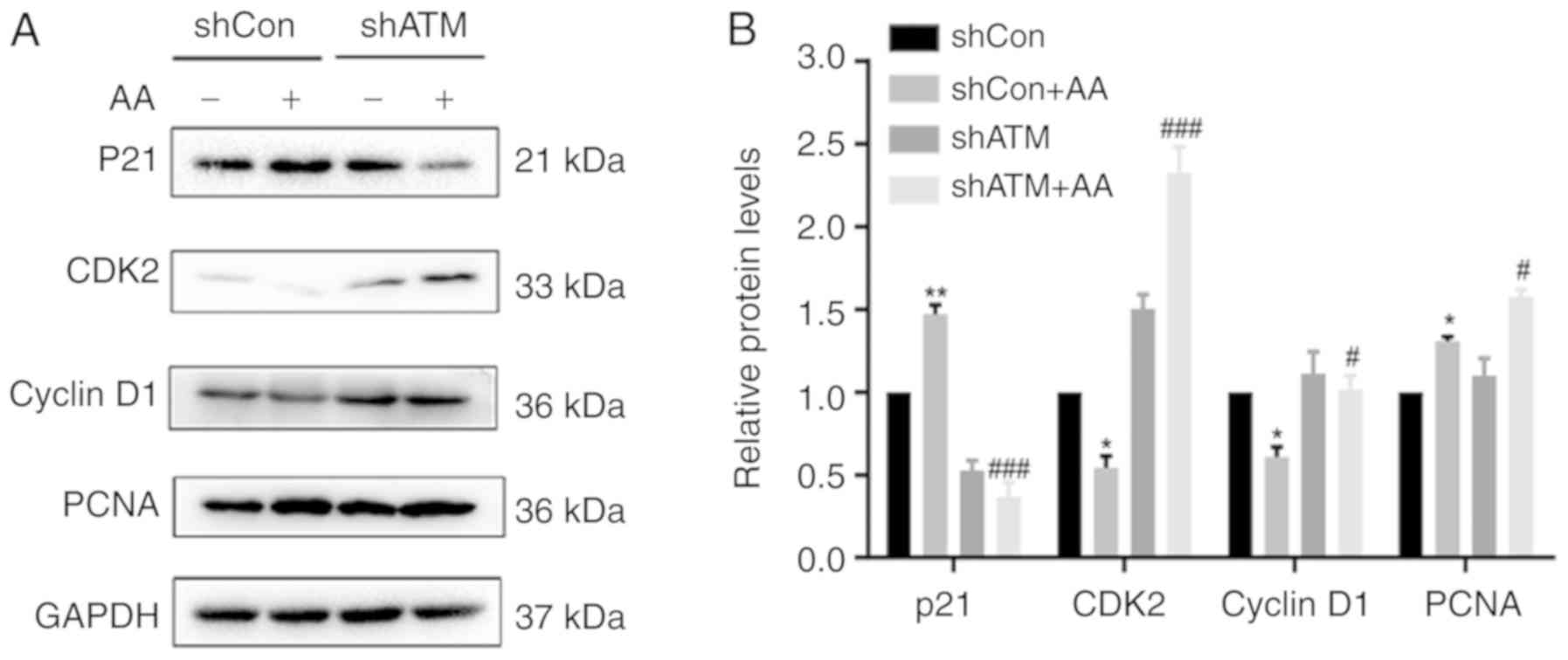 | Figure 6.Genetic knockdown of ATM suppresses
p21 activation, induces cell cycle arrest and promotes the
proliferation of HK-2 cells. (A) Protein expression levels of p21,
CDK2, cyclin D1 and PCNA were analyzed using western blotting in
cells transfected with shATM or shCon with or without AA-induced
injury. (B) Semi-quantification of data from. Data are presented as
the mean ± standard error of the mean; n=3; *P<0.05, **P<0.01
vs. shCon; #P<0.05, ###P<0.001 vs.
shCon + AA. AA, aristolochic acid; ATM, ataxia telangiectasia
mutate d; Con, control; PCNA, proliferating cell nuclear antigen;
sh, short hairpin RNA. |
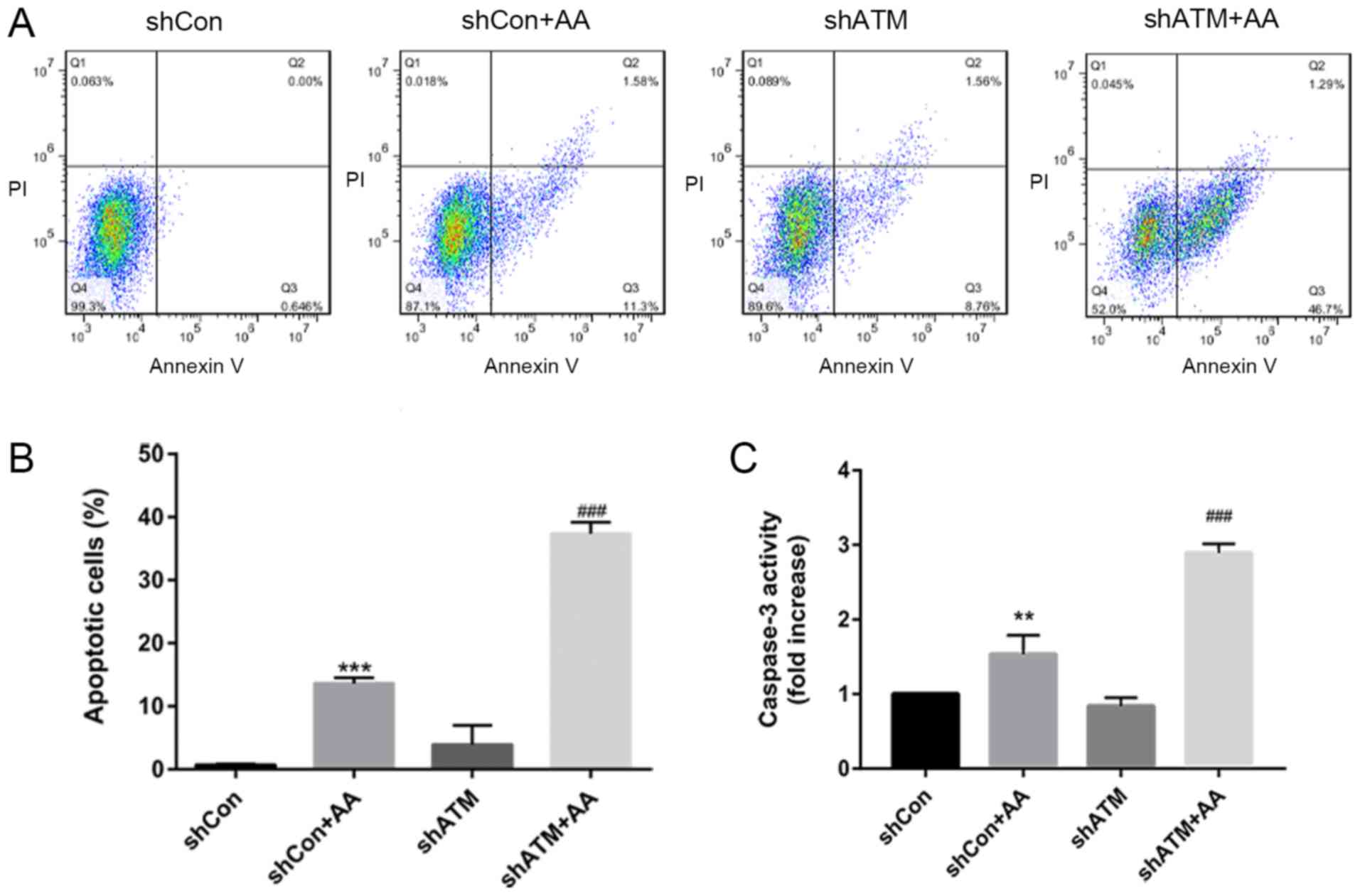
shRNA knockdown of ATM expression
levels promotes proliferation, increases caspase-3 activity and
induces apoptosis in AA-induced HK-2 cells
To further investigate whether DDR signaling affects
the proliferation and apoptosis of AA-induced HK-2 cells, the
protein expression levels of PCNA were analyzed using western
blotting (Fig. 6A and B), the
proportion of apoptotic cells were determined using flow cytometry
(Fig. 7A and B) and caspase-3
activity was analyzed using a caspase-3 activity kit (Fig. 7C). It was found that cells in the
shATM + AA group exhibited significantly increased expression
levels of PCNA and increased apoptotic rates compared with the
shCon + AA group in the presence of AA damage.
Discussion
During the vicious cycle of cell proliferation and
death, the signaling pathways within cells exist in a contradictory
state; that is, proliferative signaling pathways and cell death
signaling pathways simultaneously exist (10). Renal injury has been demonstrated
to stimulate the surviving RTECs to enter the cell cycle; however,
this has been observed to increase their sensitivity to the
external environment and subsequently promote their cell death
(17), hence the formation of the
‘proliferation-death’ cycle. Following the persistence of
injury-related factors, the inhibition of proliferation through the
mTOR and hedgehog signaling pathways has been found to protect
cells against injury factors (27,28).
At the root of TIF development should be the injury caused by the
vicious cycle; however, inflammation, invasion, RTEC
transdifferentiation and myofibroblast proliferation are only the
intermediate links. Thus, it was hypothesized that the
proliferation-death in RTECs may promote the progression of TIF
following the persistence of injury-related factors. The present
study revealed that the DDR was involved in the aberrant
proliferation and cell death cycle, in addition to suggesting that
the p21 protein may serve a major role in the ‘proliferation-death’
cycle.
Renal fibrosis is the pathological hallmark of
chronic kidney disease and it manifests as glomerulosclerosis and
tubulointerstitial fibrosis (29).
Podocyte loss and dysfunction in the glomerulus, in addition to
tubular epithelial cell atrophy and loss, has also been reported to
contribute to chronic kidney disease (30). Thus, the present study investigated
the effect of AA-induced chronic injury in human RTECs by
interfering with the expression levels of ATM and p21 proteins.
p21 regulates various p53-dependent and
p53-independent cell functions; in addition to regulating the cell
cycle, p21 regulates apoptosis, induces senescence and maintains
cellular quiescence in response to various stimuli, including
drugs, blood loss, infection, or exposure to cytotoxic agents
(31). Alongside binding to cell
cycle proteins/CDK complexes, p21 has also been discovered to
contain the COOH terminal binding site of PCNA (32,33),
which has been found to serve important roles in DNA replication
and different types of DNA repair, including nucleotide excision
repair, mismatch repair and base excision repair (34,35).
Moreover, p21 has been revealed to interact directly with PCNA and
block DNA synthesis through DNA polymerase δ (36). It has also been observed to
regulate DNA repair through its interaction with PCNA and related
proteins (37). The mechanisms of
p21-induced inhibition over cell death, including inhibiting the
promoter caspase cleavage have been investigated previously
(38); the interaction of p21 with
procaspase-3 resulted in the resistance to Fas-mediated cell death
and the stabilization of the apoptosis regulator cellular inhibitor
of apoptosis protein 1 (39,40),
whereas p21 overexpression prevented the cytoplasmic domain-induced
caspase-8 cleavage and death receptor 4 (DR4)-CD-induced apoptosis
of the DR4 receptor (38). In
addition, p21 has been discovered to contain the amino-terminus
that interacts with procaspase-3 and suppresses its activation by
inhibiting its conversion to the active protease (41). Similar to previous reports, the
results of the present study demonstrated that in the AA-treated,
p21 knocked down HK-2 cells, the inhibitory effect over CDK2 and
cyclin D1 was weakened, thus driving cell cycle progression and
promoting proliferation. Furthermore, p21 knockdown in the injured
cells stimulated caspase-3 expression and induced cell apoptosis,
whilst promoting cell cycle progression and enhancing the
sensitivity of cells to injury factors (such as AA). As expected,
in the absence of the cell cycle inhibitor protein, p21, the
proliferative activity, proportion of injured cells and the
apoptotic rate were all increased. These findings highlighted the
transition from cell proliferation to death and demonstrated that
the acceleration of the cell cycle may affect the formation and
extent of the proliferation-death cycle. The present study showed
that p21 knocked down HK-2 cells drove cell cycle progression and
promoted proliferation, stimulated caspase-3 expression and induced
cell apoptosis.
ATM regulates cellular DNA repair and serves an
important role in maintaining chromosomal integrity and genome
stability (42). DNA damage during
the cell cycle has been demonstrated to activate ATM/ataxia
telangiectasia and Rad 3-related (ATR) and their downstream
kinases, Chk2 and Cdc25 family members, which are involved in the
checkpoint pathway; this enabled cell proliferation to be halted
until damage is repaired (43),
which often involves a series of proteins, such as
BRCA1/γH2AX/E2F1/RAD (44).
However, if the damaged DNA cannot be repaired, the accumulation of
activated ATM/ATR has been found to rapidly phosphorylate the p53
protein at the Ser15 site and activate p53 (45). Activated p53 has been observed to
further induce apoptosis in p53 upregulated modulator of
apoptosis/NOXA/Bax-mediated mitochondrial pathways (46) and this process is linked to
proliferation and death (19). ATM
serves a central role in phosphorylating DDR and regulating cell
cycle-related molecules throughout the entire process (47). In the current study, acute injury
in the HK-2 cells promoted ATM signal activation and the activation
of the DDR. In brief, acute injury increased the expression levels
of ATM/p53 to activate apoptosis and p53 subsequently activated p21
to promote cell cycle arrest. Interestingly, p53 and p21 expression
levels decreased following the genetic knockdown of ATM, thus
causing increased expression levels of CDK2 and cyclin D1; and
accelerated cell cycle, promoted apoptosis and stimulated cell
proliferation. The increased rate of apoptosis may be due to the
fact that following the reduced expression levels of p21 in
response to ATM knockdown, the effect of p21 was increased and the
cells lost their p21-induced anti-apoptotic effect, leading to
increased apoptosis.
In conclusion, the present study confirmed that cell
death occurs during the progression of the cell cycle. The genetic
knockdown of p21 was found to increase cell cycle progression,
promote proliferation and cause cell death. In addition, although
proliferation and apoptosis could occur at the same time, it could
also occur periodically. It was clear that early diseases mostly
began with proliferation, so it was investigated that as the course
of the disease proceeded, whether this pattern occurred
periodically all the time. Thus, the present study discovered that
the vicious cycle of proliferation and death may be initiated
through the DDR signaling pathway. ATM, as a crucial molecule of
the DDR, has been found to serve an important role in the
regulation of persistent chronic injury (48,49).
In the present study, the genetic knockdown of ATM promoted
apoptosis and increased proliferation. The increase in apoptosis
was hypothesized to be due to the decreased expression levels of
p21 caused by the genetic knockdown of ATM. Thus, the regulatory
crosstalk between the ATM protein and p21 protein were suggested to
serve an important role in the proliferation-death cycle. These
findings provided a potential method for further pathophysiological
research into the process of chronic injury.
Acknowledgements
Not applicable.
Funding
This project was supported by grants from The
National Natural Science Foundation of China (grant no.
81572087).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BC made substantial contributions to the conception
and design of the study; LS, XZ JL and CW performed the
experiments, data analysis and interpretation; and XZ and CW were
responsible for drafting the article and critically revising it for
important intellectual content. LS, XZ and JL contributed equally
to this article. All authors read and approved the final
manuscript. All authors are accountable for all aspects of the
study in ensuring that questions related to the accuracy or
integrity of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AA
|
aristolochic acid
|
|
ATM
|
ataxia telangiectasia mutated
|
|
Chk2
|
checkpoint kinase 2
|
|
DDR
|
DNA damage response
|
|
HK-2
|
human kidney 2
|
|
PCNA
|
proliferating cell nuclear antigen
|
|
r-H2AX
|
H2AX phosphorylation at Serine 139
|
|
TIF
|
tubulointerstitial fibrosis
|
References
|
1
|
GBD 2015 Mortality and Causes of Death
Collaborators, . Global, regional, and national life expectancy,
all-cause mortality, and cause-specific mortality for 249 causes of
death, 1980-2015: A systematic analysis for the Global Burden of
Disease Study 2015. Lancet. 388:1459–1544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Duni A, Liakopoulos V, Roumeliotis S,
Peschos D and Dounousi E: Oxidative stress in the pathogenesis and
evolution of chronic kidney disease: Untangling Ariadne's thread.
Int J Mol Sci. 20:E37112019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Smeets B and Moeller MJ: Parietal
epithelial cells and podocytes in glomerular diseases. Semin
Nephrol. 32:357–367. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zeisberg M and Neilson EG: Mechanisms of
tubulointerstitial fibrosis. J Am Soc Nephrol. 21:1819–1834. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eddy AA: Overview of the cellular and
molecular basis of kidney fibrosis. Kidney Int Suppl. 4:2–8. 2014.
View Article : Google Scholar
|
|
6
|
Falke LL, Gholizadeh S, Goldschmeding R,
Kok RJ and Nguyen TQ: Diverse origins of the
myofibroblast-implications for kidney fibrosis. Nat Rev Nephrol.
11:233–244. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nangaku M: Chronic hypoxia and
tubulointerstitial injury: A final common pathway to end-stage
renal failure. J Am Soc Nephrol. 17:17–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang X, Wang L, Zhu N, Zhou Y, Gu LJ and
Yuan WJ: Hepatitis B virus X protein modulates renal tubular
epithelial cell-induced T-cell and macrophage responses. Immunol
Cell Biol. 94:266–273. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wan J, Zhou X, Cui J, Zou Z, Xu Y and You
D: Role of complement 3 in TNF-α-induced mesenchymal transition of
renal tubular epithelial cells in vitro. Mol Biotechnol. 54:92–100.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen BC, Bai YH, Tang LL, Wang BQ, Liu B,
Cai Y, Peng X, Yang YR and Zheng SL: The progression of the
tubulointerstitial fibrosis driven by stress-induced
‘proliferation-death’ vicious circle. Med Hypotheses. 82:643–647.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Monteiro MB, Ramm S, Chandrasekaran V,
Boswell SA, Weber EJ, Lidberg KA, Kelly EJ and Vaidya VS: A
High-Throughput screen identifies DYRK1A inhibitor ID-8 that
stimulates human kidney tubular epithelial cell proliferation. J Am
Soc Nephrol. 29:2820–2833. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bonventre JV: Primary proximal tubule
injury leads to epithelial cell cycle arrest, fibrosis, vascular
rarefaction, and glomerulosclerosis. Kidney Int Suppl (2011).
4:39–44. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Humphreys BD, Valerius MT, Kobayashi A,
Mugford JW, Soeung S, Duffield JS, McMahon AP and Bonventre JV:
Intrinsic epithelial cells repair the kidney after injury. Cell
Stem Cell. 2:284–291. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gniadecki R, Hansen M and Wulf HC: Two
pathways for induction of apoptosis by ultraviolet radiation in
cultured human keratinocytes. J Invest Dermatol. 109:163–169. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alenzi FQ: Links between apoptosis,
proliferation and the cell cycle. Br J Biomed Sci. 61:99–102. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pandey S and Wang E: Cells en route to
apoptosis are characterized by the upregulation of c-fos, c-myc,
c-jun, cdc2, and RB phosphorylation, resembling events of early
cell-cycle traverse. J Cell Biochem. 58:135–150. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sanz AB, Sanchez-Niño MD, Izquierdo MC,
Jakubowski A, Justo P, Blanco-Colio LM, Ruiz-Ortega M, Egido J and
Ortiz A: Tweak induces proliferation in renal tubular epithelium: A
role in uninephrectomy induced renal hyperplasia. J Cell Mol Med.
13:3329–3342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang L, Besschetnova TY, Brooks CR, Shah
JV and Bonventre JV: Epithelial cell cycle arrest in G2/M mediates
kidney fibrosis after injury. Nat Med. 16:535–543, 1p following
143. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bozzo C, Tiberio R, Graziola F, Pertusi G,
Valente G, Colombo E, Small PL and Leigheb G: A Mycobacterium
ulcerans toxin, mycolactone, induces apoptosis in primary human
keratinocytes and in HaCaT cells. Microbes Infect. 12:1258–1263.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nam AR, Jin MH, Park JE, Bang JH, Oh DY
and Bang YJ: Therapeutic targeting of the DNA damage response using
an ATR inhibitor in biliary tract cancer. Cancer Res Treat.
51:1167–1179. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee IH, Kawai Y, Fergusson MM, Rovira II,
Bishop AJ, Motoyama N, Cao L and Finkel T: Atg7 modulates p53
activity to regulate cell cycle and survival during metabolic
stress. Science. 336:225–228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shiloh Y: ATM and related protein kinases:
Safeguarding genome integrity. Nat Rev Cancer. 3:155–168. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Charen E and Harbord N: Toxicity of herbs,
vitamins, and supplements. Adv Chronic Kidney Dis. 27:67–71. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lu H, Liang Y, Guan B, Shi Y, Gong Y, Li
J, Kong W, Liu J, Fang D, Liu L, et al: Aristolochic acid
mutational signature defines the low-risk subtype in upper tract
urothelial carcinoma. Theranostics. 10:4323–4333. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak JK and Schmittgen TD: Analysis of
relative gene expression data using quantitative PCR and the
2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang HW, Chen YH, Chen YY, Huang W, Zhu
XD, Ni FB, Wu GD, Xu ZQ, Huang ZQ, Chen BC and Xiao FY: Islet
transplantation attenuates cardiac fibrosis in diabetic rats
through inhibition of TGF-β1/Smad3 pathway. Am J Transl
Res. 10:2445–2456. 2018.PubMed/NCBI
|
|
27
|
Wu MJ, Wen MC, Chiu YT, Chiou YY, Shu KH
and Tang MJ: Rapamycin attenuates unilateral ureteral
obstruction-induced renal fibrosis. Kidney Int. 69:2029–2036. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Price PM, Safirstein RL and Megyesi J: The
cell cycle and acute kidney injury. Kidney Int. 76:604–613. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garcia-Fernandez N, Jacobs-Cachá C,
Mora-Gutiérrez JM, Vergara A, Orbe J and Soler MJ: Matrix
metalloproteinases in diabetic kidney disease. J Clin Med.
9:E4722020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Srivastava T, Thiagarajan G, Alon US,
Sharma R, El-Meanawy A, McCarthy ET, Savin VJ and Sharma M: Role of
biomechanical forces in hyperfiltration-mediated glomerular injury
in congenital anomalies of the kidney and urinary tract. Nephrol
Dial Transplant. 32:759–765. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Manu KA, Cao PHA, Chai TF, Casey PJ and
Wang M: p21cip1/waf1 coordinate autophagy, proliferation and
apoptosis in response to metabolic stress. Cancers (Basel).
11:E11122019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gartel AL and Tyner AL: The role of the
cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer
Ther. 1:639–649. 2002.PubMed/NCBI
|
|
33
|
Xiong Y, Zhang H and Beach D: D type
cyclins associate with multiple protein kinases and the DNA
replication and repair factor PCNA. Cell. 71:505–514. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tsurimoto T: PCNA binding proteins. Front
Biosci. 4:D849–D858. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Warbrick E: The puzzle of PCNA's many
partners. Bioessays. 22:997–1006. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Waga S, Hannon GJ, Beach D and Stillman B:
The p21 inhibitor of cyclin-dependent kinases controls DNA
replication by interaction with PCNA. Nature. 369:574–578. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gibbs E, Kelman Z, Gulbis JM, O'Donnell M,
Kuriyan J, Burgers PM and Hurwitz J: The influence of the
proliferating cell nuclear antigen-interacting domain of p21(CIP1)
on DNA synthesis catalyzed by the human and Saccharomyces
cerevisiae polymerase delta holoenzymes. J Biol Chem.
272:2373–2381. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu SQ and El-Deiry WS: p21(WAF1/CIP1)
inhibits initiator caspase cleavage by TRAIL death receptor DR4.
Biochem Biophys Res Commun. 269:179–190. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Suzuki A, Tsutomi Y, Akahane K, Araki T
and Miura M: Resistance to Fas-mediated apoptosis: Activation of
caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene
family ILP. Oncogene. 17:931–939. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Steinman RA and Johnson DE: p21WAF1
prevents down-modulation of the apoptotic inhibitor protein c-IAP1
and inhibits leukemic apoptosis. Mol Med. 6:736–749. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Suzuki A, Tsutomi Y, Miura M and Akahane
K: Caspase 3 inactivation to suppress Fas-mediated apoptosis:
Identification of binding domain with p21 and ILP and inactivation
machinery by p21. Oncogene. 18:1239–1244. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yan S, Sorrell M and Berman Z: Functional
interplay between ATM/ATR-mediated DNA damage response and DNA
repair pathways in oxidative stress. Cell Mol Life Sci.
71:3951–3967. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee HJ, Hwang HI and Jang YJ: Mitotic DNA
damage response: Polo-like kinase-1 is dephosphorylated through
ATM-Chk1 pathway. Cell Cycle. 9:2389–2398. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cook PJ, Ju BG, Telese F, Wang X, Glass CK
and Rosenfeld MG: Tyrosine dephosphorylation of H2AX modulates
apoptosis and survival decisions. Nature. 458:591–596. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Brazina J, Svadlenka J, Macurek L, Andera
L, Hodny Z, Bartek J and Hanzlikova H: DNA damage-induced
regulatory interplay between DAXX, p53, ATM kinase and Wip1
phosphatase. Cell Cycle. 14:375–387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu J, Zhang J, Ren L, Wei J, Zhu Y, Duan
J, Jing L, Sun Z and Zhou X: Fine particulate matters induce
apoptosis via the ATM/P53/CDK2 and mitochondria apoptosis pathway
triggered by oxidative stress in rat and GC-2spd cell. Ecotoxicol
Environ Saf. 180:280–287. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ronco C, Martin AR, Demange L and Benhida
R: ATM, ATR, CHK1, CHK2 and WEE1 inhibitors in cancer and cancer
stem cells. Medchemcomm. 8:295–319. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ding H, Xu Y and Jiang N: Upregulation of
miR-101a suppresses chronic renal fibrosis by regulating KDM3A via
blockade of the YAP-TGF-β-Smad signaling pathway. Mol Ther Nucleic
Acids. 19:1276–1289. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li J, Zhang M, Mao Y, Li Y, Zhang X, Peng
X and Yu F: The potential role of aquaporin 1 on aristolochic acid
I induced epithelial mesenchymal transition on HK-2 cells. J Cell
Physiol. 233:4919–4925. 2018. View Article : Google Scholar : PubMed/NCBI
|