Introduction
Myocardial hypertrophy is an independent risk factor
of cardiovascular diseases (1). At
the molecular level, myocardial hypertrophy is caused by an
imbalance of protein synthesis and degradation. Persistent external
stimuli lead to overexpression of the signaling pathway for
myocardial protein synthesis and inhibition of the metabolic
pathway (2). Other effects include
increased myocardial cell volume, increased protein synthesis and
muscle fibre and myocardial remodeling (3). Furthermore, abnormal energy
metabolism, which turns from the oxygenolysis of fatty acids to the
utilization of glucose, is additionally accompanied (4). Such a long-term alteration will lead
to the reduction of energy production efficiency, the accumulation
of fatty acids in myocardial cells, and the increase of the
anaerobic glycolysis of glucose, which reduces the myocardial cell
contraction and accelerates the occurrence and development of heart
failure (5–7). The present study examined the
regulation of myocardial cell energy metabolism and the autophagy
signaling pathway to determine a mechanism that may inhibit
myocardial hypertrophy and may be used as a novel target of
treatment.
The primary regulatory factors in cell energy
metabolism are AMP-activated protein kinase (AMPK) and members of
the silent information regulator family (7,8).
Sirtuin3 (Sirt3) belongs to the latter and is a histone deacetylase
(HDAC) III (8). As a nicotinamide
adenine dinucleotide (NAD)-dependent HDAC primarily existing in the
mitochondria, Sirt3 not only regulates the energy metabolism of
cells; however, additionally serves an important role in apoptosis,
tumor growth and cardiovascular diseases (9). Previously, an increasing number of
studies have reported that Sirt3 serves a key role in myocardial
hypertrophy. Sundaresan et al (10) observed that Sirt3 may downregulate
mitogen-activated protein kinases/extracellular signal-regulated
kinases and the phosphoinositide 3-kinase/protein kinase B
signaling pathways through inhibition of the oxygen
radical-mediated renin activity. This inhibition occurs by
activating forkhead box protein O3 (FoxO3), manganese superoxide
dismutase and catalase, and by inhibiting myocardial hypertrophy
(10). Pillai et al
(11) determined that myocardial
hypertrophy may be inhibited through activation of the Sirt3-liver
kinase B1 (LKB1)-AMPK pathway (11). It was observed that Sirt3 gene
knockout mice demonstrated myocardial hypertrophy (12). All of these studies suggest that
Sirt3 is involved in the occurrence and development of myocardial
hypertrophy.
Autophagy is a biological phenomenon widely
available in eukaryocytes. Autophagy is additionally an important
channel of waste elimination, structure reconstruction, and growth
and development of cells (13).
Reduced autophagy has been identified in myocardial hypertrophy
since the early 1980s; Dämmrich and Pfeifer (14) identified decreased autophagy in an
aortic coarctation model. Nakai et al (15) observed significantly decreased
autophagy in a myocardial hypertrophy model induced by aortic
coarctation. A previous study published in 2014 (16) additionally demonstrated that the
autophagy level decreased significantly in an in vitro
myocardial hypertrophy model induced by angiotensin (Ang) II, which
suggests that autophagy may inhibit the development of myocardial
hypertrophy.
At present, the Sirt3-autophagy pathway is more
frequently reported, including in hepatic diseases (17–19),
the nervous system (20,21), tumors (22,23)
and skeletal muscle (24). In
myocardial ischemia reperfusion, Sirt3 may protect the heart by
promoting autophagy (25).
However, there is no study at present, to the best of the authors'
knowledge, on the Sirt3-autophagy pathway in myocardial
hypertrophy.
Materials and methods
Experimental animals
In total, 12 clean Sprague Dawley rats (6 male and 6
female) born within 1–2 days were provided by the Experimental
Animal Centre of Shantou University Medical College (Shantou,
China) from the same race and brood. The temperature was maintained
at 25°C and the humidity at was maintained at 50% with 12-h
light/dark cycle. All the rats had free access food and water.
Differences pertaining to a comparison among age, weight (average
5–6 g) and health state were not statistically significant
(P>0.05). The present study was approved by the Medical Ethics
Committee of Shantou University.
Primary reagents
Dulbecco's modified Eagle's medium (DMEM)-F12 and
fetal calf serum (FCS; HyClone; GE Healthcare Life Sciences),
trypsin and collagenase I (Gibco; Thermo Fisher Scientific, Inc.),
BrdU (Sigma-Aldrich; Merck KGaA), Ang II (AnaSpec), antibodies
against Sirt3 [Cell Signaling Technology (CST), Inc.; C73E3 Rabbit
mAb; cat. no. 2627S], light chain (LC)3I/II (CST, Inc.; D11, XP
Rabbit mAb; cat. no. 3868S), Beclin1 (CST, Inc.; D40C5, Rabbit mAb;
cat. no. 3495S), GADPH (CST, Inc.; D16H11, XP Rabbit mAb; cat. no.
5174S) and β-actin (CST, Inc.; 13E5, Rabbit mAb; cat. no. 4970T),
goat anti-rabbit immunoglobulin G-horseradish peroxidase (HRP)
secondary antibody (cat. no. R11412; Bellancom), a western blotting
kit (EMD Millipore), nuclease-free water, RNAase inhibitor,
deoxyribonucleotide mixture, reverse transcriptase and random
primers (Takara Bio, Inc.), and resveratrol (Shanghai Sangon
Pharmaceutical Co., Ltd.) were used.
Separation and culture of primary
myocardial cells of neonatal rats
The heart was removed by thoracotomy under aseptic
conditions and submerged in D-Hank's solution. Subsequent to the
bloodiness being cleaned, the connective tissue of the cardiac base
and the atrial tissue were removed. The ventricle was cut off to
remove the extravasated blood. The myocardial tissue was placed
into a 15 ml centrifuge tube and cut into blocks of 1
mm3. After 0.5 g/l collagenase type I was added, the
tube was gently oscillated in a 37°C thermostatic bath for 2 h to
lyse the tissue block. Subsequently, 0.125 g/l pancreatin was added
to digest the tissue and this was conducted 2–3 times (5 min/time).
The digested cells were collected with medium containing 100 ml/l
FCS, inoculated into a culture dish, and placed into a 5%
CO2 incubator for 1 h at 37°C. Fibroblasts were
eliminated by differential adhesion. Myocardial cells were
inoculated into a 6-well plate at a density of 1×106/ml.
A total of 0.03 g/l BrdU was added to inhibit fibroblast growth. A
total of 0.1 g/l penicillin and 0.1 g/l streptomycin were added to
prevent bacterial contamination. Subsequently, the plate was
cultured in a 50 ml/l CO2 incubator. At 24 h after
inoculation and when the cells fused and contracted synchronously,
the serum-free medium was used. After 24 h, intervention of the
different groups was conducted (26).
Protein expression is detected by
western blot analysis
Following the completion of the aforementioned
treatment, myocardial cells were lysed and the total protein was
extracted using SDS buffer (Jianglai Bio, Inc.). A total of 30 µl
of protein, determined using bicinchoninic acid method, was loaded
in each lane of a 12% of SDS-PAGE gel for electrophoresis.
Subsequent to electrophoresis, the protein was transferred to the
polyvinylidene difluoride (PVDF) membranes (80 V; 120 min); after 1
h of blocking at 25°C using 5% bovine serum albumin (Gibco; Thermo
Fisher Scientific, Inc.), the protein was incubated with the
following antibodies overnight at 4°C; Sirt3 (1:1,000), LC3I/II
(1:1,000), Beclin1 (1:1,000), medium-chain acyl-CoA dehydrogenase
(MCAD; 1:1,000), GADPH (1:20,000) and β-actin (1:10,000).
Subsequent to washing with Tris-buffered saline with 15 ml of 1X
Tween-20 the following day, the PVDF membranes were incubated with
HRP-labelled goat anti-rabbit (1:10,000) antibody for 1 h at room
temperature. Western blot analysis was conducted using enhanced
chemiluminescence substrate kit (Shanghai Yaxin Biotechnology Co.,
Ltd.). Quantity One image analysis software (version 4.5; Bio-Rad
Laboratories, Inc.) was used to detect the grey value of the
protein bands. The grey value ratio of the target band and GAPDH or
β-actin was used to indicate the expression level of the target
protein (27).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
The mRNA was extracted by routine TRIzol®
lysis (Thermo Fisher Scientific, Inc.) and reverse transcribed into
cDNA. Reverse transcription was performed using a PrimeScript RT
Reagent kit (Takara Bio, Inc.). GAPDH was used as an internal
control. The PCR reaction conditions were as follows: Initial
denaturation at 42°C for 60 min followed by 99°C for 5 min and 4°C
for 5 min. Relative expression of the target gene was calculated
using the 2−ΔΔCq method (28): ΔΔCq = Cq target gene - Cq reference
gene (experimental group), Cq target gene - Cq reference gene
(control group). Cq was the number of quantification cycles at
which the fluorescence exceeded the threshold. Each experiment was
performed in triplicate.
According to the standard curve: i) Using cDNA of
the control group as the template, the cDNA was diluted to 1:10,
1:50, 1:1,00 and 1:1,000; ii) ligand system: The total volume was
10 µl and each sample had three wells (4 µl cDNA + 0.3 µl Primer F
+ 0.3 µl Primer R + 0.4 µl H2O + 5 µl SYBR-Green (Takara
Bio, Inc.) × number of wells; iii) centrifugation: 999 × g for 4
min at 4°C; and iv) real-time PCR analysis.
The quantitative PCR was performed according to a
previous study (28). First, the
cDNA solution was diluted 50 times. Subsequently, the samples (4
µl) were loaded. The rest was the same as that above. Samples were
run in triplicate. The system was put into the Roche PCR analyzer
(Roche Molecular Diagnostics). The reaction conditions were
pre-denaturation at 95°C for 5 min followed by processing at 95°C
for 10 sec, 60°C for 10 sec and 72°C for 20 sec for a total of 45
cycles. Following completion of the reaction, DataAssist™ software
(v3.01) (Thermo Fisher Scientific, Inc.) was used for analysis. The
analysis was repeated three times. The data were used for
statistical analysis (Table
I).
 | Table I.Primer sequences for the polymerase
chain reaction assays. |
Table I.
Primer sequences for the polymerase
chain reaction assays.
| Gene | Primer | Primer
sequences |
|---|
| Sirt3 | F |
5′-TGCACGGTCTGTCGAAGGTC-3′ |
|
| R |
5′-AGGTTTCACAACGCCAGTA-3′ |
| ANP | F |
5′-CGTATACAGTGCGGTGTCCA-3′ |
|
| R |
5′-GATCTATCGGAGGGGTCCCA-3′ |
| BNP | F |
5′-TCCTTAATCTGTCGCCGCTG-3′ |
|
| R |
5′-CGCCGATCCGGTCTATCTTC-3′ |
| MCAD | F |
5′-AGCCCTGGACGAAGCTACTA-3′ |
|
| R |
5′-GCGAGCTGGTTGGCAATATC-3′ |
| GAPDH | F |
5′-TGCCACTCAGAAGACTGTGG-3′ |
|
| R |
5′-TTCAGCTCTGGGATGACCTT-3′ |
| β-actin | F |
5′-GAACCCTAAGGCCAACCGTGAAAAGAT-3′ |
|
| R |
5′-ACCGCTCGTTGCCAATAGTGATG-3′ |
| PK | F |
5′-AATCCCGGCAGATACAGACT-3′ |
|
| R |
5′-GGAGTTCCACACCCTGCTAT-3′ |
Area of myocardial cells
Following treatment with Ang II (20 µM) for 48 h at
37°C, the myocardial cells were fixed with 4% paraformaldehyde at
4°C for 30 min and imaged using a fluorescent microscope,
magnification, ×400. Fifty views of the same size were taken for
each group. The image analysis software, Image pro plus 6.0
(National Institutes of Health), was used to calculate the area of
the cells (27).
Transfection
A transfection kit was purchased from Foregene Co.,
Ltd. (TransEasy™). Ad-siRNA-Sirt3 was constructed by Sangon Biotech
(Shanghai) Co., Ltd. In the 6-well plate used to culture
1×106 myocardial cells, Ad-siRNA-Sirt3 virus
(1×108 pfu/virus) was used to infect the cells
[multiplicity of infection (MOI) =100]. The cells were collected 24
h after infection. Subsequent to lysis by the protein lysate, the
protein was extracted. A total of 50 µg protein of each group was
detected by western blot analysis. Whether the recombination of
adenovirus Ad-siRNA-Sirt3 was successful was determined utilizing
the specific Sirt3 antibody and according to the expression
alteration of the Sirt3 protein in each group.
The primary cultured myocardial cells of rats were
used. The recombinant adenovirus vector Ad-siRNA-Sirt3 was used to
infect the myocardial cells, and Ad-GFP and Ad-siRNA served as the
control. The virus solution diluted into suitable titres was added
to achieve the corresponding MOI (optimal MOI was 50 µM, according
to the preliminary experiment; data not shown). After 6 h of
culture at 37°C, the virus solution was left, and DMEM culture
medium containing a small amount of FCS was added for an additional
12 h of culture. After 48 h of treatment with 20 µM Ang II, the
mRNA expression of myocardial hypertrophy markers, atrial
natriuretic peptide (ANP), brain natriuretic peptide (BNP), MCAD (a
key enzyme of energy metabolism) and pyruvate kinase (PK), and the
protein expression of Sirt3, LC3 and Beclin1, were detected.
Statistical analysis
The SPSS 15.0 (SPSS, Inc.) statistical package was
used for the statistical analysis. The data of each group are
presented as the mean ± standard deviation. The differences between
the groups and the differences between the time-points were
analyzed by one-way analysis of variance, with the least
significant difference method as the post hoc test. P<0.05 was
considered to indicate a statistically significant difference. All
experiments were repeated three times.
Results
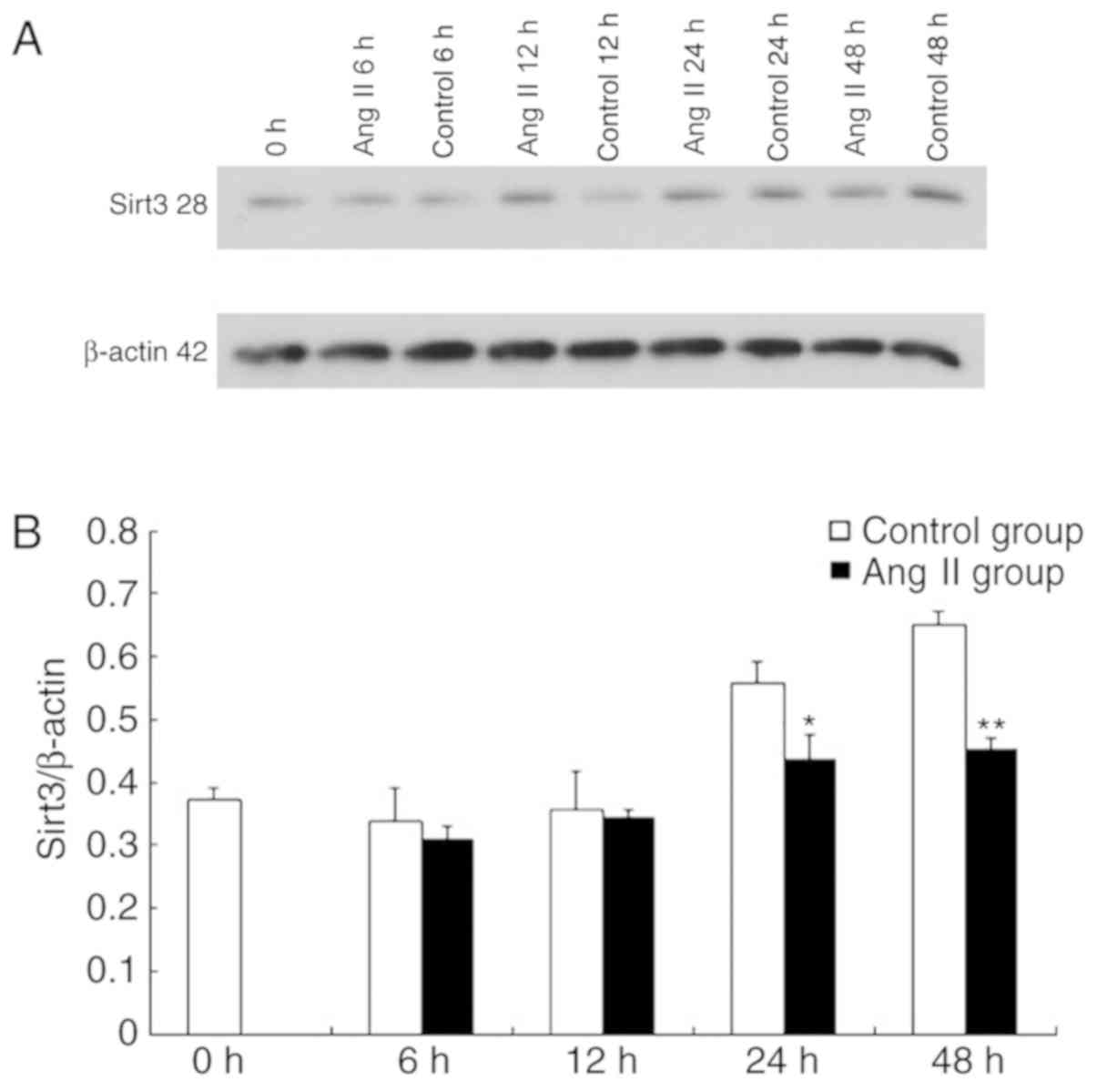
Sirt3 expression is decreased in Ang
II-induced cardiomyocyte hypertrophy
The alterations in the expression of Sirt3 were
observed in Ang II-induced cardiomyocyte hypertrophy to assess the
role of Sirt3. The cardiomyocytes were treated with Ang II in
vitro, and the expressions of Sirt3 were detected at 0, 6, 12,
24 and 48 h. The results demonstrated that compared with the
control group, the Sirt3/β-actin ratio following treatment with Ang
II decreased at different time-points (Fig. 1A and B). This suggested that Sirt3
is downregulated in Ang II-induced cardiomyocyte hypertrophy. The
expression of LC3, which is a homologue of the autophagy-associated
gene (GABA type A receptor associated protein like 2), in Ang
II-induced cardiomyocyte hypertrophy decreased (data not shown).
These results suggest that there is a potential association between
the two (Fig. 1).
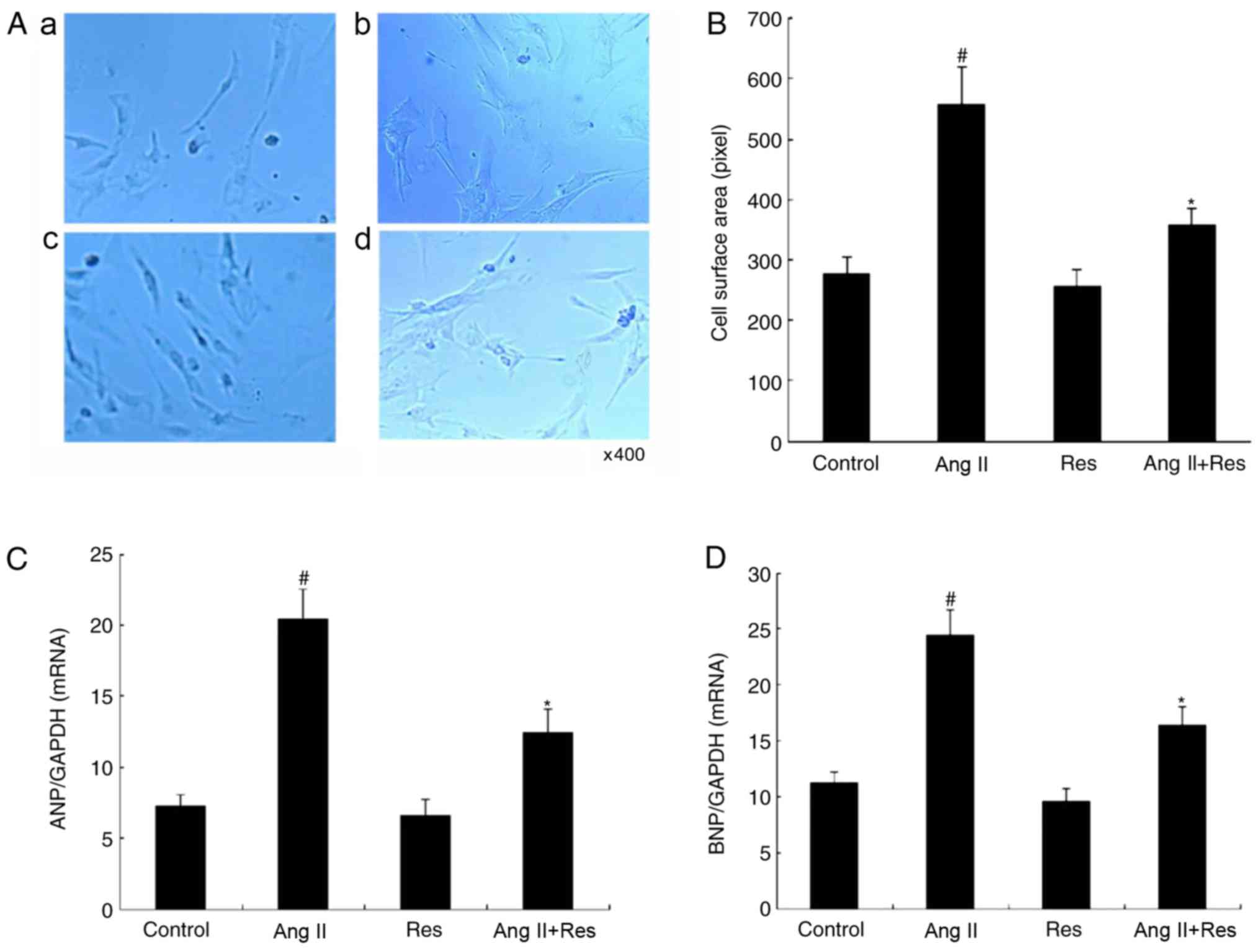
Res inhibits cardiomyocyte
hypertrophy
The Sirt3 agonist Res was used to treat
cardiomyocytes in Ang II-induced cardiomyocyte hypertrophy. It was
observed that Res significantly inhibited cardiomyocyte hypertrophy
in terms of cell surface area and the expression of
hypertrophy-associated genes compared with the Ang II group
(Fig. 2; P<0.05).
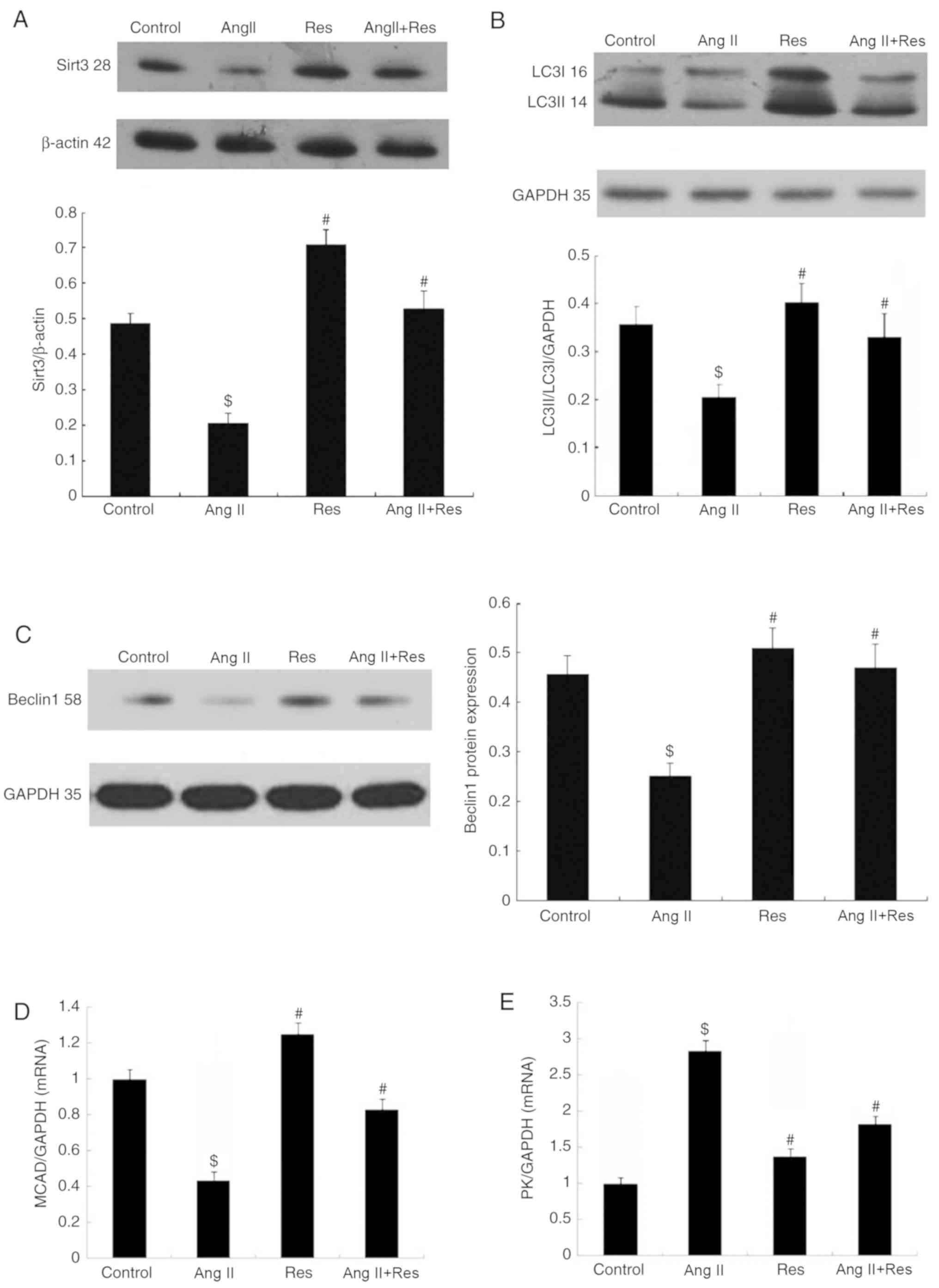
Res upregulates the expression of
Sirt3, LC3 and Beclin1, and increases the energy metabolism of
cardiomyocytes
The effects of Res on Sirt3 and autophagy were
observed in Ang II-induced cardiomyocyte hypertrophy and the
potential association between the two was assessed. The results
demonstrated that in Ang II-induced cardiomyocyte hypertrophy, Res
upregulated the expressions of Sirt3, autophagy-associated protein
LC3 and Beclin1, which counteracted the effect of Ang II to a
certain extent (Fig. 3A-C). With
regard to the energy metabolism, Res increased the mRNA expression
of MCAD while inhibiting the mRNA expression of PK (Fig. 3D and E).
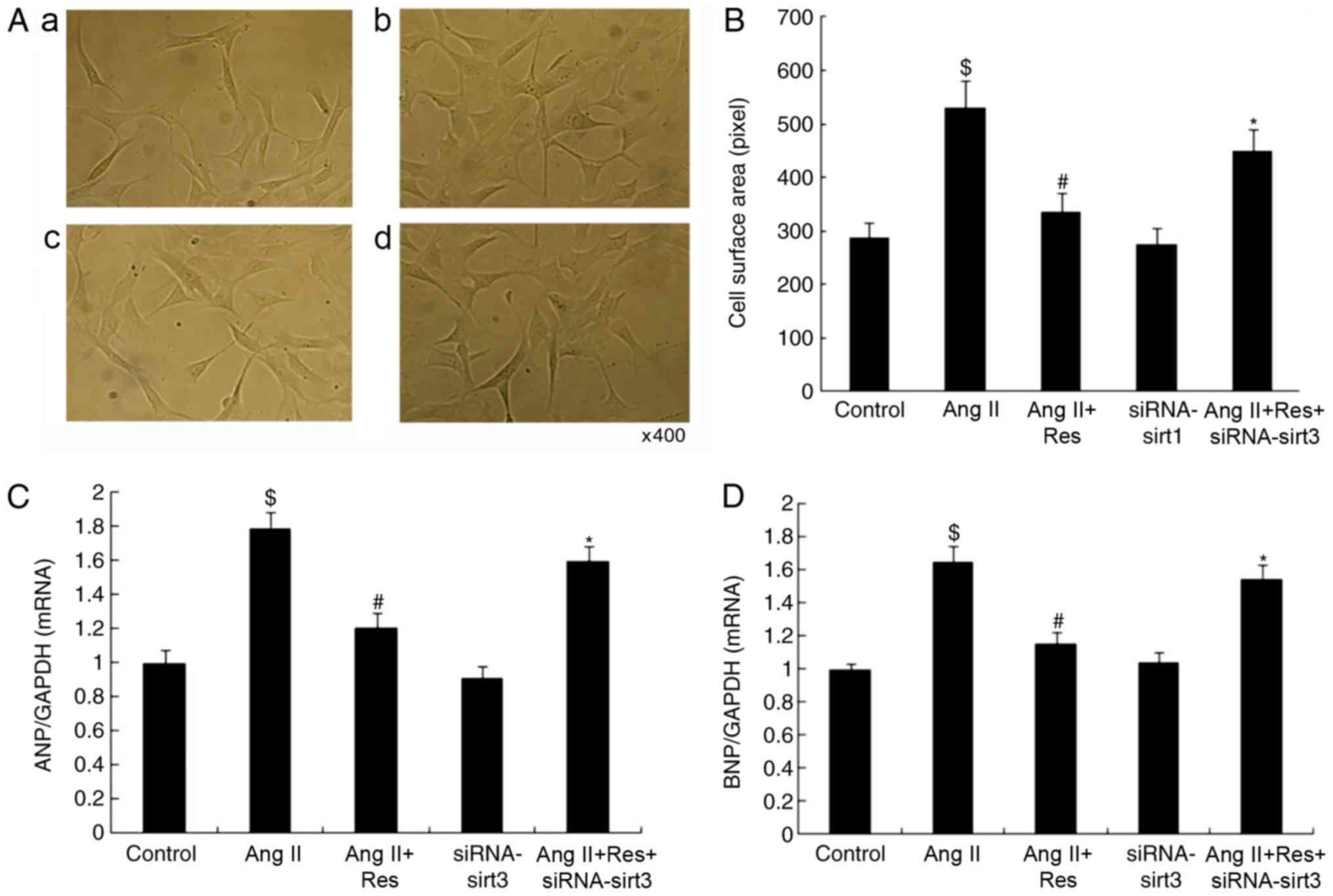
Silencing Sirt3 reverses the effects
of Res
As shown in Fig. 4,
the effect of siRNA-mediated silencing of Sirt3 on cardiomyocytes
and autophagy in Ang II-induced cardiomyocyte hypertrophy was
observed. Following silencing, the expressions of Sirt3, LC3 and
Beclin1 decreased; whereas, the mRNA expression of ANP and BNP
increased. This effect counteracted the Res-induced inhibition on
cardiomyocyte hypertrophy, increased the mRNA expression of PK and
decreased the mRNA expression of MCAD. Consequently, the effect of
Res on the energy metabolism in cardiomyocytes was reversed. These
results demonstrated that the siRNA-mediated silencing of Sirt3
counteracted the promoting effect of Res on autophagy and its
inhibitory effect on cardiomyocyte hypertrophy. From another
perspective, it was confirmed that the Sirt3-autophagy signaling
pathway served a role in cardiomyocyte hypertrophy.
Discussion
Hypertension is an important global health problem,
causing myocardial cell hypertrophy (29). At present, inhibiting myocardial
cell hypertrophy may block the signaling pathway of protein
synthesis or promote the signal pathway of proteolysis (30). The present study examined the
association between Sirt3 and autophagy in Ang II-induced
myocardial cell hypertrophy, and assessed whether Sirt3 affected
myocardial cell hypertrophy and energy metabolism through
autophagy.
Belonging to the NAD-dependent HDAC family, Sirt3 is
primarily distributed in the mitochondria (31). A previous study identified that
Sirt3 additionally exists in the cytoplasm and nuclei (32). The expression of Sirt3 is increased
in organs with high metabolic activity, including brain tissue, the
heart, liver and kidneys (33).
Exercise, hunger, cold and oxidative stress may activate the
expression of Sirt3 (34). Sirt3
expression was significantly decreased in people with a high fat
diet and those taking metformin, as well as in tumor cells
(35–37). Sirt3 is involved in the regulation
of approximately all cell metabolism-associated signaling pathways,
including reactive oxygen species scavenging (38), tricarboxylic acid cycle (39–43),
fatty acid oxidation (34,44,45),
ketogenesis (46,47), protein synthesis (48,49),
cell growth and apoptosis (50–54).
Sirt3 maintains the normal vital activities of the human body
through multisystem and multi-link regulation, energy metabolism
balance, anti-oxidative stress and the cell cycle (38). A number of previous studies
demonstrated that Sirt3 may resist myocardial hypertrophy and heart
failure through regulation of energy metabolism and reduction of
oxidative stress. Sundaresan et al (10) observed that Sirt3 knockout mice
demonstrated alterations of myocardial hypertrophy and myocardial
fibrosis. Pillai et al (11) identified that myocardial
hypertrophy may be inhibited by activating the Sirt3-LKB1-AMPK
pathway. Chen et al (55)
observed that Sirt3 may reduce myocardial fibrosis and improve the
myocardial contraction through the transforming growth factor
(TGF)-β/mothers against decapentaplegic homolog 3 pathway.
Furthermore, another previous study demonstrated that Sirt3 gene
knockout aggravates the lipid deposition of the heart and suggests
that abnormal energy metabolism may promote myocardial hypertrophy
(56).
With respect to myocardial hypertrophy, autophagy
may regulate the scavenging of cells, and maintain the mechanical
function of the myocardium and the quality of the ventricle
(55). Nakai et al
(15) observed that autophagy
related 5 (Atg5) gene knockout rats are more liable to ventricular
hypertrophy, ventricular dilatation and abnormal contraction. Ucar
et al (57) identified that
miRNA-212/132 knockout mice have upregulated autophagy and
significantly decreased myocardial hypertrophy. Another previous
study suggested that the heart volume caused by myocardial
hypertrophy is reduced by FoxO3 through upregulation of autophagy
(58). Laurent et al
(59) observed that exchanger 1
may activate the autophagy through calcium/calmodulin-dependent
protein kinase kinase β/AMPK; however, the downregulation of
3-methlyadenine and Atg5 (blockers of autophagy) may accelerate
myocardial hypertrophy. It was suggested that the increase of
autophagy is a reaction that balances cell hypertrophy to protect
the myocardial cells.
Accumulating evidence suggests that excessive
autophagy may accelerate myocardial cell death and aggravate heart
failure. Zhu et al (60)
observed that Beclin1 heterozygote (Beclin1+/−) mice
with downregulated autophagy demonstrated significantly improved
ventricular remodeling and heart failure. Kostin et al
(61) identified in heart failure
subjects that moribund myocardial cells demonstrated significantly
increased autophagy. Rawat et al (62) identified that increased active
oxygen and abnormal energy metabolism may upregulate autophagy and
accelerate the development of myocardial hypertrophy and diastolic
heart failure. Therefore, a number of researchers suggested that
cell death caused by autophagy may be the immediate cause of heart
failure. Therefore, in the process of myocardial hypertrophy,
moderate autophagy may protect the myocardial cells; whereas,
excessive autophagy will lead to the death of myocardial cells and
the progression of heart failure (63). Autophagy exhibits different effects
under the stimulation of different conditions and factors.
The present study examined the role of
Sirt3-autophagy in myocardial hypertrophy. In a previous study, it
was observed that in the process of Ang II-induced myocardial cell
hypertrophy, the activity of autophagy decreased and the expression
of the LC3 protein decreased significantly (16). It was additionally observed that a
similar trend of downregulation of the Sirt3 protein was present in
the identical cell model, which may be influenced by the
NAD/reduced NAD ratio (16). To
clarify the association between Sirt3 and autophagy, the present
study used Res, which is an agonist of Sirt3, to intervene. It was
identified that Res may increase the expression of Sirt3 and
autophagy proteins, and inhibit the expression of ANP and BNP
(myocardial cell hypertrophy factors), which suggested that Res may
resist myocardial hypertrophy through activation of Sirt3 to induce
autophagy. To further verify the observations, the present study
combined adenovirus transfected siRNA-Sirt3 with Res intervention.
It was observed that the expression of autophagy proteins was
significantly decreased and the expression of ANP and BNP
(myocardial cell hypertrophy factors) was significantly increased,
which suggests that Sirt3 inhibited retinal neovascularization by
regulating the migration-, neovascularization- and
autophagy-associated factors expression (64). Sirt3 exerts protective effects in
response to various damage factors involved in endothelial
dysfunction, including Ang II, TGF-β and high glucose (65). The reduced microvascular formation
and VEGF expression in cardiac tissue was accompanied by a loss of
mitochondrial Sirt3 during Ang II-induced cardiac remodeling
(65).
In the present study, the expression of MCAD and PK
was detected, and it was identified that the expression of MCAD was
significantly decreased; however, the expression of PK was
significantly increased in hypertrophic myocardial cells, which is
consistent with a previous study (2). These results suggest that the energy
utilization method of myocardial cells is altered and the
efficiency of energy utilization decreases. Res intervention may
reverse the alteration of MCAD and PK. However, the effect of Res
decreases significantly following the combination of siRNA-Sirt3
and Res intervention. This result suggests that Sirt3 may promote
fatty acid oxidation in myocardial cells, reduce the anaerobic
glycolysis of glucose, lower the toxic effect of the intermediate
product of glycolysis on myocardial cells and protect the heart
accordingly (56).
The previous studies and the present results suggest
that Sirt3 activation may improve autophagy and inhibit myocardial
hypertrophy. In addition, Sirt3 protects the cell by reversing the
abnormal energy metabolism caused by Ang II. As Sirt3 is vital in
regulating cellular energy metabolism, cell growth and apoptosis,
an increasing number of studies have identified that Sirt3 is
closely associated with cardiovascular diseases, and Sirt3 may
become a novel target for the treatment of cardiovascular diseases.
Sirt3 is highly valuable for scientific research with strong
potential for clinical application. The present study demonstrated
that Sirt3 may inhibit myocardial cell hypertrophy by regulating
autophagy; however, further investigations are required to
determine the specific molecular mechanism of Sirt3-mediated
regulation of autophagy.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
Guangdong Medical Research Fund (Guangzhou, China, grant no.
B2012249).
Availability of data and materials
The data generated in the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
HNW and JLL designed the study and performed the
experiments. TX and HQY performed the statistical analysis, GHC and
JH were involved in designing the study, drafting the manuscript
and conducted important revisions to the manuscript. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Medical Ethics
Committee of Shantou University (Shantou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vanberg P and Atar D: Androgenic anabolic
steroid abuse and the cardiovascular system. Handb Exp Pharmacol.
195:411–457. 2010. View Article : Google Scholar
|
|
2
|
Ruppert M, Korkmaz-Icöz S, Loganathan S,
Jiang W, Lehmann L, Oláh A, Sayour AA, Barta BA, Merkely B, Karck
M, et al: Pressure-volume analysis reveals characteristic
sex-related differences in cardiac function in a rat model of
aortic banding-induced myocardial hypertrophy. Am J Physiol Heart
Circ Physiol. 315:H502–H511. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sugden PH and Clerk A: Cellular mechanisms
of cardiac hypertrophy. J Mol Med (Berl). 76:725–746. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
den Besten G, van Eunen K, Groen AK,
Venema K, Reijngoud DJ and Bakker BM: The role of short-chain fatty
acids in the interplay between diet, gut microbiota, and host
energy metabolism. J Lipid Res. 54:2325–2340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chatham JC and Young ME: Metabolic
remodeling in the hypertrophic heart: Fuel for thought. Circ Res.
111:666–668. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shen W, Asai K, Uechi M, Mathier MA,
Shannon RP, Vatner SF and Ingwall JS: Progressive loss of
myocardial ATP due to a loss of total purines during the
development of heart failure in dogs: A compensatory role for the
parallel loss of creatine. Circulation. 100:2113–2118. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ingwall JS: Energy metabolism in heart
failure and remodelling. Cardiovasc Res. 81:412–419. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blander G and Guarente L: The Sir2 family
of protein deacetylases. Annu Rev Biochem. 73:417–435. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Giralt A and Villarroya F: SIRT3, a
pivotal actor in mitochondrial functions: Metabolism, cell death
and aging. Biochem J. 444:1–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sundaresan NR, Gupta M, Kim G, Rajamohan
SB, Isbatan A and Gupta MP: Sirt3 blocks the cardiac hypertrophic
response by augmenting Foxo3a-dependent antioxidant defense
mechanisms in mice. J Clin Invest. 119:2758–2771. 2009.PubMed/NCBI
|
|
11
|
Pillai VB, Sundaresan NR, Kim G, Gupta M,
Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A and Gupta
MP: Exogenous NAD blocks cardiac hypertrophic response via
activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol
Chem. 285:3133–3144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yue Z, Ma Y, You J, Li Z, Ding Y, He P, Lu
X, Jiang J, Chen S and Liu P: NMNAT3 is involved in the protective
effect of SIRT3 in Ang II-induced cardiac hypertrophy. Exp Cell
Res. 347:261–273. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shintani T and Klionky DJ: Autophagy in
health and disease: a double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dämmrich J and Pfeifer U: Cardiac
hypertrophy in rats after supravalvular aortic constriction. II.
Inhibition of cellular autophagy in hypertrophying cardiomyocytes.
Virchows Arch B Cell Pathol Incl Mol Pathol. 43:287–307. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakai A, Yamaguchi O, Takeda T, Higuchi Y,
Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et
al: The role of autophagy in cardiomyocytes in the basal state and
in response to hemodynamic stress. Nat Med. 13:619–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang HN, Liu C, Li YH, Chen Z, Sun G, Xue
RC and Dong YG: Role of autophagy in myocardial hypertrophy induced
by angiotensin II. J Zhongshan Univ. 33:440–443. 2014.
|
|
17
|
Hindle AG, Grabek KR, Epperson LE,
Karimpour-Fard A and Martin SL: Metabolic changes associated with
the long winter fast dominate the liver proteome in 13-lined ground
squirrels. Physiol Genomics. 46:348–361. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pi H, Xu S, Reiter RJ, Guo P, Zhang L, Li
Y, Li M, Cao Z, Tian L, Xie J, et al: SIRT3-SOD2-mROS-dependent
autophagy in cadmium-induced hepatotoxicity and salvage by
melatonin. Autophagy. 11:1037–1051. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tong W, Ju L, Qiu M, Xie Q, Cheng Y, Shen
W, Sun W, Wang W and Tian J: Liraglutide ameliorate non-alcohol
fatty liver disease by enhancing mitochondrial architecture and
promoting autophagy through SIRT1/SIRT3-FOXO3a pathway. Hepatol
Res. 46:933–943. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Auburger G, Gispert S and Jendrach M:
Mitochondrial acetylation and genetic models of Parkinson's
disease. Prog Mol Biol Transl Sci. 127:155–182. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marques-Aleixo I, Santos-Alves E, Balça
MM, Moreira PI, Oliveira PJ, Magalhães J and Ascensão A: Physical
exercise mitigates doxorubicin-induced brain cortex and cerebellum
mitochondrial alterations and cellular quality control signaling.
Mitochondrion. 26:43–57. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fang Y, Wang J, Xu L, Cao Y, Xu F, Yan L,
Nie M, Yuan N, Zhang S, Zhao R, et al: Autophagy maintains
ubiquitination-proteasomal degradation of Sirt3 to limit oxidative
stress in K562 leukemia cells. Oncotarget. 7:35692–35702. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qiao A, Wang K, Yuan Y, Guan Y, Ren X, Li
L, Chen X, Li F, Chen AF, Zhou J, et al: Sirt3-mediated mitophagy
protects tumor cells against apoptosis under hypoxia. Oncotarget.
7:43390–43400. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shi L, Zhang T, Zhou Y, Zeng X, Ran L,
Zhang Q, Zhu J and Mi M: Dihydromyricetin improves skeletal muscle
insulin sensitivity by inducing autophagy via the AMPK-PGC-1α-Sirt3
signaling pathway. Endocrine. 50:378–389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mukherjee S, Ray D, Lekli I, Bak I, Tosaki
A and Das DK: Effects of Longevinex (modified resveratrol) on
cardioprotection and its mechanisms of action. Can J Physiol
Pharmacol. 88:1017–1025. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen GQ, Liu C, Zhang CX, Meng RS, Chen
BL, Xiong ZJ and Dong YG: The role of HO-1 in inhibiting cardiac
hypertrophy by AMPK. Journal of Sun Yat-sen University. 31:614–618.
2010.
|
|
27
|
Yin R, Dong YG, Li HL and Liu D: Activated
AMPK attenuates cardiac hypertrophy in rats through increasing
myocardial fatty acid oxidation. Chin J Pathophysiol. 23:1258–1262.
2007.
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔC(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Watson CJ, Horgan S, Neary R, Glezeva N,
Tea I, Corrigan N, McDonald K, Ledwidge M and Baugh J: Epigenetic
therapy for the treatment of hypertension-induced cardiac
hypertrophy and fibrosis. J Cardiovasc Pharmacol Ther. 21:127–137.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lu L, Wu D, Li L and Chen L: Apelin/APJ
system: A bifunctional target for cardiac hypertrophy. Int J
Cardiol. 230:164–170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cooper HM and Spelbrink JN: The human
SIRT3 protein deacetylase is exclusively mitochondrial. Biochem J.
411:279–285. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Scher MB, Vaquero A and Reinberg D: SirT3
is a nuclear NAD+-dependent histone deacetylase that
translocates to the mitochondria upon cellular stress. Genes Dev.
21:920–928. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jin L, Galonek H, Israelian K, Choy W,
Morrison M, Xia Y, Wang X, Xu Y, Yang Y, Smith JJ, et al:
Biochemical characterization, localization, and tissue distribution
of the longer form of mouse SIRT3. Protein Sci. 18:514–525. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shi T, Wang F, Stieren E and Tong Q:
SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial
function and thermogenesis in brown adipocytes. J Biol Chem.
280:13560–13567. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li H, Feng Z, Wu W, Li J, Zhang J and Xia
T: SIRT3 regulates cell proliferation and apoptosis related to
energy metabolism in non-small cell lung cancer cells through
deacetylation of NMNAT2. Int J Oncol. 43:1420–1430. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bell EL, Emerling BM, Ricoult SJ and
Guarente L: SirT3 suppresses hypoxia inducible factor 1α and tumor
growth by inhibiting mitochondrial ROS production. Oncogene.
30:2986–2996. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jing E, Emanuelli B, Hirschey MD, Boucher
J, Lee KY, Lombard D, Verdin EM and Kahn CR: Sirtuin-3 (Sirt3)
regulates skeletal muscle metabolism and insulin signaling via
altered mitochondrial oxidation and reactive oxygen species
production. Proc Natl Acad Sci USA. 108:14608–14613. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jacobs KM, Pennington JD, Bisht KS,
Aykin-Burns N, Kim HS, Mishra M, Sun L, Nguyen P, Ahn BH, Leclerc
J, et al: SIRT3 interacts with the daf-16 homolog FOXO3a in the
mitochondria, as well as increases FOXO3a dependent gene
expression. Int J Biol Sci. 4:291–299. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fernandez-Marcos PJ, Jeninga EH, Canto C,
Harach T, de Boer VC, Andreux P, Moullan N, Pirinen E, Yamamoto H,
Houten SM, et al: Muscle or liver-specific Sirt3 deficiency induces
hyperacetylation of mitochondrial proteins without affecting global
metabolic homeostasis. Sci Rep. 2:4252012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Finley LW, Haas W, Desquiret-Dumas V,
Wallace DC, Procaccio V, Gygi SP and Haigis MC: Succinate
dehydrogenase is a direct target of sirtuin 3 deacetylase activity.
PLoS One. 6:e232952011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ahn BH, Kim HS, Song S, Lee IH, Liu J,
Vassilopoulos A, Deng CX and Finkel T: A role for the mitochondrial
deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad
Sci USA. 105:14447–14452. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen Y, Zhang J, Lin Y, Lei Q, Guan KL,
Zhao S and Xiong Y: Tumour suppressor SIRT3 deacetylates and
activates manganese superoxide dismutase to scavenge ROS. EMBO Rep.
12:534–541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Finley LW, Carracedo A, Lee J, Souza A,
Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish
CB, et al: SIRT3 opposes reprogramming of cancer cell metabolism
through HIF1α destabilization. Cancer Cell. 19:416–428. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hallows WC, Yu W, Smith BC, Devries MK,
Ellinger JJ, Someya S, Shortreed MR, Prolla T, Markley JL, Smith
LM, et al: Sirt3 promotes the urea cycle and fatty acid oxidation
during dietary restriction. Mol Cell. 41:139–149. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hirschey MD, Shimazu T, Goetzman E, Jing
E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S,
Ilkayeva OR, et al: SIRT3 regulates mitochondrial fatty-acid
oxidation by reversible enzyme deacetylation. Nature. 464:121–125.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hirschey MD, Shimazu T, Capra JA, Pollard
KS and Verdin E: SIRT1 and SIRT3 deacetylate homologous substrates:
AceCS1,2 and HMGCS1,2. Aging (Albany NY). 3:635–642. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shimazu T, Hirschey MD, Hua L,
Dittenhafer-Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt
FW, Denu JM, et al: SIRT3 deacetylates mitochondrial
3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body
production. Cell Metab. 12:654–661. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kendrick AA, Choudhury M, Rahman SM,
McCurdy CE, Friederich M, Van Hove JL, Watson PA, Birdsey N, Bao J,
Gius D, et al: Fatty liver is associated with reduced SIRT3
activity and mitochondrial protein hyperacetylation. Biochem J.
433:505–514. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yang Y, Cimen H, Han MJ, Shi T, Deng JH,
Koc H, Palacios OM, Montier L, Bai Y, Tong Q, et al:
NAD+-dependent deacetylase SIRT3 regulates mitochondrial
protein synthesis by deacetylation of the ribosomal protein MRPL10.
J Biol Chem. 285:7417–7429. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hafner AV, Dai J, Gomes AP, Xiao CY,
Palmeira CM, Rosenzweig A and Sinclair DA: Regulation of the mPTP
by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses
age-related cardiac hypertrophy. Aging (Albany NY). 2:914–923.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Iwahara T, Bonasio R, Narendra V and
Reinberg D: SIRT3 functions in the nucleus in the control of
stress-related gene expression. Mol Cell Biol. 32:5022–5034. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Allison SJ and Milner J: SIRT3 is
pro-apoptotic and participates in distinct basal apoptotic
pathways. Cell Cycle. 6:2669–2677. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cooper HM, Huang JY, Verdin E and
Spelbrink JN: A new splice variant of the mouse SIRT3 gene encodes
the mitochondrial precursor protein. PLoS One. 4:e49862009.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shulga N and Pastorino JG: Ethanol
sensitizes mitochondria to the permeability transition by
inhibiting deacetylation of cyclophilin-D mediated by sirtuin-3. J
Cell Sci. 123:4117–4127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen T, Li J, Liu J, Li N, Wang S, Liu H,
Zeng M, Zhang Y and Bu P: Activation of SIRT3 by resveratrol
ameliorates cardiac fibrosis and improves cardiac function via the
TGF-β/Smad3 pathway. Am J Physiol Heart Circ Physiol.
308:H424–H434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen T, Liu J, Li N, Wang S, Liu H, Li J,
Zhang Y and Bu P: Mouse SIRT3 attenuates hypertrophy-related lipid
accumulation in the heart through the deacetylation of LCAD. PLoS
One. 10:e01189092015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ucar A, Gupta SK, Fiedler J, Erikci E,
Kardasinski M, Batkai S, Dangwal S, Kumarswamy R, Bang C, Holzmann
A, et al: The miRNA-212/132 family regulates both cardiac
hypertrophy and cardiomyocyte autophagy. Nat Commun. 3:10782012.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang X and Su H: FoxO3 hastens autophagy
and shrinks the heart but does not curtail pathological hypertrophy
in adult mice. Cardiovasc Res. 91:561–562. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Laurent AC, Bisserier M, Lucas A, Tortosa
F, Roumieux M, De Régibus A, Swiader A, Sainte-Marie Y, Heymes C,
Vindis C, et al: Exchange protein directly activated by cAMP 1
promotes autophagy during cardiomyocyte hypertrophy. Cardiovasc
Res. 105:55–64. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhu H, Tannous P, Johnstone JL, Kong Y,
Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA and Hill
JA: Cardiac autophagy is a maladaptive response to hemodynamic
stress. J Clin Invest. 117:1782–1793. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kostin S, Pool L, Elsässer A, Hein S,
Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klövekorn
WP, et al: Myocytes die by multiple mechanisms in failing human
hearts. Circ Res. 92:715–724. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Rawat DK, Alzoubi A, Gupte R, Chettimada
S, Watanabe M, Kahn AG, Okada T, McMurtry IF and Gupte SA:
Increased reactive oxygen species, metabolic maladaptation, and
autophagy contribute to pulmonary arterial hypertension-induced
ventricular hypertrophy and diastolic heart failure. Hypertension.
64:1266–1274. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
De Meyer GR, De Keulenaer GW and Martinet
W: Role of autophagy in heart failure associated with aging. Heart
Fail Rev. 15:423–430. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Mao XB, You ZP, Wu C and Huang J:
Potential suppression of the high glucose and insulin-induced
retinal neovascularization by Sirtuin 3 in the human retinal
endothelial cells. Biochem Biophys Res Commun. 482:341–345. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wei T, Huang G, Gao J, Huang C, Sun M, Wu
J, Bu J and Shen W: Sirtuin 3 deficiency accelerates hypertensive
cardiac remodeling by impairing angiogenesis. J Am Heart Assoc.
6:e0061142017. View Article : Google Scholar : PubMed/NCBI
|