Introduction
Neonatal hyperbilirubinemia primarily occurs in term
newborns and in >84% of late preterm newborns (1). Extreme neonatal hyperbilirubinemia
may lead to the accumulation of bilirubin in the brain stem nuclei,
cerebellum and basal ganglia, and subsequently cause acute
bilirubin encephalopathy (ABE) (2). ABE is a serious disease that can
cause death or a lifelong neurodevelopmental disorder, known as
chronic bilirubin encephalopathy or kernicterus (3). Although the incidence of ABE has
declined, the reported incidence rate in developed countries in
2009–2018 ranged from 0.4–2.7 cases per 100,000 births (4,5).
Moreover, the reported incidence rates of ABE in Asia, the Middle
East and Africa have been reported to be notably higher than in
Western developed countries (6–8).
ABE is a clinical bilirubin-induced neurological
dysfunction (BIND) characterized by poor feeding, lethargy,
hypotonia and opisthotonus (9,10).
The neurotoxicity is due to the diffusion of unconjugated bilirubin
(UCB) into brain cells after traversing the blood-brain barrier
(10). High concentrations of UCB
exert neurotoxic effects by inducing a number of cellular and
molecular events (e.g., neuronal excitotoxicity, mitochondrial
energy failure, activation of apoptotic or necrosis pathways)
(11). The in vitro exposure of
neurons to UCB has been identified to induce cellular macroscopic
changes, such as decreased dendritic and axonal arborization,
neurite extension and ramification, and cell proliferation
(12). Non-neuronal cells, such as
astrocytes, microglia and oligodendrocytes, have also demonstrated
sensitivity to UCB, which triggered the secretion of inflammatory
mediators, including tumor necrosis factor (TNF) and interleukin
(IL)-1 (13,14), upregulated the activity of
matrix-metalloproteinases 2 and 9 (15) and increased caspase 3-mediated
apoptosis (16). In addition,
cultured vascular endothelial cells of the blood-brain barrier and
the blood-cerebrospinal fluid (CSF) barrier were observed to
exhibit upregulated P-glycoprotein/ABCB1 and downregulated
multidrug resistance-associated protein 1/ABC, C1 expression
levels, respectively, upon the exposure to high concentrations of
bilirubin (17). Moreover,
bilirubin-induced endoplasmic reticulum stress was previously
demonstrated to contribute to inflammatory processes and apoptosis
in neuronal cells, which indicated the involvement of endoplasmic
reticulum stress in UCB-induced neurotoxicity (18).
However, despite numerous cell and animal
experiments, the basic mechanisms of ABE are not fully understood.
Over previous years, there has been an increasing interest in
extracellular microvesicles/exosomes (MV/E), which are released
from a number of cells, including glial cells, neurons and
astrocytes in the brain (19).
MV/E are membrane-bound nanovesicles that serve as important
mediators of intercellular communication by transporting molecular
signals, including proteins, mRNA and microRNAs, between cells
(20,21). High-throughput proteomics
technology has been used to identify underlying mechanisms
associated with the initiation and development of numerous types of
cerebral disease (22–24). However, to the best of our
knowledge, few studies have investigated the molecular mechanism of
bilirubin-induced neurotoxicity based on proteomics techniques or
extracellular MV/E (12,25,26).
Therefore, focusing on the proteomics of MV/E may elucidate the
mechanisms of bilirubin-induced neurological neurotoxicity. The aim
of the present study was to obtain protein profiles of
extracellular vesicles from the CSF of patients with ABE. These
results may help determine the pathogenesis of ABE and provide
novel perspectives for the early diagnosis and treatment of
ABE.
Materials and methods
Participant characteristics and CSF
collection
The study protocol was approved by the Ethics
Committee of Affiliated First People's Hospital of Chenzhou,
Southern Medical University, University of South China (Chenzhou,
China). Written informed consent was obtained from
parents/caregivers of all subjects prior to the start of the study.
All experimental procedures were performed in accordance with the
Declaration of Helsinki (27).
All subjects in the present study were full-term
newborns(n=30) recruited between January 2017 and November 2019
from the Department of Neonatology of the Affiliated First People's
Hospital of Chenzhou, Southern Medical University. Patient
characteristics are presented in Table
I. Patients with ABE were diagnosed according to the clinical
BIND score; BIND scores of 1–3, 4–6 and 7–9 are indicative of mild,
moderate and severe ABE, respectively (28). The inclusion criteria for patients
with ABE in the present study were: Newborns, aged ≤14 days who
required exchange transfusion and had clinical BIND scores of >4
(3,28). Patients with ABE were divided into
moderate (n=10) and severe groups (n=10). The control group (n=10)
comprised full-term newborns who required lumbar puncture to
exclude sepsis and meningitis, but who were not diagnosed with
sepsis or meningitis due to normal CSF and peripheral blood test
results. A total of four patients in the control group were
diagnosed with pneumonia; six were diagnosed with enteritis. The
demographic and clinical characteristics of the subjects are
presented in Table I. Newborns
with congenital cerebral malformation or secondary brain injury due
to other causes (such as hypoglycemic encephalopathy,
hypoxic-ischemic encephalopathy, intracranial hemorrhage, epilepsy,
sepsis, meningitis or inherited metabolic diseases) were excluded
from the present study.
 | Table I.Characteristics of the study
population. |
Table I.
Characteristics of the study
population.
|
| Patients with acute
bilirubin encephalopathy |
|
|
|---|
|
|
|
|
|
|---|
| Variables | Moderate group | Severe group | Control group | P-value |
|---|
| Number | 10 | 10 | 10 |
|
| Female/male | 4/6 | 3/7 | 5/5 |
|
| Gestational age,
weeks |
38.60±1.40 |
38.20±1.70 |
38.40±1.34 |
0.930 |
| Birth weight,
g |
3,513.50±188.18 |
3,498.30±135.13 |
3,505.40±240.86 |
0.985 |
| Age at admission,
days |
5.70±0.95 |
5.50±1.08 |
6.40±1.57 |
0.248 |
| Peak serum TB
levels, µmol/l |
485.40±20.30 |
587.50±57.50 |
77.21±19.70 | <0.001 |
| Bilirubin-induced
neurological dysfunction score at admission |
4.80±0.91 |
8.50±0.71 |
| <0.001 |
| Routine test of
cerebrospinal fluid |
| Number
of cells, 106/l |
7.60±2.01 |
7.50±1.58 |
6.90±1.96 |
0.666 |
| Protein
concentration, mg/l |
810.50±94.96 |
790.26±107.76 |
745.90±131.77 |
0.289 |
| Peripheral blood
test |
| White
blood cells, 109/l |
8.99±1.03 |
9.11±1.16 |
8.54±1.44 |
0.553 |
|
C-reactive protein, mg/l |
2.41±1.040 |
2.46±1.15 |
2.52±0.78 |
0.970 |
|
Procalcitonin, ng/ml |
0.18±0.09 |
0.23±0.12 |
0.19±0.15 |
0.617 |
| Cause of
hyperbilirubinemia |
|
Hemolysis, yes/no | 3/7 | 4/6 |
|
|
|
Idiopathic, yes/no | 1/9 | 1/9 |
|
|
|
Glucose-6-phosphate
1-dehyrodgenase deficiency, yes/no | 6/4 | 5/5 |
|
|
| Treatment | ET + PT | ET + PT +
intravenous albumin |
|
|
CSF samples were obtained at the time of diagnosis
of ABE, while samples from controls were obtained prior to the
administration of antibiotics at admission. CSF samples (2.5 ml)
were collected via lumbar puncture and centrifuged at 2,000 × g for
20 min at 4°C and stored at −80°C until analysis.
Isolation of MV/E via
co-precipitation
A volume of 1 ml supernatant was pipetted into an
Eppendorf tube and mixed with 330 µl Ribo™ exosome isolation
reagent (cat. no. C10120; Guangzhou Ribobio Co., Ltd.). The tube
was subsequently placed in a refrigerator at 4°C for 30 min, then
centrifuged at 15,000 × g for 2 min at 4°C. The resultant
precipitate was the crude MV/E-enriched fraction, which was
resuspended and further diluted with1 ml 1X PBS to achieve the
detectable concentration (106 particles/ml) for further
analysis.
Nanoparticle tracking analysis
(NTA)
In order to characterize MV/E, NTA was performed
using the NanoSight NS300 instrument (Malvern Panalytical). The
crude MV/E-enriched fraction were diluted with 1 ml 1X PBS to
achieve a suitable concentration (106 particles/ml), and
then injected into the detection chamber (flow rate, 35 µl/second;
temperature 23.1–23.7°C) equipped with a 405 nm (purple) laser. The
parameters were adjusted to the best condition according to the
manufacturer's instructions, and samples were measured three times.
Data were analyzed using NTA 3.2 Dev Build 3.2.16 software (Malvern
Panalytical) and SOP Standard Measurement. The NTA technology
combines a video imaging system and laser light scattering
microscopy to obtain the nanoparticle size distribution of samples
in liquid suspension (29).
Brownian motion of each particle was measured and analyzed
individually. A light beam is used to illuminate the particles in
the sample. As the particles scatter light and undergo Brownian
motion, a scientific CMOS camera recorded the path of each particle
to determine the mean velocity and diffusivity. The particle
concentration and size distribution were calculated according to a
modified Stokes-Einstein relationship (30).
Protein extraction, reduction,
alkylation, digestion and isobaric tagging for relative and
absolute quantification (iTRAQ)-4 plex labeling
For protein extraction, all pooled MV/E samples were
removed from ice. Each sample was added to ×10 the volume of
protein lysis buffer (8 M urea, 1% SDS, 1X protease inhibitor
cocktail) with an ultrasonic cell disruptor (25 kHz) for 2 min in
an ice bath and then allowed to react for 30 min on ice.
Subsequently, precooled 100% acetone (acetone:sample ratio, 5:1)
was added and incubated at −20°C overnight to precipitate the
protein. The sample was centrifuged for 30 min at 12,000 × g at
4°C, and the precipitate was cleaned using 90% acetone and
centrifuged again for 30 min at 12,000 × g at 4°C. Then, the
supernatant was discarded, and the precipitate was fully dissolved
in protein lysis buffer and centrifuged for 30 min at 12,000 × g at
4°C.
Proteins were quantified via BCA assay. For protein
reduction and alkylation, protein lysis buffer was added to 50 µg
protein samples to a volume of 150 µl. Then, 10 mM Bond-Breaker™
Tris (2-carboxyethyl) phosphine solution (Thermo Fisher Scientific,
Inc.) was added to the sample and incubated for 60 min at 37°C.
Following the incubation, 10 μl solution (40 mM iodoacetamide in 40
mM NH4HCO3) was added to the sample and
incubated for 40 min at 37°C in the dark. Precooled 100% acetone
(acetone: sample volume ratio, 6:1) was added to each tube for
precipitation at −20°C for 4 h. Then, the sample was centrifuged at
10,000 × g for 20 min at 4°C and the precipitate was completely
dissolved in 100 µl triethylammonium bicarbonate (TEAB; 100
mM).
For subsequent protein tryptic digestion, 2 µl
trypsin (0.5 µg/ml) was added to each sample. Each sample was
vortexed briefly, covered with parafilm to prevent evaporation and
incubated overnight at 37°C. Subsequently, samples were desalted
using a Sep-Pak C18 cartridge (particle size, 55–105 µm) (Waters
Corporation) according to the manufacturer's instructions, and then
vacuum dried. For iTRAQ-4 plex labeling, 100 μg digested peptides
were dissolved in 0.5 M TEAB and labeled with the iTRAQ reagents
4-plex kit (cat. no. 43374321; Shanghai AB SCIEX Analytical
Instrument Trading Co.), according to the manufacturer's protocol.
iTRAQ reagents 114, 115 and 116 from the iTRAQ reagents 4-plex kit
were used to label digested peptides of the control, moderate and
severe groups, respectively. After reacting for 2 h at room
temperature, 40 µg labeled peptides in each group were mixed
together in a tube and vacuum concentrated for high-performance
liquid chromatography analysis (HPLC).
Ultimate HPLC (UPLC)
In order to decrease sample complexity,
first-dimensional separation of the iTRAQ-labeled mixed peptides
was performed using the ACQUITY UPLC system (Waters Corporation).
The peptide mixture (120 µg) was reconstituted withsolvent A (20 mM
ammonium formate; 2% acetonitrile; pH 10.0) to 25 µl and loaded
onto the ACQUITY UPLC BEH C18 column (grain size, 1.7 µm; 2.1×150.0
mm; temperature, 37°C; Waters Corporation) through an autosampler
(temperature, 10°C). The peptides were separated at 200 µl/min for
37 min via a linear gradient, which was established by mixing
solvents A and B (20 mM ammonium formate; 80% acetonitrile; pH
10.0) as follows: 0–5 min, 0.0–3.8% solvent B in solvent A; 6–16
min, 4–43% solvent B in solvent A; 17–37 min, 100% solvent B in
solvent A. The ultraviolet detection wavelength was set at 214 nm.
A total of 5 fractions were collected based on peak area, then
vacuum dried and stored at −80°C for further liquid chromatography
with tandem mass spectrometry (LC-MS/MS) analysis.
Reverse-phase LC-MS/MS
Second-dimensional reverse phase LC-MS/MS was
performed at room temperature to separate and identify the digested
samples (1 µg/fraction). The chromatographic separation was
performed using the EASY-nLC 1200 system on a C18 column (75 µm ×
25 cm; Thermo Fisher Scientific, Inc.). The mobile phase contained
solvents A (2% acetonitrile with 0.1% formic acid) and B (80%
acetonitrile with 0.1% formic acid). The samples were resuspended
in 40 µl solvent A to a concentration of 0.5 µg/μl and were
fractionated sequentially. The total fractionation time was 120 min
at a flow rate of 300 nl/min via a linear gradient profile, which
was established by mixing solvent A and solvent B as follows: 0–63
min, 0–23% solvent B in solvent A; 64–88 min, 23–48% solvent B in
solvent A; 89–120 min, 100% solvent B in solvent A.
A Q-Exactive hybrid quadrupole-Orbitrap mass
spectrometer (Thermo Fisher Scientific, Inc.) equipped with an
EASY-nLC™ 1200 nanoflow LC system (Thermo Fisher Scientific, Inc.)
was used for qualitative analysis using electrospray ionization in
the positive mode. The conditions for MS were: Source temperature,
350°C; spray voltage, −3 kv; nebulizer gas pressure, 35 psi; and
nebulizer nitrogen flow rate, 10 l/min. The data-dependent MS
acquisition mode was used to select the top 20 most abundant ions
for subsequent analysis. The mass spectrum scanning range (m/z) was
set at 350–1,300 m/z and the mass spectrum resolution was 70,000.
For MS/MS, the top 20 intense precursor ions were sequentially
subjected to higher energy collision dissociation. The fragment
ions were detected at a mass resolution of 17,500. Dynamic
exclusion was set for 18 sec. Thermo Xcalibur™ 4.0 (Thermo Fisher
Scientific, Inc.) software was used for data acquisition. A total
of three biological repeats was performed to minimize the
experimental variation.
Data analysis and bioinformatics
The raw data was sifted using Proteome Discoverer™
2.1 software (Thermo Fisher Scientific, Inc.) and was searched
against the UniProt Homo sapiens database
(uniprot-proteome-UP000005640-Homo sapiens-20180626-73045s.fasta)
using Mascot software (version 2.3.0; Matrix Science, Inc.). The
search parameters were as follows: i) Dynamic modification:
Oxidation (M), acetyl (Protein N-Terminus), iTRAQ4plex (Y); ii)
static modification: iTRAQ4plex (K), iTRAQ4plex (N-Terminus),
carbamidomethyl (C); iii) enzyme, trypsin; iv) maximum missed
cleavage sites, 2; v) precursor mass tolerance, 20 ppm; vi)
fragment mass tolerance, 0.05 Da; and vii) requirements for protein
identification: Two peptides or ≥1 unique peptide to match. The
cut-off values for differentially expressed proteins (DEPs) were
set at fold change >1.2 or <0.83 and P<0.05. All P-values
were corrected for multiple hypotheses testing by controlling the
false discovery rate (FDR). P<0.05 and FDR <0.05 were
considered to indicate a statistically significant difference.
Gene Ontology (GO) consortium (geneontology.org/) was used for functional term
annotation and enrichment analysis using the UniProt accession
numbers of the identified DEPs. The Kyoto Encyclopedia of Genes and
Genomes (KEGG) database (kegg.jp/) was used for signaling pathway
enrichment analysis. In addition, a protein-protein interaction
(PPI) network of the DEPs was constructed using the Search Tool for
the Retrieval of Interacting Genes/Proteins (STRING) software
(version 10.5; string-db.org/) (confidence score
≥0.4). Fisher's exact values and Q-values (corrected using FDR)
<0.05 were considered significant for the GO functional term and
KEGG signaling pathway enrichment analysis.
Western blotting validation
A total of four DEPs [S100A9, S100A7, lactoferrin
(LTF) and α defensin 1 (DEFA1)] were selected for further
validation. The selection criteria for validation were: i) Fold
change >1.5 or <0.67 and P<0.05; ii) DEPs were upregulated
or downregulated in both the ABE and control groups; iii) results
of the bioinformatics analysis (GO functional annotation and KEGG
signaling pathways analysis of DEPs); iv) potential association
with ABE pathogenesis, based on existing knowledge; and v) not
previously studied in patients with ABE.
Proteins from harvested MV/E of CSF were extracted
using protein extraction buffer [50 Tris-HCl (pH, 6.8), 100 mM DTT,
2% SDS, 0.1% bromophenol blue, 10% glycerol] and quantified via BCA
assay. Proteins samples were separated by 12% SDS-PAGE at room
temperature using 30 mA constant current electrophoresis and 50 μg
protein was loaded per lane. The separated proteins were
subsequently transferred onto PVDF membranes and blocked with 5%
non-fat milk in TBST (0.1% Tween-20) for 60 min at room
temperature. The membranes were washed three times with TBST for 5
min and incubated with primary antibodies at 4°C overnight.
Following the primary antibody incubation, the membranes were
washed three times with TBST for 5 min and incubated for 60 min at
room temperature with a horseradish peroxidase-conjugated secondary
antibody (1:1,000) as follows: Goat anti-mouse (cat. no. SC-3697;
Santa Cruz Biotechnology, Inc.), goat anti-rabbit (cat. no.
G-21234; Thermo Fisher Scientific, Inc.), rabbit anti-goat (cat.
no. 81-1620; Thermo Fisher Scientific, Inc. The chromogenic
reaction was developed using a chemiluminescent substrates kit
(Amresco, LLC). Images were visualized using a KodakImage Station
4000R digital imaging system (Kodak). The Quantity One software
(version 4.6.7; Bio-Rad Laboratories, Inc) was used for
densitometric analysis. The primary antibodies (all 1:1,000) were
as follows: Monoclonal anti-CD9 and anti-LTF (cat. nos. sc-59140
and sc-53498, respectively; both Santa Cruz Biotechnology, Inc.),
polyclonal anti-DEFA1 (cat. no. PA5-19228; Thermo Fisher
Scientific, Inc.) and polyclonal anti-S100A7 and anti-S100A9 (cat.
nos. PA5-75689 and PA1-46489, respectively; both Thermo Fisher
Scientific, Inc.).
Statistical analysis
Statistical analysis was performed using the SPSS
statistical package (version 20.0; IBM Corp.). Continuous variables
are presented as the mean ± SD of three experimental repeats.
One-way ANOVA and Bonferroni's post hoc test were performed to
evaluate the statistical differences between multiple group
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
NTA of MV/E from patients with ABE and
controls
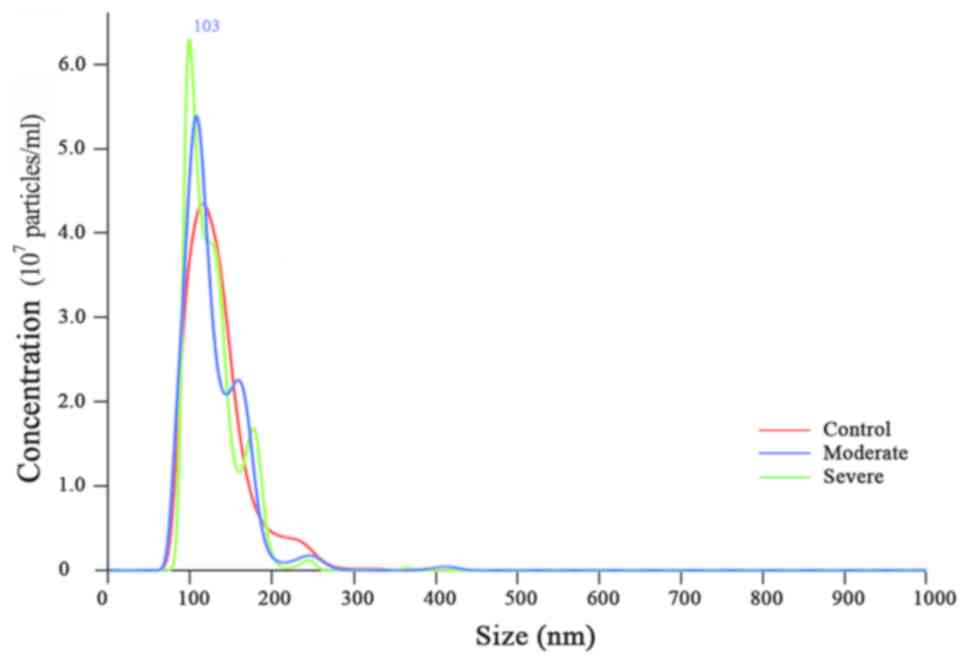
Representative particle concentration distribution
and size scattering intensity analysis of MV/E samples is presented
in Fig. 1. The particle size of
MV/E from patients with ABE and controls ranged from 75–200 nm in
diameter and the peak concentration of the particle of MV/E from
patients with ABE and controls was at 103 nm in diameter. The three
curves overlapped, which indicated that the three groups of samples
were homogeneous and that there were few impurities, which
suggested the successful isolation of MV/E from the CSF.
Qualitative results of dysregulated
proteins
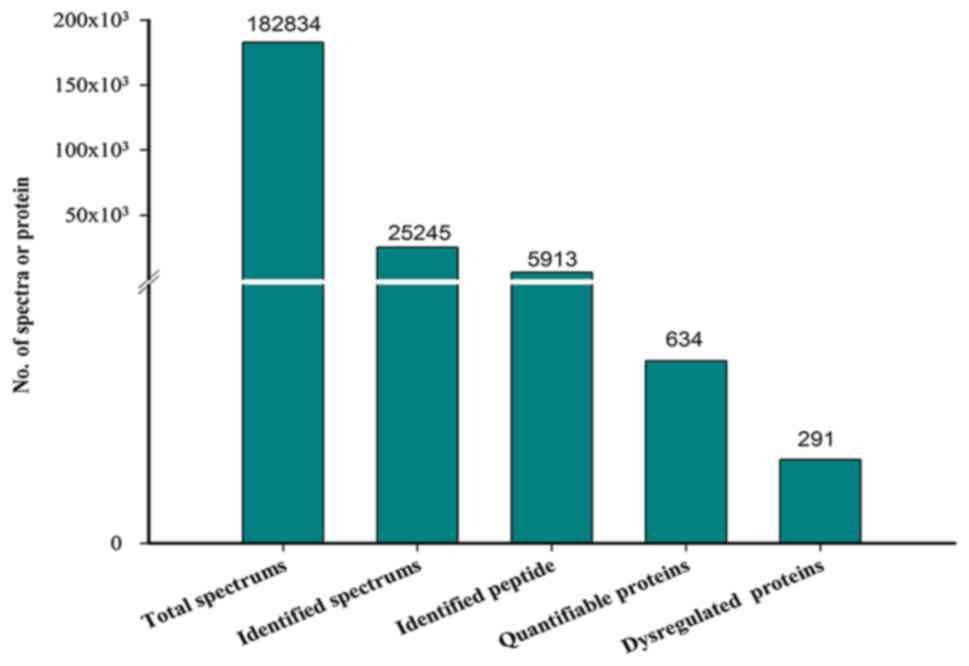
A total of 182,834 MS/MS spectra were obtained via
iTRAQ-based proteomics and 25,245 peptide spectrum matches (PSMs)
were matched to 5,913 peptides. A total of 634 proteins were
identified and 291 dysregulated proteins were selected for further
study (Fig. 2; Table SI). Of the 291 proteins, 186 DEPs
(75 upregulated and 111 downregulated proteins) were identified
between the severe and moderate groups (Table SII). A total of 119 DEPs (37
upregulated and 82 downregulated proteins) were identified between
the severe and control groups (Table
SIII) and 194 DEPs (92 up- and 102 downregulated proteins) were
identified between the moderate and control groups (Table SIV). Certain DEPs [serum amyloid
A-1 protein (SAA1), amyloid-β precursor protein (APP),
immunoglobulin-like domains (immunoglobulin heavy constant γ4 and
immunoglobulin heavy variable 2–5), complement components (C4B and
C5), S100A9, S100A7, DEFA1 and LTF] were identified between the ABE
and control groups (Table
SI).
GO annotation and functional term
enrichment analysis of DEPs
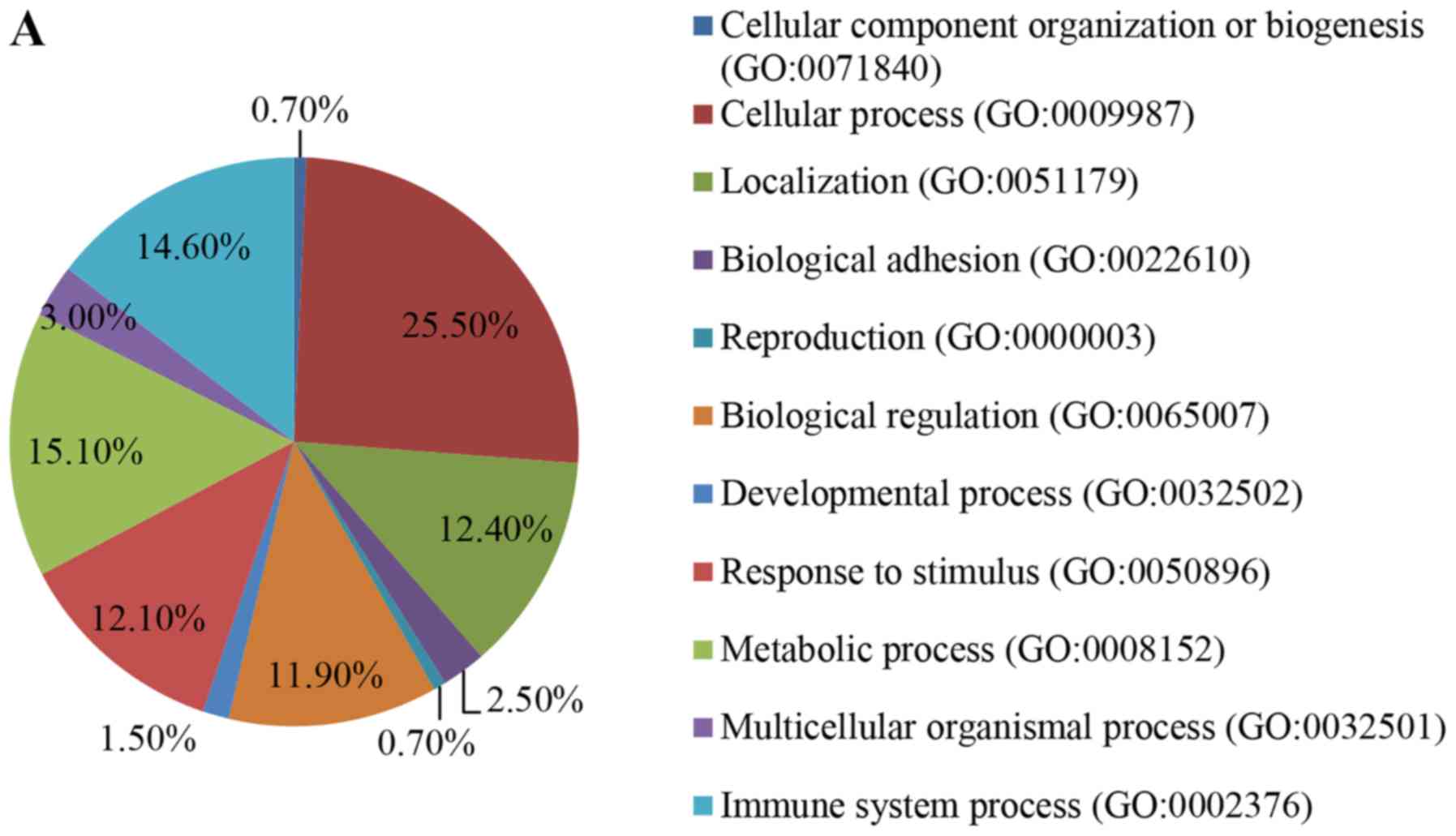
GO annotation of all DEPs was categorized according
to biological process (BP), molecular function (MF) and cellular
components (CC). The majority of DEPs were associated with
‘cellular process’, ‘immune system process’ and ‘metabolic process’
(Fig. 3A) and the identified MFs
indicated that the DEPs were primarily involved in ‘binding’ and
‘catalytic activity’ (Fig. 3B). In
addition, the DEPs were revealed to be primarily located in the
‘cell’ and ‘extracellular region’ (Fig. 3C).
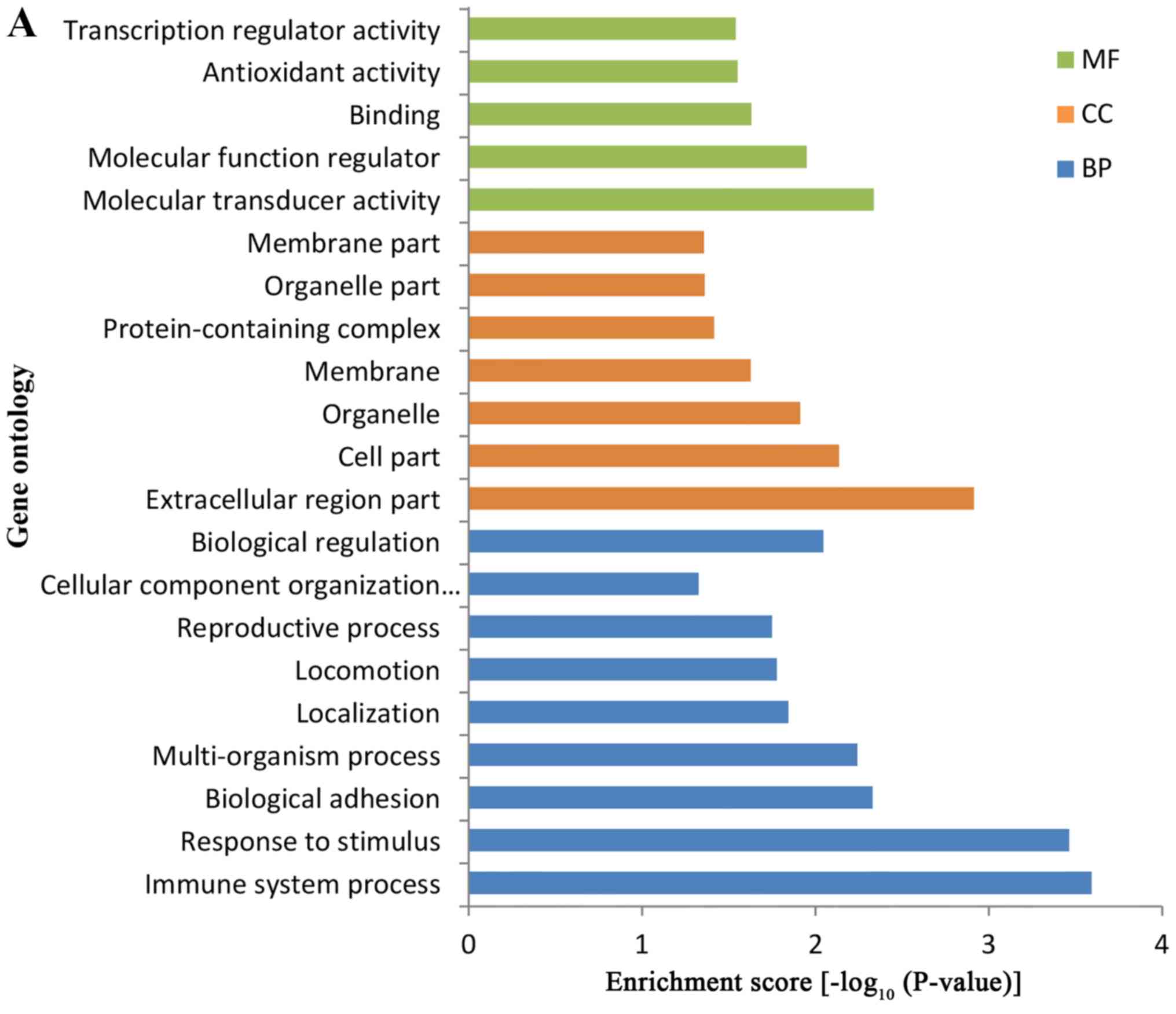
GO enrichment analysis revealed that the majority of
the DEPs were primarily enriched in ‘immune system process’,
‘response to stimulus’ and ‘biological regulation’ in the BPs. In
addition, the MFs of the DEPs were primarily associated with
‘molecular transducer activity’, ‘molecular function regulator’ and
‘transcription regulator activity’, whilst the DEPs were identified
to be primarily located in ‘extracellular region part’ and ‘cell
part’ (Fig. 4A).
KEGG signaling pathway enrichment
analysis
KEGG signaling pathway enrichment analysis was
performed to profile the functional differences of the proteomes of
MV/E isolated from the CSF of patients with ABE and the controls.
Significantly enriched signaling pathways of patients with ABE
included ‘Ras signaling pathway’, ‘PI3K-AKT signaling pathway’,
‘MAPK signaling pathway’, ‘cytokine-cytokine receptor interaction’,
‘Rap1 signaling pathway’, ‘mTOR signaling pathway’ and ‘NF-κB
signaling pathway’ (Fig. 4B).
Other enriched signaling pathways were identified, including ‘HIF-1
signaling pathway’, ‘mTOR signaling pathway’, ‘NF-κB signaling
pathway’, ‘Neuroactive ligand-receptor interaction’, and
‘ECM-receptor interaction’.
PPI of DEPs
STRING software was used to construct a potential
PPI of the identified DEPs. The robust and cross-talking
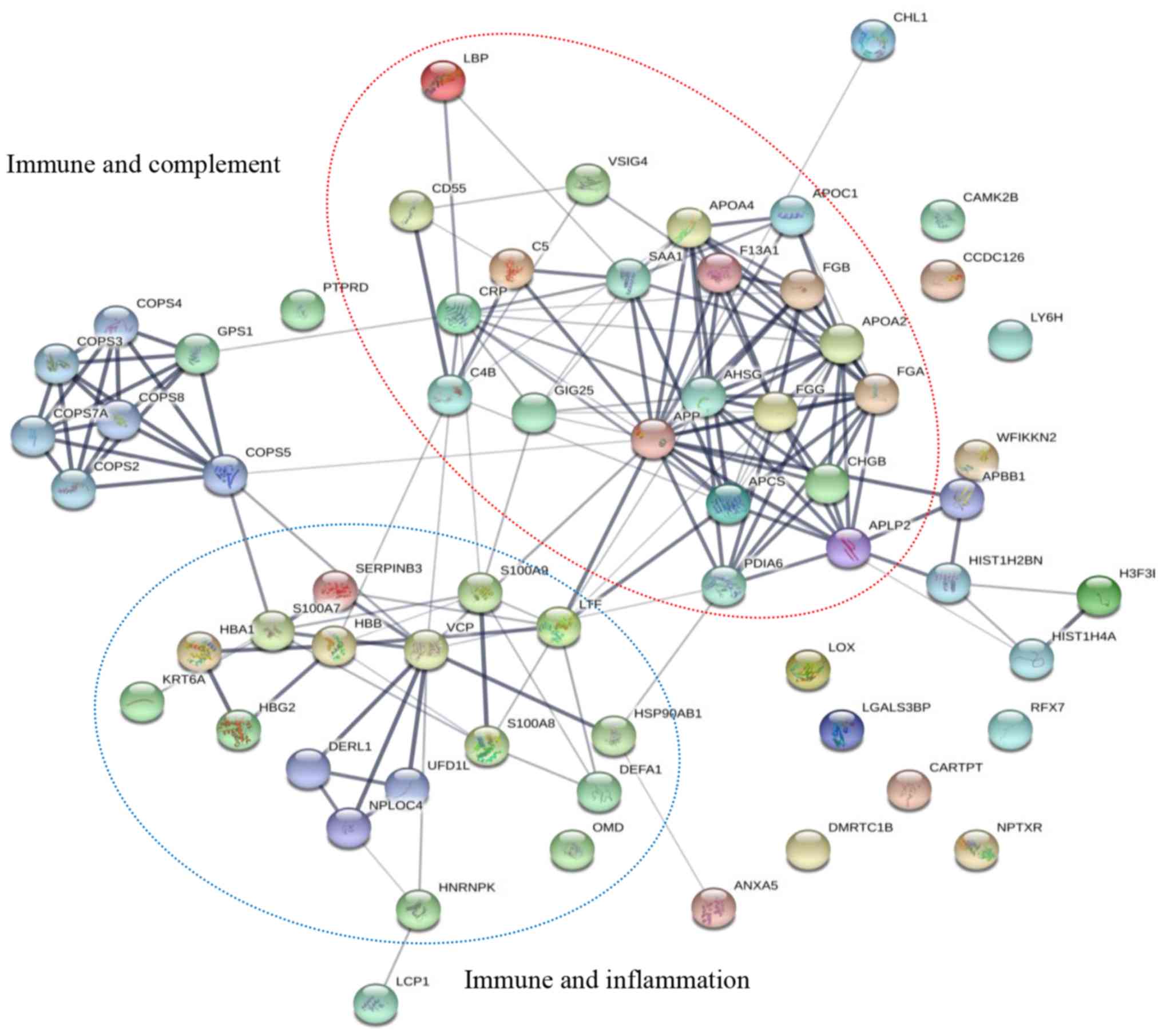
interactions are mapped in Fig. 5.
The DEPs were discovered to be associated with the immune system
and inflammation. In particular, the functions of serum amyloid A-1
protein (SAA1), amyloid-β precursor protein (APP), complement C4-B
(C4B), complement C5 (C5), DEFA1, LTF, C-reactive protein (CRP),
S100A7, S100A9 and lipopolysaccharide-binding protein (LBP) were
demonstrated to be associated with each other.
Verification of DEPs using western
blotting
Based on the comprehensive analysis of the
pathogenesis and bioinformatics of ABE, four candidate DEPs
(S100A9, S100A7, LTF and DEFA1) were further verified by western
blotting and an additional 12 participants were enrolled for the
validation. These participants included 4 patients with moderate
ABE, 4 with severe ABE and 4 controls. The expression levels of LTF
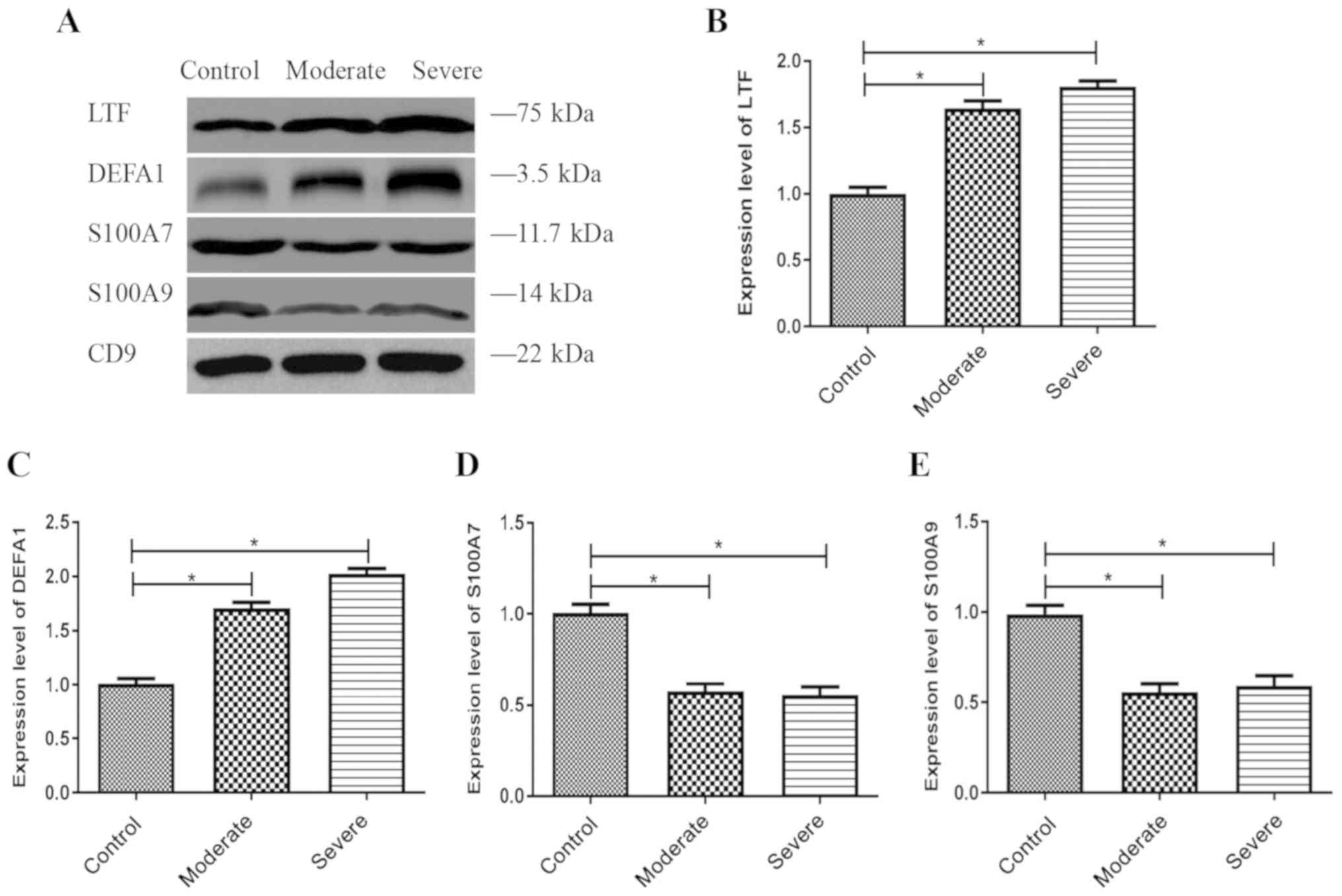
and DEFA1 were significantly upregulated (Fig. 6A-C), while the expression levels of
S100A7 and S100A9 were significantly downregulated (Fig. 6A, D and E) in both the moderate and
severe patients with ABE compared with the control groups; these
findings were consistent with the LC-MS/MS data.
Discussion
The incidence of clinical ABE is high, particularly
in developing countries, and subclinical BE (hypobilirubin
encephalopathy) is also seen in clinical practice (6–8). The
pathogenesis of BE is still unclear. In the present study,
iTRAQ-based quantitative proteomic technology combined with
LC/MS/MS was used to identify the proteomic profile of MV/E
obtained from the CSF of patients with ABE. Certain DEPs [SAA1,
APP, LBP, CRP, immunoglobulin, complement components (C4B and C5),
S100A9, S100A7, DEFA1 and LTF] associated with the
immune-inflammatory response were identified in the extracellular
vesicles, which indicated that these extracellular vesicles may
serve an important role in the pathophysiology of bilirubin-induced
neurological neurotoxicity by regulating and transmitting complex
communication among neurocytes.
Since the discovery of circulating extracellular
vesicles in 1979 (31), there has
been a rapid increase in scientific research on vesicles. Previous
studies have suggested that extracellular vesicles may serve a
crucial role in the pathophysiology of neurologic disorders by
regulating the communication among neurocytes via complex
mechanisms (32,21). Exosomes have been demonstrated to
promote neurite growth and enhance axonal regeneration in injured
neurons (33). Moreover, the cargo
of extracellular vesicles has been discovered to be involved in the
mechanisms underlying traumatic brain injury (32). However, to the best of our
knowledge, the role of extracellular vesicles in bilirubin-induced
neurological injury is not well characterized. In the present
study, MV/E were isolated from the CSF of patients with ABE and
changes in the proteomic profiles in these extracellular vesicles
were identified. Certain DEPs were associated with
immune-inflammation; these included SAA1, APP, LBP, CRP,
immunoglobulin, complement components (C4B and C5), S100A9, S100A7,
DEFA1 and LTF. In addition, GO annotation and enrichment analysis
of the DEPs revealed that the majority of the DEPs were primarily
associated with ‘immune system process’, ‘biological regulation’ or
‘molecular transducer activity’. KEGG signaling pathway enrichment
analysis also demonstrated that these DEPs were primarily enriched
in immune and inflammatory signaling pathways (‘PI3K-AKT signaling
pathway’, ‘NF-κB signaling pathway’ and ‘MAPK signaling pathway’).
In particular, the NF-κB signaling pathway and MAPK signaling
pathway are important immune-inflammatory pathways, which have been
reported to be activated by UCB at an early stage of brain
development (15). These findings
indicated that the DEPs identified in the extracellular vesicles of
patients with CSF may serve a critical role in the pathogenesis of
bilirubin-induced cerebral injury.
Previous studies have demonstrated that hazardous
levels of UCB may induce the acute and chronic activation of
microglia and astrocytes via the upregulation of proinflammatory
gene expression levels, subsequently triggering the release of
inflammatory biomarkers, such as IL-1β, TNF-α and IL-6 (14,34–37).
Hazardous levels of UCB were discovered to induce the unfolded
protein response and endoplasmic reticulum stress, which
contributed to neuronal inflammation by inducing IL-8 expression
levels via the regulation of the upstream NF-кB/PKR-like
endoplasmic reticulum kinase axis (18,38,39).
These results are consistent with the findings of the present
bioinformatics analysis, which revealed the enrichment of the
‘NF-kappa B signaling pathway’. These bilirubin-induced
immune-inflammatory signaling pathways and biomarkers have been
identified previously to contribute to neuronal apoptosis, necrosis
or pyroptosis (40). Altogether,
these results suggested that bilirubin-induced neurological
neurotoxicity may be associated with certain immune-inflammatory
processes.
The present study demonstrated differential
expression levels of certain proteins in the CSF of patients with
ABE associated with immune-inflammation, including APP, LBP and
CRP. In a previous study, APP protein was demonstrated to induce a
predominantly proinflammatory phenotype of microglia in Alzheimer's
disease (41). Moreover, LBP and
CRP, which are non-specific markers of inflammation, were found to
be significantly increased in the serum of patients with traumatic
brain injury (42,43). Although these non-specific
immune-inflammatory markers have been studied in the context of
traumatic brain injury and neurodegenerative diseases, to the best
of our knowledge, their role in the context of UCB-induced
neurological dysfunction is not well characterized. The significant
increase in APP, LBP and CRP expression levels in extracellular
vesicles isolated from the CSF in the present study may represent
the physiological and compensatory immune-inflammatory response to
UCB. However, further investigations are required to verify the
association between these proteins and bilirubin-induced
neurological neurotoxicity.
The preset study also demonstrated the significant
upregulation of complement-associated proteins, such as C4B and C5,
in patients with ABE. Previous studies have reported that both
neurons and glial cells are capable of synthesizing complement
receptors (44,45). The complement system is
hypothesized to serve an important role in brain homeostasis,
neural development, central nervous system (CNS) repair,
regeneration, neuroprotection, neurogenesis and the progression of
pathology in neurodegenerative disorders, stroke and traumatic
brain injury (46–48). These studies showed that the levels
of complement components were upregulated. The present results are
consistent with these studies. However, to the best of our
knowledge, no previous studies have investigated the association
between complement and bilirubin-induced brain injury. The present
study demonstrated the significant upregulation of numerous
immunoglobulin-like domains, such as immunoglobulin heavy constant
γ4 and immunoglobulin heavy variable 2–5 in MV/E isolated from the
CSF of patients with ABE. Immunoglobulins are an essential
component of the immune response (49). These immunoglobulin-like domains
have been suggested to serve as neural cell adhesion molecules that
mediate interactions among nerve cells in the brain (50). Thus, extreme hyperbilirubinemia may
trigger the complement system and upregulate certain
immunoglobulin-like domains. However, further investigations are
required to assess whether the upregulation of
complement-associated proteins and immunoglobulin-like domains
observed in CSF vesicles serve an important role in ABE.
S100A9, is a Ca2+-binding protein
belonging to the S100 family, which is primarily derived from
immunocytes (neutrophils, macrophages and myeloid-derived dendritic
cells), but also expressed in astrocytes and neurons (51). S100A9 has been reported to activate
multiple immune-inflammatory regulation signaling pathways,
including Ras/ERK/NF-κB and PI3K/Akt in astrocytes (52,53).
Moreover, the overexpression of S100A9 in neurons was identified to
be involved in the infiltration of microglia and the growth of
primary astrocytes in vitro via the activation of ‘Ras signaling
pathways’ (54). Thus, these
studies suggested that S100A9 may be an important immune
inflammatory biomarker in the nervous system, which participates in
the inflammatory process.
Although the upregulation of S100A9 may be involved
in the immune-inflammatory response in numerous types of neuronal
disorder, the present results demonstrated a significant
downregulation of S100A9 expression levels in MV/E isolated from
the CSF of patients with ABE. These results indicated that the
balance of neurological immune-inflammatory regulation functions of
S100A9 may be disrupted by hyperbilirubinemia; alternatively, the
downregulation of S100A9 expression levels in the MV/E may be an
intrinsic protective mechanism to trigger tissue proliferation or
repair activity following the exposure to hyperbilirubinemia.
S100A9 serves multiple biological roles depending on the
concentration, as well as the proximal microenvironment (55). Numerous studies have demonstrated
that S100A9 is involved in the modulation of cellular
proliferation, differentiation and apoptosis, as well as
proinflammatory and anti-inflammatory functions (56,57).
At high concentrations, extracellular S100A9 exhibited
proinflammatory functions and growth-inhibitory activity, induced
cell apoptosis, and exerted deleterious effects by stimulating
leukocyte migration and recruitment, enhancing
leukocyte-endothelial contact and increasing vascular permeability
(58–61). However, at low levels,
extracellular S100A9 was reported to exert either anti-inflammatory
properties to avoid tissue damage, or tissue proliferation/repair
activity at local inflammatory sites (55). Thus, the downregulated expression
levels of S100A9 may be associated with its intrinsic protective
mechanism to trigger tissue proliferation or repair activity
following exposure to hyperbilirubinemia. However, the association
between the downregulation of S100A9 and its anti-inflammatory or
intrinsic protective effects in bilirubin-induced brain injury
requires further investigation. Additionally, since the S100A9
protein in the present study was detected in extracellular
vesicles, it may differ from studies in which the protein was
detected in total CSF (62,63).
The S100A9 in extracellular vesicles represents the storage of
extracellular S100A9 in the brain (64). Physiologically, there is sufficient
storage of S100A9 in astrocytes or neurons; however, trauma, heat,
stress, infection and inflammation have all been identified to
trigger its upregulation and active release (55). Therefore, further investigations
are required to determine whether the downregulated expression
levels of S100A9 in extracellular vesicles are due to the release
of S100A9 from extracellular vesicles into the CSF. Whether the
downregulated expression levels of S100A9 and increased severity of
bilirubin-induced brain damage are due to impaired neuronal
function or gene suppression also remains to be elucidated.
S100A7 is also an important member of the S100
family, which contains the EF-hand calcium-binding protein
(65). S100A7 is a proinflammatory
protein expressed in immune cells and in neurons, microglia and
astrocytes in the brain (66).
S100A7 was also observed to be expressed in glial and meningeal
cells, which supports the hypothesis that S100A7 may be associated
with immune-inflammatory processes in the CNS (66). A previous study documented
increased levels of S100A7 in the CSF of patients with Alzheimer's
disease (67). However, the
present study demonstrated a significant downregulation in the
expression levels of S100A7 in the MV/E isolated from the CSF of
patients with ABE. A study by Sharma et al (68) also reported a significant decrease
in the serum levels of S100A7 in patients with acute ischemic
stroke. Similar to S100A9, S100A7 expression differs between
tissues and organs or in different disease states; this indicates
the multifaceted nature of S100A7 function (66). The downregulated expression levels
of S100A7 in the present study indicated that S100A7 may
participate in BIND; however, the exact mechanism of action and
function of S100A7 in this disease remains unclear. Further
investigations are required to confirm the results of the present
study.
In addition to the downregulation of S100A9 and
S100A7 expression levels, the present study also identified the
upregulation of the expression levels of certain bioactive
proteins, including defensins and LTF, which regulate
immune-inflammatory responses, as well as antioxidant and
neuroprotective processes (69,70).
The present study identified the significant upregulation of DEFA1
expression levels in the MV/E isolated from the CSF of patients
with ABE. Defensins are antimicrobial peptides that serve
multifaceted roles and exhibit immunomodulatory and
anti-inflammatory properties (71,72).
Variable expression levels of defensins have been identified in
cerebral microglia and astrocytes in both the mouse and human
brain, where they have been observed to serve complex roles in
immunomodulatory processes (73,74).
Moreover, following the injury to the CNS, microglia and astrocytes
provided immune defense in a stimulus-dependent manner via the
production and release of defensins (75). As ABE is a form of secondary brain
injury caused by hyperbilirubinemia, the upregulated expression
levels of DEFA1 indicated that DEFA1 may serve an important
immunomodulatory role in the pathogenesis of bilirubin-induced
brain injury. Additionally, as defensins are anti-inflammatory
peptides (76), the upregulated
expression levels of DEFA1 may have had an anti-inflammatory
neuroprotective function by preventing the excessive inflammation
in brain lesions. Thus, the findings of the present study suggested
that DEFA1 may be important in the immunomodulatory and
anti-inflammatory processes of bilirubin-induced neurological
neurotoxicity; however, further investigations are required to
elucidate the underlying mechanism.
The present study also demonstrated that the
expression levels of LTF were significantly upregulated in the MV/E
of patients with ABE. LTF is an iron-binding glycoprotein that
belongs to the transferrin family and serves numerous beneficial
biological functions, such as immunomodulatory, antioxidant and
neuroprotective effects (77).
Previously, LTF was observed to modulate the migration, maturation
and function of immune cells (78,79).
In addition, the expression levels of LTF in biological fluids were
significantly upregulated in patients with inflammatory diseases
(80). Moreover, in addition to
the immune-inflammatory mechanisms, oxidative stress is also
hypothesized to be an important pathogenetic mechanism of bilirubin
encephalopathy (81). The
upregulated expression levels of LTF indicated that it may be
involved in maintaining hemostasis between oxidation and
anti-oxidation. Previous studies have shown that ABE is partly
caused by oxidative stress and brain cell damage induced by high
bilirubin levels (11,81). LTF has been demonstrated to possess
antioxidant properties (77).
Therefore, the upregulated expression levels of LTF in the present
study may be involved in maintaining homeostasis between oxidation
and anti-oxidation.
LTF has been previously demonstrated to exhibit
antioxidant properties, in addition to decreasing ROS generation
and removing ROS from the brain; LTF was also reported to help
maintain the levels of ascorbate and glutathione, which are
important endogenous antioxidants (70,82).
These previous findings indicated that the upregulated expression
levels of LTF may improve the antioxidant capacity of the brain.
Furthermore, LTF has been demonstrated to exert neuroprotective
effects on the immature brain in rodent models of intrauterine
growth restriction and cerebral hypoxia/ischemia (83,84).
A number of studies have also revealed that upregulated expression
levels of LTF served as an important neuroprotective factor,
potentially due to its capacity to stimulate cell cycle progression
and induce erythropoietin synthesis (77,85).
Overall, the upregulated expression levels of LTF in ABE indicated
that it may either modulate immune-inflammatory processes or exert
antioxidant or neuroprotective effects in the pathological process
of BIND. However, the mechanism linking LTF with BIND still needs
to be evaluated.
In conclusion, the present study determined the
proteomic profile of extracellular vesicles (MV/E) isolated from
the CSF of patients with ABE. The findings of the present study
provided an improved understanding of the pathophysiology of BIND.
Certain proteins and signaling pathways involved in the MV/E were
primarily associated with the immune-inflammatory response. This
indicated that extracellular vesicles may serve an important role
in the development of bilirubin-induced neurological neurotoxicity
via immune-inflammatory proteins and signaling pathways. Moreover,
extracellular vesicles in the CSF are derived from neurocytes;
thus, their cargo (S100A9, S100A7, DEFA1 and LTF) represents the
microenvironment of the brain and offers information about the
pathophysiology of cerebral injury for patients with ABE (86). Altered DEP levels in extracellular
vesicles of CSF results in changes to the microenvironment of the
brain and the adaptation process of nerve cells due to
hyperbilirubinemia. The results therefore suggested that
extracellular vesicles and the identified proteins may serve as
biomarkers and provide a critical advantage over traditional
biomarkers in the early diagnosis of ABE. Currently, the diagnosis
of ABE primarily depends on clinical manifestations and cranial MRI
scans. Extracellular vesicles and proteins may serve as objective
biomarkers. Furthermore, due to the lack of specific drug
therapeutic options for ABE, extracellular vesicles may present a
novel therapeutic target; the immune-inflammatory targets involved
in extracellular vesicles may be engineered to regulate
intercellular communication and improve the outcomes for patients
with ABE, which could provide novel therapeutic perspectives. Thus,
further investigations are required to determine the usefulness of
MV/E as early diagnostic biomarkers and novel therapeutic targets
for ABE.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present work was supported by the Hunan
Provincial Science and Technology Department, China (grant no.
2019JJ80059) and the Health and Family Planning Commission of Hunan
Province, China (grant no. B20180259).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BW and NT designed the study and wrote the
manuscript. SH, ZW and BW wrote the manuscript. NT, SH, ZW and ZH
performed the proteome experiments and western blotting analysis.
NT, ZW and BW revised the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Ethics
Committee of Affiliated First People's Hospital of Chenzhou,
Southern Medical University (approval no. 2019049; Chenzhou,
China). Written informed consent was obtained from
parents/caregivers of all subjects prior to the start of the study.
All experimental procedures were performed in accordance with the
Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ABE
|
acute bilirubin encephalopathy
|
|
BIND
|
bilirubin-induced neurological
dysfunction
|
|
CSF
|
cerebrospinal fluid
|
|
DEPs
|
differentially expressed proteins
|
|
GO
|
Gene Ontology
|
|
iTRAQ
|
isobaric tagging for relative and
absolute quantification
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
MV/E
|
microvesicles/exosomes
|
|
MS/MS
|
tandem mass spectrometry
|
|
NTA
|
nanoparticle tracking analysis
|
|
UPLC
|
ultimate high-performance liquid
chromatography
|
References
|
1
|
Bhutani VK, Stark AR, Lazzeroni LC, Poland
R, Gourley GR, Kazmierczak S, Meloy L, Burgos AE, Hall JY and
Stevenson DK: Predischarge screening for severe neonatal
hyperbilirubinemia identifies infants who need phototherapy. J
Pediatr. 162:477–482.e1. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bech LF, Donneborg ML, Lund AM and Ebbesen
F: Extreme neonatal hyperbilirubinemia, acute bilirubin
encephalopathy, and kernicterus spectrum disorder in children with
galactosemia. Pediatr Res. 84:228–232. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bahr TM, Christensen RD, Agarwal AM,
George TI and Bhutani VK: The neonatal acute bilirubin
encephalopathy registry (NABER): Background, aims, and protocol.
Neonatology. 115:242–246. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Christensen RD, Agarwal AM, George TI,
Bhutani VK and Yaish HM: Acute neonatal bilirubin encephalopathy in
the state of utah 2009–2018. Blood Cells Mol Dis. 72:10–13. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McGillivray A and Evans N: Severe neonatal
jaundice: Is it a rare event in Australia? J Paediatr Child Health.
48:801–807. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Olusanya BO, Kaplan M and Hansen TWR:
Neonatal hyperbilirubinaemia: A global perspective. Lancet Child
Adolesc Health. 2:610–620. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Diala UM, Wennberg RP, Abdulkadir I,
Farouk ZL, Zabetta CDC, Omoyibo E, Emokpae A, Aravkin A, Toma B,
Oguche S, et al On behalf of the Stop Kernicterus In Nigeria (SKIN)
study group, : Patterns of acute bilirubin encephalopathy in
Nigeria: A multicenter pre-intervention study. J Perinatol.
38:873–880. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Morioka I, Nakamura H, Koda T, Yokota T,
Okada H, Katayama Y, Kunikata T, Kondo M, Nakamura M, Hosono S, et
al: Current incidence of clinical kernicterus in preterm infants in
Japan. Pediatr Int. 57:494–497. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Radmacher PG, Groves FD, Owa JA, Ofovwe
GE, Amuabunos EA, Olusanya BO and Slusher TM: A modified
Bilirubin-induced neurologic dysfunction (BIND-M) algorithm is
useful in evaluating severity of jaundice in a resource-limited
setting. BMC Pediatr. 15:282015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morioka I, Iwatani S, Koda T, Iijima K and
Nakamura H: Disorders of bilirubin binding to albumin and
bilirubin-induced neurologic dysfunction. Semin Fetal Neonatal Med.
20:31–36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Watchko JF: Kernicterus and the molecular
mechanisms of bilirubin-induced CNS injury in newborns.
Neuromolecular Med. 8:513–529. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deganuto M, Cesaratto L, Bellarosa C,
Calligaris R, Vilotti S, Renzone G, Foti R, Scaloni A, Gustincich
S, Quadrifoglio F, et al: A proteomic approach to the
bilirubin-induced toxicity in neuronal cells reveals a protective
function of DJ-1 protein. Proteomics. 10:1645–1657. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brites D: The evolving landscape of
neurotoxicity by unconjugated bilirubin: Role of glial cells and
inflammation. Front Pharmacol. 3:882012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Watchko JF and Tiribelli C:
Bilirubin-induced neurologic damage - mechanisms and management
approaches. N Engl J Med. 369:2021–2030. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Silva SL, Vaz AR, Barateiro A, Falcão AS,
Fernandes A, Brito MA, Silva RF and Brites D: Features of
bilirubin-induced reactive microglia: From phagocytosis to
inflammation. Neurobiol Dis. 40:663–675. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brites D: Bilirubin injury to neurons and
glial cells: New players, novel targets, and newer insights. Semin
Perinatol. 35:114–120. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Daood M, Tsai C, Ahdab-Barmada M and
Watchko JF: ABC transporter (P-gp/ABCB1, MRP1/ABCC1, BCRP/ABCG2)
expression in the developing human CNS. Neuropediatrics.
39:211–218. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qaisiya M, Brischetto C, Jašprová J, Vitek
L, Tiribelli C and Bellarosa C: Bilirubin-induced ER stress
contributes to the inflammatory response and apoptosis in neuronal
cells. Arch Toxicol. 91:1847–1858. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chivet M, Javalet C, Laulagnier K, Blot B,
Hemming FJ and Sadoul R: Exosomes secreted by cortical neurons upon
glutamatergic synapse activation specifically interact with
neurons. J Extracell Vesicles. 3:247222014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Izadpanah M, Seddigh A, Ebrahimi Barough
S, Fazeli SAS and Ai J: Potential of extracellular vesicles in
neurodegenerative diseases: Diagnostic and therapeutic indications.
J Mol Neurosci. 66:172–179. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Paolicelli RC, Bergamini G and Rajendran
L: Cell-to-cell communication by extracellular vesicles: Focus on
microglia. Neuroscience. 405:148–157. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiong XG, Liang Q, Zhang C, Wang Y, Huang
W, Peng W, Wang Z and Xia ZA: Serum proteome alterations in
patients with cognitive impairment after traumatic brain injury
revealed by itraq-based quantitative proteomics. BioMed Res Int.
2017:85725092017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Núñez Galindo A, Macron C, Cominetti O and
Dayon L: Analyzing cerebrospinal fluid proteomes to characterize
central nervous system disorders: A highly automated mass
spectrometry-based pipeline for biomarker discovery. Methods Mol
Biol. 1959:89–112. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Adav SS, Park JE and Sze SK: Quantitative
profiling brain proteomes revealed mitochondrial dysfunction in
Alzheimer's disease. Mol Brain. 12:82019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bortolussi G, Codarin E, Antoniali G,
Vascotto C, Vodret S, Arena S, Cesaratto L, Scaloni A, Tell G and
Muro AF: Impairment of enzymatic antioxidant defenses is associated
with bilirubin-induced neuronal cell death in the cerebellum of
Ugt1 KO mice. Cell Death Dis. 6:e17392015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chiasserini D, van Weering JRT, Piersma
SR, Pham TV, Malekzadeh A, Teunissen CE, de Wit H and Jiménez CR:
Proteomic analysis of cerebrospinal fluid extracellular vesicles: A
comprehensive dataset. J Proteomics. 106:191–204. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
World Medical Association: World Medical
Association Declaration of Helsinki, . Ethical Principles for
Medical Research Involving Human Subjects. JAMA. 310:2191–2194.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bhutani VK and Johnson-Hamerman L: The
clinical syndrome of bilirubin-induced neurologic dysfunction.
Semin Fetal Neonatal Med. 20:6–13. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gardiner C, Ferreira YJ, Dragovic RA,
Redman CW and Sargent IL: Extracellular vesicle sizing and
enumeration by nanoparticle tracking analysis. J Extracell
Vesicles. 2:22013. View Article : Google Scholar
|
|
30
|
Shao H, Im H, Castro CM, Breakefield X,
Weissleder R and Lee H: New technologies for analysis of
extracellular vesicles. Chemical reviews. 118:1917–1950. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Taylor DD and Doellgast GJ: Quantitation
of peroxidase-antibody binding to membrane fragments using column
chromatography. Anal Biochem. 98:53–59. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Osier N, Motamedi V, Edwards K, Puccio A,
Diaz-Arrastia R, Kenney K and Gill J: Exosomes in acquired
neurological disorders: New insights into pathophysiology and
treatment. Mol Neurobiol. 55:9280–9293. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang BL: Promoting axonal regeneration
through exosomes: An update of recent findings on exosomal PTEN and
mTOR modifiers. Brain Res Bull. 143:123–131. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fernandes A, Barateiro A, Falcão AS, Silva
SL, Vaz AR, Brito MA, Silva RF and Brites D: Astrocyte reactivity
to unconjugated bilirubin requires TNF-α and IL-1β receptor
signaling pathways. Glia. 59:14–25. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li M, Song S, Li S, Feng J and Hua Z: The
blockade of nf-kappab activation by a specific inhibitory peptide
has a strong neuroprotective role in a sprague-dawley rat
kernicterus model. J Biol Chem. 290:30042–30052. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Watchko JF: Bilirubin-induced
neurotoxicity in the preterm neonate. Clin Perinatol. 43:297–311.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Watchko JF and Maisels MJ: The enigma of
low bilirubin kernicterus in premature infants: Why does it still
occur, and is it preventable? Semin Perinatol. 38:397–406. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vodret S, Bortolussi G, Jašprová J, Vitek
L and Muro AF: Inflammatory signature of cerebellar
neurodegeneration during neonatal hyperbilirubinemia in
Ugt1−/− mouse model. J Neuroinflammation. 14:642017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kitamura M: Control of NF-κB and
inflammation by the unfolded protein response. Int Rev Immunol.
30:4–15. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Feng J, Li M, Wei Q, Li S, Song S and Hua
Z: Unconjugated bilirubin induces pyroptosis in cultured rat
cortical astrocytes. J Neuroinflammation. 15:232018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhu M, Wang X, Schultzberg M and Hjorth E:
Differential regulation of resolution in inflammation induced by
amyloid-β42 and lipopolysaccharides in human microglia. J
Alzheimers Dis. 43:1237–1250. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Davies MG and Hagen PO: Systemic
inflammatory response syndrome. Br J Surg. 84:920–935. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Anada RP, Wong KT, Jayapalan JJ, Hashim OH
and Ganesan D: Panel of serum protein biomarkers to grade the
severity of traumatic brain injury. Electrophoresis. 39:2308–2315.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gasque P, Fontaine M and Morgan BP:
Complement expression in human brain. Biosynthesis of terminal
pathway components and regulators in human glial cells and cell
lines. Immunol. 154:4726–4733. 1995.
|
|
45
|
Nataf S, Levison SW and Barnum SR:
Expression of the anaphylatoxin C5a receptor in the oligodendrocyte
lineage. Brain Res. 894:321–326. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hammad A, Westacott L and Zaben M: The
role of the complement system in traumatic brain injury: A review.
J Neuroinflammation. 15:242018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Brennan FH, Anderson AJ, Taylor SM,
Woodruff TM and Ruitenberg MJ: Complement activation in the injured
central nervous system: Another dual-edged sword? J
Neuroinflammation. 9:1372012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bellander BM, Olafsson IH, Ghatan PH, Bro
Skejo HP, Hansson LO, Wanecek M and Svensson MA: Secondary insults
following traumatic brain injury enhance complement activation in
the human brain and release of the tissue damage marker S100B. Acta
Neurochir (Wien). 153:90–100. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Stoll BJ, Lee FK, Hale E, Schwartz D,
Holmes R, Ashby R, Czerkinsky C and Nahmias AJ: Immunoglobulin
secretion by the normal and the infected newborn infant. J Pediatr.
122:780–786. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zinn K and Özkan E: Neural immunoglobulin
superfamily interaction networks. Curr Opin Neurobiol. 45:99–105.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Denstaedt SJ, Spencer-Segal JL, Newstead
MW, Laborc K, Zhao AP, Hjelmaas A, Zeng X, Akil H, Standiford TJ
and Singer BH: S100a8/a9 drives neuroinflammatory priming and
protects against anxiety-like behavior after sepsis. J Immunol.
200:3188–3200. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hermani A, De Servi B, Medunjanin S,
Tessier PA and Mayer D: S100A8 and S100A9 activate MAP kinase and
NF-kappaB signaling pathways and trigger translocation of RAGE in
human prostate cancer cells. Exp Cell Res. 312:184–197. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ghavami S, Rashedi I, Dattilo BM, Eshraghi
M, Chazin WJ, Hashemi M, Wesselborg S, Kerkhoff C and Los M:
S100A8/A9 at low concentration promotes tumor cell growth via RAGE
ligation and MAP kinase-dependent pathway. J Leukoc Biol.
83:1484–1492. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ryu MJ, Liu Y, Zhong X, Du J, Peterson N,
Kong G, Li H, Wang J, Salamat S, Chang Q, et al: Oncogenic Kras
expression in postmitotic neurons leads to S100A8-S100A9 protein
overexpression and gliosis. J Biol Chem. 287:22948–22958. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang S, Song R, Wang Z, Jing Z, Wang S and
Ma J: S100A8/A9 in Inflammation. Front Immunol. 9:12982018.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Low D, Subramaniam R, Lin L, Aomatsu T,
Mizoguchi A, Ng A, DeGruttola AK, Lee CG, Elias JA, Andoh A, et al:
Chitinase 3-like 1 induces survival and proliferation of intestinal
epithelial cells during chronic inflammation and colitis-associated
cancer by regulating S100A9. Oncotarget. 6:36535–36550. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Donato R, Cannon B, Sorci G, Riuzzi F, Hsu
K, Weber D and Geczy C: Functions of s100 proteins. Curr Mol Med.
13:24–57. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ryckman C, Vandal K, Rouleau P, Talbot M
and Tessier PA: Proinflammatory activities of s100: Proteins
s100a8, s100a9, and s100a8/a9 induce neutrophil chemotaxis and
adhesion. J Immunol. 170:3233–3242. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Pruenster M, Kurz AR, Chung KJ, Cao-Ehlker
X, Bieber S, Nussbaum CF, Bierschenk S, Eggersmann TK, Rohwedder I,
Heinig K, et al: Extracellular MRP8/14 is a regulator of β2
integrin-dependent neutrophil slow rolling and adhesion. Nat
Commun. 6:69152015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nishikawa Y, Kajiura Y, Lew JH, Kido JI,
Nagata T and Naruishi K: Calprotectin induces il-6 and mcp-1
production via toll-like receptor 4 signaling in human gingival
fibroblasts. J Cell Physiol. 232:1862–1871. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ma L, Sun P, Zhang JC, Zhang Q and Yao SL:
Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling
pathways in BV-2 microglial cells. Int J Mol Med. 40:31–38. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Horvath I, Jia X, Johansson P, Wang C,
Moskalenko R, Steinau A, Forsgren L, Wågberg T, Svensson J,
Zetterberg H, et al: Pro-inflammatory s100a9 protein as a robust
biomarker differentiating early stages of cognitive impairment in
alzheimer's disease. ACS Chem Neurosci. 7:34–39. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wache C, Klein M, Ostergaard C, Angele B,
Häcker H, Pfister HW, Pruenster M, Sperandio M, Leanderson T, Roth
J, et al: Myeloid-related protein 14 promotes inflammation and
injury in meningitis. J Infect Dis. 212:247–257. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Meldolesi J: Exosomes and ectosomes in
intercellular communication. Curr Biol. 28:R435–R444. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Fritz G, Botelho HM, Morozova-Roche LA and
Gomes CM: Natural and amyloid self-assembly of S100 proteins:
Structural basis of functional diversity. FEBS J. 277:4578–4590.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Jansen S, Podschun R, Leib SL, Grötzinger
J, Oestern S, Michalek M, Pufe T and Brandenburg LO: Expression and
function of psoriasin (S100A7) and koebnerisin (S100A15) in the
brain. Infect Immun. 81:1788–1797. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Qin W, Ho L, Wang J, Peskind E and
Pasinetti GM: S100A7, a novel Alzheimer's disease biomarker with
non-amyloidogenic alpha-secretase activity acts via selective
promotion of ADAM-10. PLoS One. 4:e41832009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sharma R, Gowda H, Chavan S, Advani J,
Kelkar D, Kumar GS, Bhattacharjee M, Chaerkady R, Prasad TS, Pandey
A, et al: Proteomic signature of endothelial dysfunction identified
in the serum of acute ischemic stroke patients by the itraq-based
lc-ms approach. J Proteome Res. 14:2466–2479. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kim C and Kaufmann SH: Defensin: A
multifunctional molecule lives up to its versatile name. Trends
Microbiol. 14:428–431. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Park YG, Jeong JK, Lee JH, Lee YJ, Seol
JW, Kim SJ, Hur TY, Jung YH, Kang SJ and Park SY: Lactoferrin
protects against prion protein-induced cell death in neuronal cells
by preventing mitochondrial dysfunction. Int J Mol Med. 31:325–330.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Semple F and Dorin JR: β-Defensins:
Multifunctional modulators of infection, inflammation and more? J
Innate Immun. 4:337–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tollner TL, Bevins CL and Cherr GN:
Multifunctional glycoprotein DEFB126 - a curious story of
defensin-clad spermatozoa. Nat Rev Urol. 9:365–375. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kazakos EI, Kountouras J, Polyzos SA and
Deretzi G: Novel aspects of defensins' involvement in virus-induced
autoimmunity in the central nervous system. Med Hypotheses.
102:33–36. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Williams WM, Torres S, Siedlak SL,
Castellani RJ, Perry G, Smith MA and Zhu X: Antimicrobial peptide
β-defensin-1 expression is upregulated in Alzheimer's brain. J
Neuroinflammation. 10:1272013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Rocha-Ferreira E and Hristova M:
Antimicrobial peptides and complement in neonatal hypoxia-ischemia
induced brain damage. Front Immunol. 6:562015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Miles K, Clarke DJ, Lu W, Sibinska Z,
Beaumont PE, Davidson DJ, Barr TA, Campopiano DJ and Gray M: Dying
and necrotic neutrophils are anti-inflammatory secondary to the
release of alpha-defensins. J Immunol. 183:2122–2132. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zakharova ET, Sokolov AV, Pavlichenko NN,
Kostevich VA, Abdurasulova IN, Chechushkov AV, Voynova IV,
Elizarova AY, Kolmakov NN, Bass MG, et al: Erythropoietin and Nrf2:
Key factors in the neuroprotection provided by apo-lactoferrin.
Biometals. 31:425–443. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Legrand D, Elass E, Carpentier M and
Mazurier J: Lactoferrin: A modulator of immune and inflammatory
responses. Cell Mol Life Sci. 62:2549–2559. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Legrand D: Lactoferrin, a key molecule in
immune and inflammatory processes. Biochem Cell Biol. 90:252–268.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Bennett RM and Kokocinski T: Lactoferrin
content of peripheral blood cells. Br J Haematol. 39:509–521. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Qaisiya M, Coda Zabetta CD, Bellarosa C
and Tiribelli C: Bilirubin mediated oxidative stress involves
antioxidant response activation via Nrf2 pathway. Cell Signal.
26:512–520. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ginet V, van de Looij Y, Petrenko V,
Toulotte A, Kiss J, Hüppi PS and Sizonenko SV: Lactoferrin during
lactation reduces lipopolysaccharide-induced brain injury.
Biofactors. 42:323–336. 2016.PubMed/NCBI
|
|
83
|
van de Looij Y, Ginet V, Chatagner A,
Toulotte A, Somm E, Hüppi PS and Sizonenko SV: Lactoferrin during
lactation protects the immature hypoxic-ischemic rat brain. Ann
Clin Transl Neurol. 1:955–967. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Somm E, Larvaron P, van de Looij Y,
Toulotte A, Chatagner A, Faure M, Métairon S, Mansourian R, Raymond
F, Gruetter R, et al: Protective effects of maternal nutritional
supplementation with lactoferrin on growth and brain metabolism.
Pediatr Res. 75:51–61. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lee SH, Pyo CW, Hahm DH, Kim J and Choi
SY: Iron-saturated lactoferrin stimulates cell cycle progression
through PI3K/Akt pathway. Mol Cells. 28:37–42. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Thompson AG, Gray E, Heman-Ackah SM, Mäger
I, Talbot K, Andaloussi SE, Wood MJ and Turner MR: Extracellular
vesicles in neurodegenerative disease - pathogenesis to biomarkers.
Nat Rev Neurol. 12:346–357. 2016. View Article : Google Scholar : PubMed/NCBI
|