Introduction
Age-related cataracts (ARCs) are characterized by an
increase in the opacity of the lens with age; however, there are
currently no effective drugs that can inhibit or reverse the
progression of ARCs (1). The exact
mechanism of ARC development is not fully understood, but it has
been confirmed that oxidative stress serves a key role in its
pathogenesis (2). UV rays,
nutrient starvation, H2O2 in aqueous humor,
amongst other factors can damage the lens by oxidative stress,
particularly via oxidative damage to human lens epithelial cells
(HLECs) (3). HLECs are a thin
layer of cells in the anterior lens capsule that are responsible
for the defense of the lens against oxidative stress and are
therefore vulnerable to oxygen free radicals (3). Under oxidative stress, reactive
oxygen species (ROS), a byproduct of metabolism, accumulate in
HLECs and cause oxidative damage to nucleic acids, lipids and
organelles, eventually leading to apoptosis (4–6).
Furthermore, apoptosis of HLECs is considered to be an early event
in the development of cataracts (4,5,7–9). In
response to oxidative stress, particularly the increase in ROS
production in HLECs, lens proteins are denatured, modified and
aggregated, which eventually leads to cataract formation (10). In addition, oxidative stress has
been shown to induce lens opacification in experimental animal
models and in cultured rat lenses (11).
Humanin (HN) was the first identified
mitochondrial-derived peptide (MDP), which comprises 21–24 amino
acids expressed by the open reading frame of mitochondrial 16S rRNA
(12). HN is expressed in various
tissues, including neuronal cells, skeletal muscle cells (13), retinal pigment epithelial cells
(14) and blood-derived cells
(15). Previous studies have
reported that HN may serve a role in metabolic regulation and
antioxidant injury in nerve cells, cardiomyocytes, epidermal stem
cells and other cell types, as well as in pathological models, such
as Alzheimer's disease, type 2 diabetes mellitus (T2DM) and
age-related macular degeneration (AMD). These effects include
H2O2-induced cell death (16), β-amyloid toxicity (17), serum starvation-induced neuronal
cell death (18) and protective
effects of oxidized low-density lipoproteins-induced vascular
endothelial cell death (19).
Furthermore, knockdown of endogenous HN increased the sensitivity
of cells to apoptosis induced by oxidative stress (12). However, to the best of our
knowledge, there have been no previous studies examining the
expression and function of HN in HLECs, and the role of HN in the
prevention or treatment of ARCs.
Considering the important role of oxidative stress
and ROS in the pathogenesis of cataracts, the present study
investigated the role of HN in HLECs under oxidative stress and its
underlying mechanisms. Moreover, the present study examined the
relationship between HN and the formation of ARCs, by administering
exogenous HN or knocking down endogenous HN to assess whether HN
can reduce oxidative damage and apoptosis.
Materials and methods
Cell culture and treatment
HLECs (HLE-B3; American Type Culture Collection)
were grown and maintained in 1:1 DMEM and Ham's F-12 medium
(DMEM/F-12; cat. no. 10-092-CV; Corning, Inc.), which was
supplemented with 10% FBS (Biological Industries) and 100 units
penicillin and streptomycin (Gibco; Thermo Fisher Scientific, Inc.)
at 37°C with 5% CO2. HN peptides were chemically
synthesized and purified to >95% purity (Nanjing Taiye Chemical
Industry Co., Ltd.). The HN group was incubated with DMEM
containing 50 µM HN at 37°C for 2 h before receiving type B UV
(UVB) radiation. In the small interfering RNA (siRNA) HN group, HN
siRNA was transfected into cells using Lipofectamine®
3000 (Invitrogen; Thermo Fisher Scientific, Inc.) for 48 h, after
which, cells received UVB radiation. For the rescue experiment, the
siHN group was incubated with DMEM containing 50 µM HN at 37°C for
2 h prior to UVB radiation. For serum starvation, after
pretreatment with 50 µM HN for 2 h or transfection with siHN for 48
h, cells were incubated in serum-free medium after being rinsed
twice with PBS, for 72 h before experiments. Cells incubated in
DMEM containing 10% FBS were considered the nutrients group.
For UVB irradiation, cells were treated as
aforementioned, rinsed with PBS and treated with a given dose of
UVB (0, 10, 20, 30 and 50 mJ/cm2; 37°C; air). Cells were
exposed to UVB light (from the bottom) in PBS (37°C; air) for 2.5
min using a Spectroline ‘medium wave’ UV lamp (Spectroline) at a
distance of 0.8 cm. Control cells were treated similarly, but were
not exposed to UVB light. UVB levels reaching the cells were
determined with a radiometer (UVX Digital; UVP, Inc.) equipped with
a UVB sensor at 312 nm (UVX-31). Following UVB exposure, cells were
rinsed twice with PBS and cultured for 24 h in DMEM containing 10%
FBS. The morphological analysis of HLECs was conducted using a
fluorescence microscope (Leica Microsystems GmbH; magnification,
×400).
RNA interference
The siRNAs were designed and synthesized by
Guangzhou Ribobio Co., Ltd. The sequence of HN-specific siRNA was:
5′-GGGUUCAGCUGUCUCUUAC-3′. A nonsilencing siRNA (cat. no.
SI03650318; Qiagen, Inc.) was used as a negative control. At 50%
confluence, cells were transfected with siRNA (5 nmol/l) using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.). A total of 48 h post-transfection, subsequent
experiments were performed. Control transfection was also performed
using Lipofectamine® 3000 without nucleic acids (Mock).
The knockdown of HN was confirmed by reverse
transcription-quantitative PCR (RT-qPCR) and western blotting.
RT-qPCR
Total RNA was extracted from cells using
TRIzol® reagent (Thermo Fisher Scientific, Inc.). Total
RNA concentration was estimated by spectrophotometry. Total RNA was
reverse transcribed into cDNA using the PrimeScript RT reagent kit
(Takara Biotechnology Co., Ltd.). qPCR was performed using Power
SYBR Green PCR Master Mix (cat. no. 4367659; Thermo Fisher
Scientific, Inc.) and the StepOnePlus RT PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) to investigate the
expression levels of apoptosis, autophagy, endoplasmic reticulum
(ER) stress and antioxidant-associated genes. Each sample was run
in triplicate. The following thermocycling conditions were used for
qPCR: Initial denaturation for 1 min at 94°C, followed by 30 cycles
of 30 sec at 94°C, 30 sec at 60°C, 2 min at 72°C; and a final
extension step at 72°C for 5 min. RT-qPCR data were analyzed using
the 2−ΔΔCq method (20). ΔCq was the difference between the
quantification cycle (Cq) of the target gene and Cq of the
housekeeper gene (reference gene). ΔΔCq was calculated by
subtracting ΔCq of each experimental group from ΔCq of the control
group. Fold change was calculated using the following formula:
Fold-change=2−ΔΔCq.
The primers used for RT-qPCR were as follows: Human
NADH dehydrogenase 1 (ND1) forward, 5′-ATACCCATGGCCAACCTCCT-3′ and
reverse, 5′-GGGCCTTTGCGTAGTTGTAT-3′; human β-globin forward,
5′-GTGCACCTGACTCCTGAGGAGA-3′ and reverse,
5′-CCTTGATACCAACCTGCCCAG-3′; β-actin, forward,
5′-GAGAGGGAAATCGTGCGTGAC-3′ and reverse,
5′-CTGCTGGAAGGTGGACAGTGAG-3′; HN, forward,
5′-CTCCACGAGGGTTCAGCTGT-3′ and reverse, 5′-TTATGTCCGCCTCTTCACGG-3′;
and human GAPDH, forward, 5′-GGTGAAGGTCGGAGTCAACG-3′ and reverse,
5′-CAAAGTTGTCATGGATGHACC-3′.
Cell viability assay
Cytotoxicity was assessed using a Cell Counting
Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc.).
Briefly, 24 h before UVB treatment, 1×104 cells were
seeded in each well of a 96-well plate. Cells were rinsed twice
with PBS and treated with a given dose of UVB (0, 10, 20, 30 and 50
mJ/cm2; 37°C; air). Following incubation for 24 h, the
medium was removed, and the cells were washed with PBS. Each well
was then refilled with 90 µl DMEM/F12 supplemented with 10% FBS and
10 µl CCK-8 reagents, which was incubated at 37°C for 3 h. Cell
viability was evaluated at an optical density of 450 nm using a
96-well microplate reader (Bio-Rad Laboratories, Inc.).
Lactate dehydrogenase (LDH)
cytotoxicity assay
The percentage of living cells was determined using
a LDH Cytotoxicity assay kit (Beyotime Institute of Biotechnology).
Briefly, 1×104 cells were seeded in each well of a
96-well plate and incubated for 24 h. Cells were rinsed twice with
PBS and treated with a given dose of UVB (0 or 30
mJ/cm2; 37°C; air). After incubation for 24 h, the
culture media was collected and centrifuged for 5 min at 2,442 × g
at room temperature. The supernatant was dispensed into a 96-well
plate and LDH assay reagent was added to each well. The plate was
incubated at room temperature for 30 min. Absorbance values were
measured at 450 nm using a microplate reader. The living cells (%)
was calculated as follows: Cytotoxicity (%) = (absorbance of test
sample-absorbance of low control)/(absorbance of maximum enzyme
activity-absorbance of low control) ×100.
Measurement of intracellular ROS
ROS were measured using 2′,7′-dichlorofluorescein
diacetate (DCFH-DA, Beyotime Institute of Biotechnology). Following
incubation for 24 h after UVB exposure, HLECs were washed three
times with PBS. DCFH-DA, diluted to a final concentration of 10 µM,
was added to HLECs and incubated for 30 min at 37°C in the dark.
After the cells were washed three times with serum-free medium, the
fluorescence intensity was detected with a multi-detection
microplate reader with excitation at 488 nm and emission at 530 nm
within 15 min. Fluorescence signals were captured using a
fluorescence microscope (Leica Microsystems GmbH). Intracellular
levels of ROS were calculated by the average fluorescence intensity
as analyzed by Image-Pro Plus software (version 6.0; National
Institutes of Health). The measured fluorescence values were
expressed as a percentage of the fluorescence in control cells.
Mitochondrial membrane potential (ΔΨm)
measurement
JC-1 was used to measure the ΔΨm of HLECs. Briefly,
5×105 cells were collected into 2 ml tubes and incubated
with 10 µg/ml JC-1 for 20 min at 37°C. The fluorescence intensity
was detected with a flow cytometer (BD FACSAria I cell sorter;
Becton, Dickinson and Company). The wavelengths of excitation and
emission were 514 and 529 nm, for detection of the monomeric form
of JC-1. In addition, 585 and 590 nm were used to detect
aggregation of JC-1. The ratio of aggregated JC-1 and monomeric
JC-1 represented the ΔΨm of HLECs. The data were analyzed using
Quantity One software (version 4.6.6; National Institutes of
Health). Images were captured by fluorescence microscopy (Leica
Microsystems GmbH; magnification, ×400).
Transmission electron microscopy
(TEM)
Cells were fixed for 2 h at 4°C in glutaraldehyde
and paraformaldehyde (3 and 4%, respectively) in 0.1 M cacodylate
buffer (pH 7.2). After washing in 0.1 M cacodylate buffer, cells
were post-fixed with 1% osmium tetroxide in 0.1 M cacodylate buffer
(pH 7.2) for 1 h at room temperature. Cells were dehydrated using a
graded acetone series and embedded in EPON-82 resin (Sigma-Aldrich;
Merck KGaA) for 72 h at 60°C. Ultra-thin sections (80 nm) were
stained with uranyl acetate (5%; 10 min) and lead citrate (2%; 30
min) at room temperature and observed using a JEM-1200EX TEM
(magnification, ×25,000; JEOL Ltd.) at 120 V. Data were analyzed
using Image-Pro Plus software (version 6.0; National Institutes of
Health).
Cell apoptosis assay
Cells of each experimental group (5×106
cells/ml) were cultured in 35 mm2 dishes and incubated
for 24 h. Cells were rinsed twice with PBS and treated with a given
dose of UVB (0 or 30 mJ/cm2; 37°C; air). After 24 h
incubation, cells were collected by trypsinization and
centrifugation at 2442 × g for 5 min. The cell pellet was washed
twice with cold PBS and resuspended in 1X Annexin-binding buffer
(BD Biosciences). Subsequently, the cell suspension (100 µl) was
incubated with 5 µl Annexin V and 1 µl 100 µg/ml PI working
solution (BD Biosciences) at room temperature for 15 min in the
dark. After the incubation, 400 µl 1X Annexin-binding buffer was
added. After mixing, the fluorescence intensity was detected with a
flow cytometer (CyAn ADP; Beckman Coulter, Inc.) at 530 and 575 nm
emission, and 488 nm excitation. BD FACSdiva Software (version
8.0.1; BD Biosciences) was used to analyze the data. The percentage
of cells stained by AnnexinV+/PI−, which
indicates early apoptosis, was presented as a bar chart.
Determination of mitochondrial DNA
(mtDNA) copy number
The mtDNA copy number was defined as the total mtDNA
copies divided by the total nuclear DNA (nDNA) copies. qPCR was
performed to measure the mtDNA and nDNA copies of each experimental
group. Total DNA of the cells in each experimental group was
extracted using the DNA/RNA Isolation kit (Qiagen GmbH) according
to the manufacturer's instructions. For each qPCR, 1 µl sample DNA
(10 ng/µl) was amplified in a 10-µl reaction mixture that contained
0.25 µl each primer (20 µM; human ND1 for mtDNA; human β-globin for
nDNA; β-actin reference gene), 5 µl SensiFAST SYBR Hi-ROX premix
(Thermo Fisher Scientific, Inc.) and 3.5 µl PCR-grade water. In
each experiment, 1 µl HLECs DNA (1 ng/µl) and PCR-grade water were
used as the positive and negative controls, respectively. The
following thermocycling conditions were used for qPCR: Initial
denaturation for 30 sec at 95°C; followed by 45 cycles of
denaturation for 15 sec at 95°C, annealing for 20 sec at 60°C,
extension for 20 sec at 72°C. After obtaining Cq values, the mtDNA
copies and nDNA copies of the sample DNA (10 ng) relative to those
of HLECs were determined according to the aforementioned
equations.
Western blotting
After treatment, total protein was extracted from
HLECs using RIPA lysis buffer (cat. no. r0020; Beijing Solarbio
Science & Technology Co, Ltd.) and a bicinchoninic acid protein
assay kit (cat. no. B9643-1L; Sigma-Aldrich; Merck KGaA) was used
to determine the total protein concentration. Equal concentrations
of total protein samples (8 µl/lane) were loaded into the wells of
4–12% Bolt mini gels (Thermo Fisher Scientific, Inc.) followed by
SDS-PAGE. The proteins were then transferred onto PVDF membranes.
Following transfer, membranes were blocked with 5% fat-free milk in
TBS for 30 min at room temperature and then incubated with primary
antibodies against HN (cat. no. LS-C109400; 1:2,000; LifeSpan
BioSciences), Bcl-2 (cat. no. 4223; 1:2,000; Cell Signaling
Technology, Inc), Bax (cat. no. 5023; 1:2,000; Cell Signaling
Technology, Inc.), cleaved caspase 3 (cat. no. 9665; 1:2,000; Cell
Signaling Technology, Inc.) and β-actin (cat. no. sc-47778;
1:5,000; Santa Cruz Biotechnology, Inc.) overnight at 4°C.
Subsequently, goat anti-mouse IgG H&L (cat. no. ab205719;
1:5,000; Abcam) and goat anti-rabbit IgG H&L (cat. no.
ab205718; 1:5,000; Abcam) horseradish peroxidase-conjugated
secondary antibodies or 2 h at room temperature. Protein bands were
visualized using the SuperSignal™ Western Blot Substrate bundle
(Pierce; Thermo Fisher Scientific, Inc.). Data were analyzed using
Image-Pro Plus 6.0 software (version 6.0; National Institutes of
Health).
Statistical analysis
Statistical analysis was performed using one-way
ANOVA followed by Tukey post hoc test using GraphPad InStat
(version 3.05; GraphPad Software, Inc.). Data are presented as the
mean ± SD. P<0.05 was considered to indicate a statistically
significant difference. Each experiment was repeated at least three
times.
Results
HN expression in HLECs is associated
with oxidative stress
It was revealed that 24 h after UVB treatment, the
morphology of HLECs changed (Fig.
1A); UVB induced cell loss, disordered intracellular structure
and enhanced the appearance of apoptotic bodies. Moreover, the
higher the UVB dose used to treat HLECs, the more significant the
change in cell morphology, indicating that the cells were under
high oxidative stress. To further investigate the relationship
between endogenous HN expression and oxidative stress, RT-qPCR was
used to detect the mRNA expression levels of HN in HLECs treated
with various doses of UVB (0, 10, 30 and 50 mJ/cm2;
Fig. 1B). It was demonstrated that
the expression levels of HN were increased 12 h after UVB
irradiation and HN expression was positively associated with UVB
exposure. Moreover, the mRNA expression levels of HN were
upregulated by 62% (P<0.05) at the dose of 10 mJ/cm2,
138% (P<0.01) at 30 mJ/cm2 and 219% (P<0.01) at 50
mJ/cm2 compared with the control group. Therefore, the
present results suggested that UVB irradiation may induce
upregulation of HN in HLECs, and that HN may be involved in the
response of cells to oxidative stress.
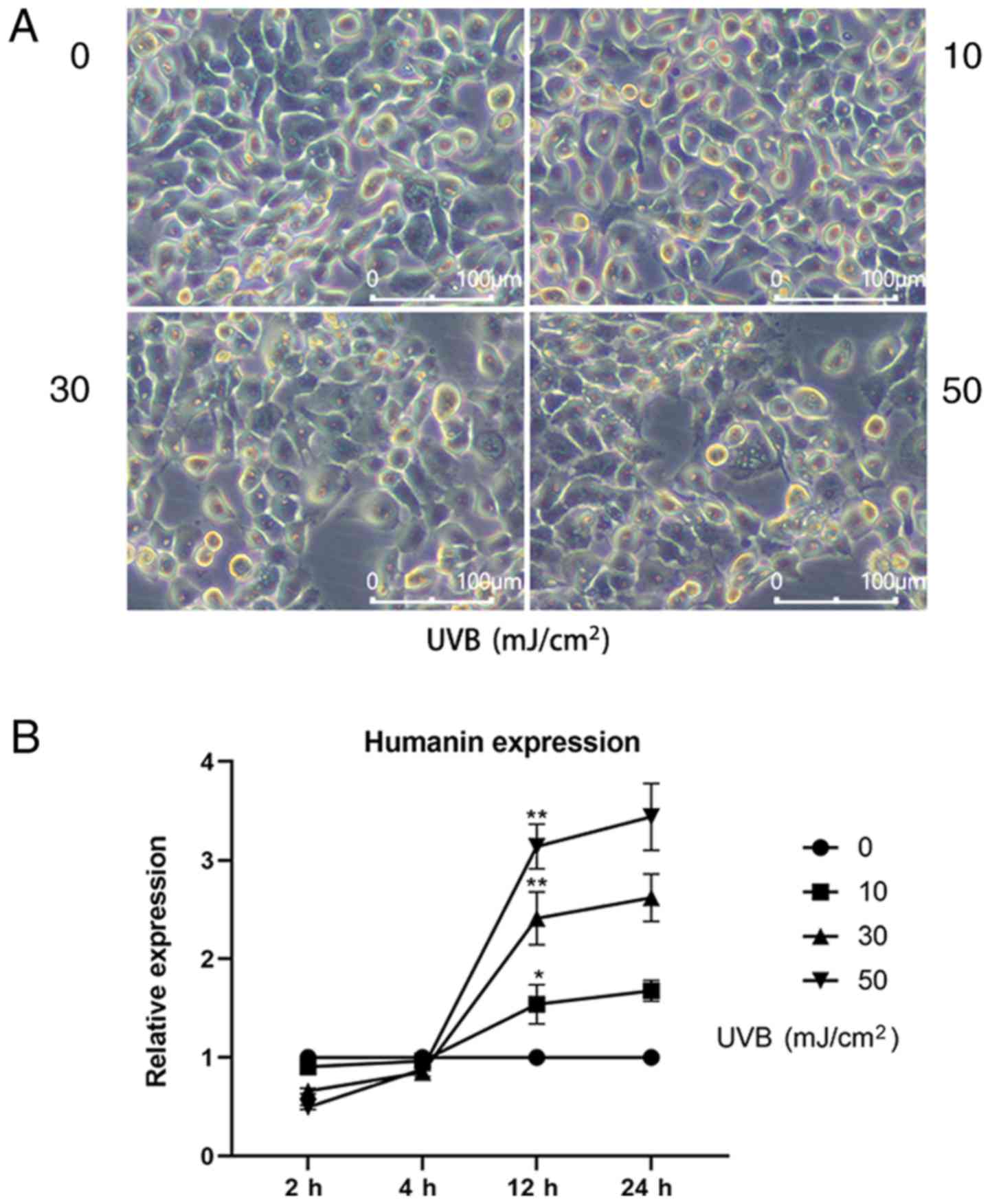 | Figure 1.Expression of HN in HLECs at
different levels of oxidative stress. (A) Images of HLECs 24 h
after exposure to a 0, 10, 30 or 50 mJ/cm2 dose of UVB.
(B) HLECs were subjected to different doses of UVB (0, 10, 30 or 50
mJ/cm2), and the mRNA expression levels of HN were
detected by reverse transcription-quantitative PCR at 2, 4, 12 and
24 h. Data are presented as the mean ± SD, n=3. *P<0.05,
**P<0.01 vs. 12 h 0 mJ/cm2 UVB. HN, humanin; HLECs,
human lens epithelial cells; UVB, type B UV. |
HN protects HLECs from oxidative
damage
To investigate whether HN has a protective effect on
HLECs under oxidative stress, a HN knockdown cell line was
established by gene silencing. qPCR and western blot analysis of HN
were performed and it was confirmed by qPCR analysis that the
expression of HN was downregulated by 83% compared with the
notransfection group (Fig. 2A and
B). In addition, a CCK-8 assay was used to detect the viability
of HLECs (Fig. 2C). The results
revealed that knockdown of HN significantly decreased the viability
of HLECs (P<0.05). In addition, the present study treated the HN
knockdown group with different levels of UVB; the results
demonstrated that cell viability was reduced by ~50% in the control
group treated with UVB irradiation (30 mJ/cm2) and by
77% (P<0.01) in the HN knockdown group compared with the control
group (0 UVB; Fig. 2C). Therefore,
30 mJ/cm2 was used as the UVB irradiation dose in
follow-up experiments. The viability of HLECs pretreated with
various doses of exogenous HN 24 h after UVB irradiation was then
measured (Fig. 2D). It was
demonstrated that the cellular protective effect of HN was
dose-dependent, but there was no significant increase in response
to 100 µM HN compared with 50 µM HN; therefore, the present study
used 50 µM HN (P<0.01) as the concentration of exogenous HN in
the follow-up experiments. Furthermore, 30 mJ/cm2 UVB
exerted significant cytotoxicity on HLECs (P<0.01). However,
pretreatment with HN reduced UVB cytotoxicity (P<0.05) and
reduced LDH activity by 20% (Fig.
2E), whereas knockdown of HN significantly increased the
sensitivity of HLECs to UVB cytotoxicity (P<0.01). Collectively,
the present results indicated that HN may effectively protect HLECs
from oxidative damage induced by UVB.
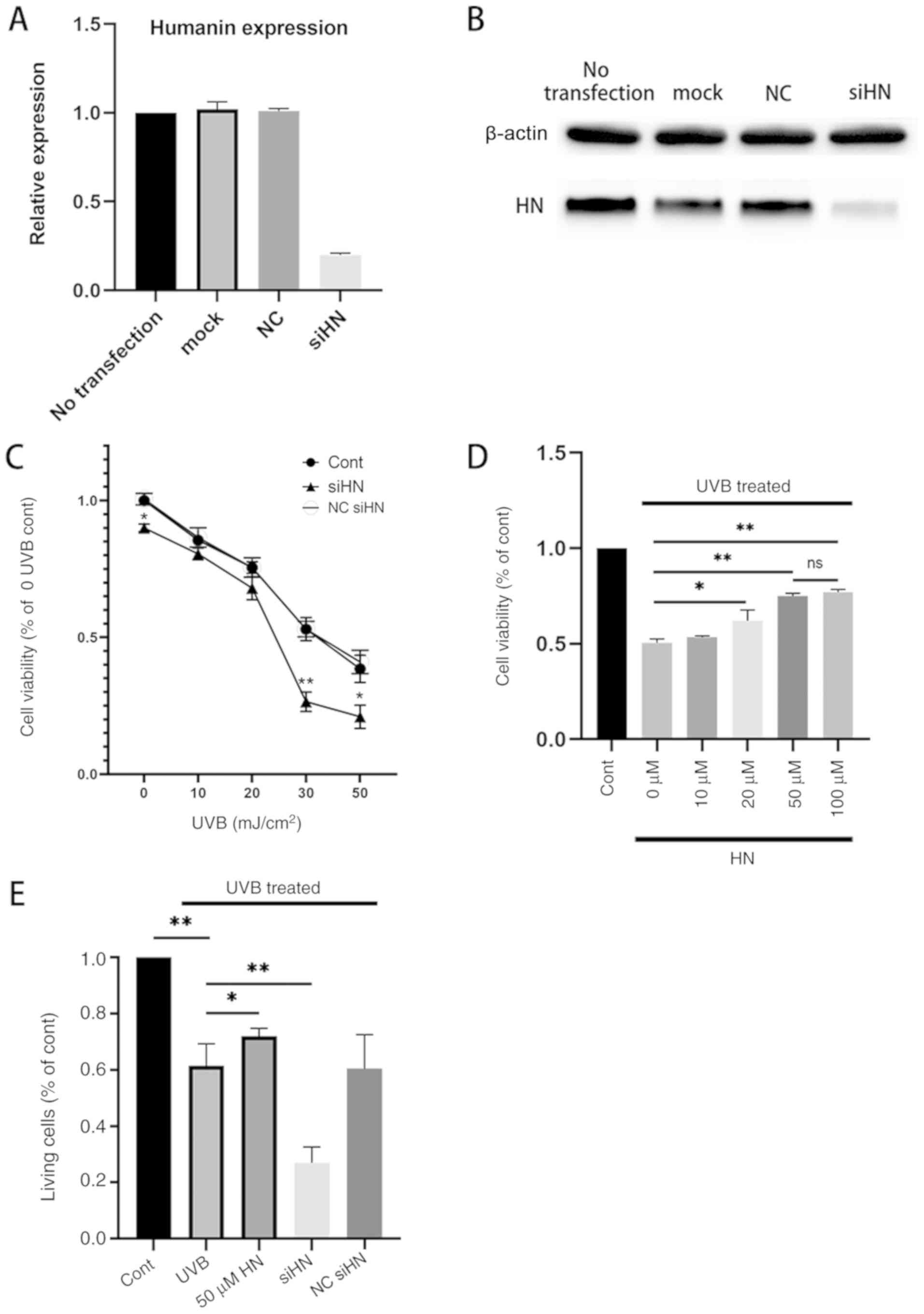 | Figure 2.Effects of HN on the viability and
LDH cytotoxicity of HLECs subjected to UVB. (A and B) Cells were
transfected with siHN for 48 h. NC siHN group cells were
transfected with NC siRNA for 48 h. Mock transfection was performed
using Lipofectamine® 3000 without nucleic acids. (A)
mRNA expression levels of HN were detected by reverse
transcription-quantitative PCR. (B) Protein expression levels of HN
were examined using western blotting. (C and D) Cell Counting Kit-8
assay. (C) After transfection with siHN and NC siRNA for 48 h, the
cells were washed twice with PBS, and HLECs were subjected to
different doses (0, 10, 20, 30 and 50 mJ/cm2) of UVB.
(D) After incubation with a given concentration (0, 10, 20, 50 or
100 µM) of HN for 2 h, the cells were washed twice with PBS, and
HLECs were treated with UVB at a dose of 30 mJ/cm2. (E)
After pretreatment with 50 µM HN for 2 h or transfection with siHN
or NC siRNA for 48 h, the cells were washed twice with PBS, and
then HLECs were subjected to UVB radiation at a dose of 30
mJ/cm2. After 24 h, LDH release was determined using a
commercial kit. Data are presented as the mean ± SD, n=3.
*P<0.05 and **P<0.01 vs. Cont. HN, humanin; LDH, lactate
dehydrogenase; HLECs, human lens epithelial cells; UVB, type B UV;
siHN, HN siRNA; siRNA, small interfering RNA; NC, negative control;
Cont, control. |
HN inhibits ROS production in
HLECs
Under oxidative stress, the level of ROS in cells
increases significantly (4). ROS
derived from damaged mitochondria are important physiological
signaling molecules that regulate gene expression, apoptosis and
cell proliferation (5,6). Excessive ROS can further impair
mitochondrial function and affect cell viability (21,22).
Therefore, the present study hypothesized that HN may reduce the
production of ROS in HLECs and act as an antioxidant. The present
study detected intracellular ROS using the fluorescent probe
DCFH-DA, and it was found that the level of ROS in HLECs increased
under oxidative stress (Fig. 3).
In addition, the level of ROS in the HN siRNA group was
significantly higher compared with the control group, regardless of
whether there was UVB treatment. However, HLECs pretreated with 50
µM HN under oxidative stress showed lower ROS levels compared with
the UVB-treated control group (P<0.05). Thus, the present
results suggested that HN can significantly reduce ROS production
in HLECs under oxidative stress.
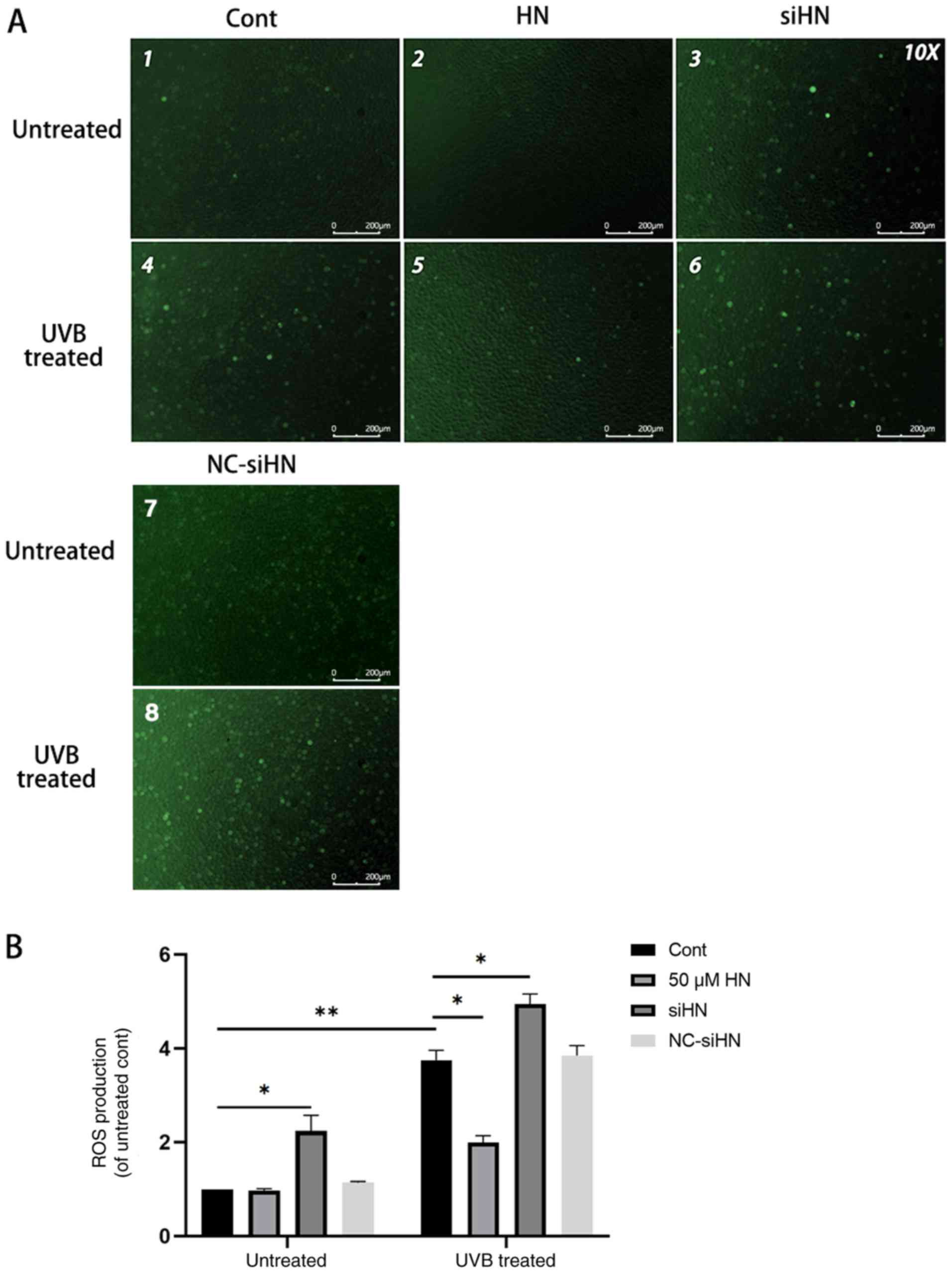 | Figure 3.Endogenous HN inhibits UVB-induced
ROS production in HLECs. After pretreatment with 50 µM HN for 2 h
or transfection with siHN or NC siRNA for 48 h, HLECs were
subjected to 30 mJ/cm2 UVB radiation. After 24 h,
intracellular ROS levels were determined by
2′,7′-dichlorofluorescein diacetate. (A) Observation under a
fluorescence microscope and (B) detection by a fluorescence
microplate reader. Data are presented as the mean ± SD, n=3.
*P<0.05, **P<0.01. HN, humanin; HLECs, human lens epithelial
cells; UVB, type B UV; ROS, reactive oxygen species; siHN, HN
siRNA; siRNA, small interfering RNA; NC, negative control; Cont,
control. |
HN protects mitochondria from
oxidative stress in HLECs
The present study identified a significant increase
in ROS levels in UVB-treated HLECs, which aggravated mitochondrial
damage under oxidative stress. Due to the poor repair ability of
mtDNA, damage to mtDNA can cause energy metabolism disorders,
apoptosis and cell necrosis (23).
To further investigate whether HN is involved in mitochondrial
protection, the present study detected the Δψm using JC-1 staining.
JC-1 aggregates in normal mitochondria and has red fluorescence. It
was found that UVB treatment increased green fluorescence in HLECs,
indicating a decrease in Δψm and an increase in depolarizing
mitochondria (Fig. 4A). A decrease
in Δψm is also considered an important event during early apoptosis
(23). Moreover, the
HN-administered group exhibited a higher red-green fluorescence
ratio after UVB treatment compared with the control group
(P<0.01), whereas the HN siRNA group had a significantly lower
red-green fluorescence ratio compared with the control group and
the HN knockdown group supplemented with exogenous HN (P<0.01;
Fig. 4B). The present study also
examined the mtDNA copy number to assess the degree of mtDNA damage
and biosynthesis (Fig. 4C). It was
demonstrated that HN knockdown resulted in a decrease in
mitochondrial copy number under oxidative stress (P<0.01).
However, exogenous HN significantly reversed the damage to
mitochondria in the siHN group (P<0.01). Moreover, the number of
mitochondria per cell was counted after 24 h of UVB irradiation by
TEM (Fig. 4D). Consistent with the
results of mtDNA copy number, exogenous HN could increase
mitochondrial number in HLECs induced by oxidative stress (Fig. 4E; P<0.05). Furthermore, a
significant increase in mitochondrial autophagosomes was identified
(Fig. 4D) in HLECs administered
with exogenous HN compared with the UVB control cells, thus HN may
enhance mitophagy (Fig. 4F). This
allows HLECs to remove damaged mitochondria in time to prevent ROS
accumulation within cells (24).
In summary, the present results indicated that HN has a beneficial
effect on mitochondrial damage and biosynthesis in HLECs under
oxidative stress.
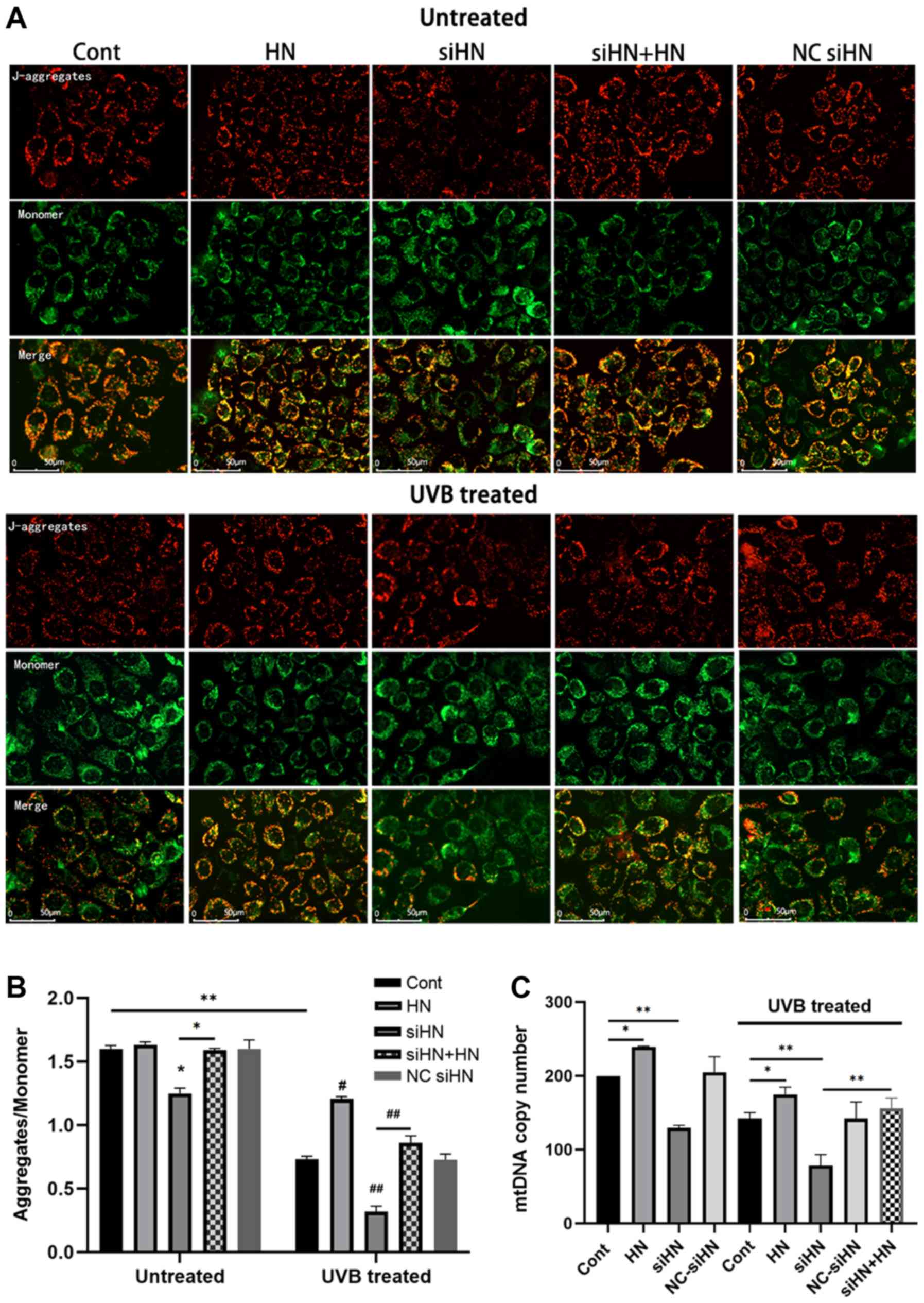 | Figure 4.HN protects mitochondria from
oxidative stress in HLECs. After pretreatment with 50 µM HN for 2 h
or transfection with siRNA for 48 h, HLECs were subjected to 30
mJ/cm2 UVB radiation. (A) Red fluorescence represents
the mitochondrial aggregate form of JC-1, indicating an intact ΔΨm.
Green fluorescence represents the monomeric form of JC-1,
indicating dissipation of ΔΨm. (B) Detection by a fluorescence
microplate reader. The ratio of red to green fluorescence indicates
the ratio of JC-1 aggregates/monomer. Data are presented as the
mean ± SD, n = 6. (C) Determination of mtDNA copy number. After 24
h of cultivation, ND1 and β-actin expression levels were detected
by reverse transcription-quantitative PCR. HN protects mitochondria
from oxidative stress in HLECs. After pretreatment with 50 µM HN
for 2 h or transfection with siRNA for 48 h, HLECs were subjected
to 30 mJ/cm2 UVB radiation. ((D) Mitochondria and
mitophagosomes were detected by TEM (magnification, ×25,000X). The
thin arrow indicates mitochondria and the thick arrow indicates
mitochondrial autophagosomes. (E) Average number of mitochondria
per cell. Record the number of mitochondria in 15 cells and get the
average value. (F) Average number of mitophagosomes per cell.
Record the number of mitochondria in 15 cells and get the average
value. Data are presented as the mean ± SD, n=3. *P<0.05,
**P<0.01. ΔΨm, mitochondrial membrane potential; mtDNA,
mitochondrial DNA; HN, humanin; HLECs, human lens epithelial cells;
UVB, type B UV; siHN, HN siRNA; siRNA, small interfering RNA; NC,
negative control; Cont, control. |
HN protects HLECs from oxidative
stress-induced apoptosis
Under oxidative stress, elevated levels of
intracellular ROS and damaged mitochondria can lead to apoptosis,
which is also considered to be an early event in the development of
cataracts (2). To investigate the
effect of HN on oxidative stress-induced apoptosis of HLECs, cells
were stained with Annexin V-FITC/PI to analyze the apoptotic rate
of HLECs induced by UVB (Fig. 5A)
or serum starvation (Fig. S1)
using flow cytometry. HLECs exhibited significant apoptosis after
UVB irradiation or serum starvation (P<0.001), and apoptosis in
the HN siRNA group was more significant compared with the control
group with or without oxidative stress (Fig. 5A and B; P<0.01). Under UVB
irradiation, HN-pretreated cell apoptosis was decreased by 11%
compared with the control group (P<0.05), while under serum
starvation, HN-pretreated cell apoptosis was decreased by 42%
compared with the control group (P<0.01). Thus, exogenous HN may
serve a moderate protective role in UVB-induced HLEC apoptosis.
Compared with the control group, oxidative stress significantly
increased the protein expression levels of Bax and cleaved
caspase-3, and decreased the expression levels of Bcl-2 (P<0.01;
Fig. 5C). Furthermore, under UVB
treatment, the HN-administered group displayed higher Bcl-2
expression levels compared with the control group (37.5%;
P<0.05; Fig. 5D), but 17
(P<0.05) and 18% (P<0.05) lower Bax and cleaved caspase3
expression levels compared with the control group. Collectively,
the present results suggested that HN may inhibit apoptosis by
regulating the expression levels of Bcl-2 and Bax, and the
activation of caspase-3.
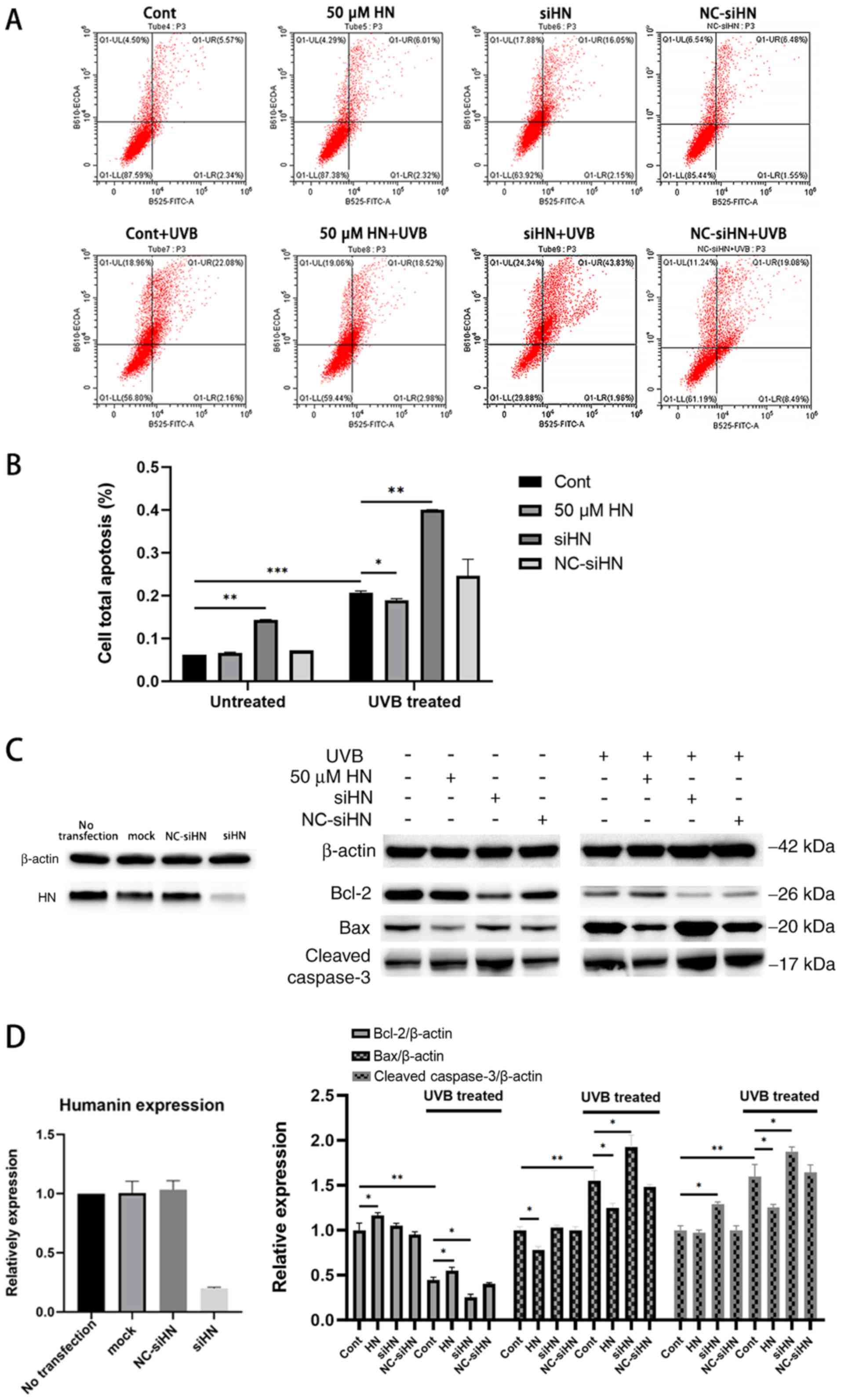 | Figure 5.Apoptosis assay of HLECs under
oxidative stress induced by UVB. After pretreatment with 50 µM HN
for 2 h or transfection with siRNA for 48 h, HLECs were subjected
to 30 mJ/cm2 UVB radiation. (A) Annexin V/PI staining
detected by flow cytometry. (B) Quantification of flow cytometry
results. (C) Western blotting and (D) semi-quantification of the
expression levels of the apoptosis-related proteins Bcl-2, Bax and
cleaved caspase-3. Data are presented as the mean ± SD, n=3.
*P<0.05, **P<0.01, ***P<0.001. HN, humanin; HLECs, human
lens epithelial cells; UVB, type B UV; siHN, HN siRNA; siRNA, small
interfering RNA; NC, negative control; Cont, control. |
Discussion
Epidemiology has linked oxidative stress to the
development of cataracts, and oxidative stress-induced apoptosis of
HLECs has been reported as a key early event of ARC development
(22,25,26).
The in vitro experiments suggested that HN may act as an
antioxidant MDP in HLECs. To the best of our knowledge, the present
study was the first to identify that exogenous HN enhanced the
resistance of HLECs to oxidative stress and reduced apoptosis by
inhibiting the production of ROS, preventing the decrease of Δψm,
increasing mitochondrial membrane biosynthesis and enhancing the
autophagy of mitochondria. Furthermore, it was revealed that HN
knockdown increased the sensitivity of HLECs to oxidative damage
and led to increased apoptosis.
The present study revealed that HN was expressed in
HLECs and responded to oxidative stress. The level of ROS in HLECs
has previously been reported to be positively associated with the
dose of UVB and induced different degrees of oxidative stress in
cells (27). The results indicated
that 4–12 h after UVB irradiation, HN expression increased, which
was associated with UVB irradiation. Thus, HN, as a highly
conserved cytoprotective peptide, may have an important role in the
survival mechanism of HLECs under oxidative stress. HN expression
in various tissues and cells, as well as age-related pathological
models, has been reported to be associated with oxidative stress
resistance (13,19,28).
It was previously shown that HN expression was elevated in
polarized retinal pigment epithelium monolayers, which increased
their resistance to oxidative stress (14). Moreover, HN levels were
downregulated in patients with T2DM, which reduced their
antioxidant capacity (29).
Under oxidative pressure (29,30),
characteristic changes in cells include a significant increase in
intracellular ROS and mitochondrial depolarization (31). High concentrations of ROS in cells
may cause oxidative injury to lipids, proteins and nucleic acids,
and could damage the integrity of various biomolecules (23). In addition, excessive ROS may have
a central role in the pathogenesis of various human diseases, such
as cataracts, cardiovascular disease, AMD, diabetes and aging
(32,33). The oxidative phosphorylation
process of depolarized mitochondria is a major source of
intracellular ROS (34,35). Furthermore, mitochondria are
sensitive to oxidative stress, and the repair of mtDNA is slower
compared with that of nDNA (23).
Decreased resistance to oxidative damage or lack of rapid removal
of damaged mitochondria may result in a significant accumulation of
ROS within the cell, which may cause more severe oxidative damage
to mitochondria and various biomolecules (36), and eventually lead to apoptotic
cell death (31,34). Therefore, reducing ROS production
and protecting mitochondrial function are critical for HLEC
resistance to oxidative stress. The present study revealed that
knockdown of HN resulted in a significant increase in the levels of
ROS in HLECs, particularly under oxidative stress, whereas
exogenous HN significantly reduced the levels of ROS in HLECs under
oxidative stress. The present study hypothesized that the primary
reason for this was that the expression of endogenous HN may be
important for cells to maintain a low level of ROS, and under
oxidative stress HN reduces the production of endogenous ROS via
its protective effect against mitochondrial damage. Moreover, it
was found that HN effectively enhanced mitochondrial resistance to
oxidative stress, and that exogenous HN pretreatment significantly
increased Δψm in UVB-treated HLECs. In addition, it was
demonstrated that HN co-treatment rescued the increased sensitivity
of mitochondria to oxidative stress. Furthermore, the present
results suggested that the changes in HN altered mitochondrial
biosynthesis and mitochondrial numbers. In HLECs with HN knockdown,
the mtDNA copy number was significantly reduced compared with the
control group, whereas exogenous HN could rescue this reduction in
mtDNA copy number, as verified by the TEM results. Sreekumar et
al (15) reported that HN
upregulated the expression of mitochondrial transcription factor A,
a key biogenesis regulator protein, to regulate mitochondrial
transcription initiation (37,38).
Similarly, the present results indicated that HN increased the copy
number of mtDNA with or without oxidative stress. Therefore, HN may
maintain mitochondrial germination and normal function by promoting
the initiation of mtDNA transcription, thereby increasing the
resistance of HLECs to oxidative stress and preventing oxidation of
lipids and proteins, which is important for maintaining the
transparency of the lens (39).
HN may promote the removal of damaged depolarized
mitochondria from cells (15).
Mitochondrial damage caused by oxidative stress induces mitophagy
to restore and maintain cellular energy metabolism, reduces
mitochondria-mediated cell and tissue damage, and induces apoptosis
(40). The present TEM results
identified that exogenous HN pretreatment significantly enhanced
mitophagy in HLECs under oxidative stress. However, as shown by
results from previous studies, the relationship between HN and
autophagy is controversial. Gong et al (41) reported that HN induced
chaperone-mediated autophagy as opposed to macroautophagy. However,
Han et al (42) found that
HN induced macroautophagy in the nervous system. Cataracts are
associated with lens epithelial cells and lens fiber cells.
Moreover, lens fibroblasts are differentiated from lens epithelial
cells. The programmed removal of organelles from differentiating
lens fiber cells contributes towards lens transparency via the
formation of an organelle-free zone (OFZ). Disruptions in OFZ
formation are accompanied by the persistence of organelles in lens
fiber cells and can contribute towards cataracts (43). Furthermore, mitochondrial autophagy
can eliminate damaged or unnecessary mitochondria in HLECs, which
ensures the continuous differentiation of lens epithelial cells
into lens fibroblasts, and plays an important role in lens
development and the maintenance of lens transparency (44). Costello et al (45) reported that the loss of mitophagy
may result in retention of mitochondria in the OFZ, leading to
light scattering and cataract formation. Mutations in the autophagy
gene FYVE and coiled coil domain containing 1 have been confirmed
to cause congenital cataracts (46). In addition, in lens fiber cells,
crystallin is denatured, modified and aggregated, which can
eventually lead to cataracts (10). Therefore, further research on lens
fiber cells is necessary. To the best of our knowledge, the present
study is the first to use TEM to demonstrate that HN significantly
promoted mitochondrial autophagy in HLECs under oxidative stress.
However, further investigation is required into the relationship
between HN and mitochondrial autophagy, and its role in cataract
formation. Future studies will detect autophagy-related proteins,
such as microtubule-associated protein 1A/1B-light chain 3, and
study the specific molecular pathways of HN to promote
mitochondrial autophagy via the use of various autophagy
regulators. Overall, the present results identified the mechanisms
of HN in reducing intracellular ROS and clearing damaged
mitochondria, which is of great significance for understanding the
repair of oxidative damage and maintaining cell homeostasis.
Apoptotic cell death of HLECs under oxidative stress
may involve complex mechanisms, including the mitochondrial
pathway, Bcl-2 protein family and caspase-3 activation. In damaged
mitochondria, the decrease in Δψm, massive production of ROS and
the release of nucleotide-modified mitochondrial proteins from
mitochondria into the cytosol or nucleus play a key role in
apoptosis (47). The present study
demonstrated that under oxidative stress, HN may have an important
role in maintaining HLEC viability, reducing cytotoxic sensitivity
and preventing apoptosis. Moreover, it was found that exogenous HN
pretreatment alleviated apoptosis of HLECs induced by UVB or serum
starvation, while knockdown of HN increased apoptosis in HLECs. In
addition, Gross et al (48)
found a common checkpoint in the mammalian cell death pathway,
which is at the level of the pro-apoptotic Bax and anti-apoptotic
Bcl-2. The present study found that exogenous HN treatment
upregulated Bcl-2 protein levels and reduced the expression of Bax
in HLECs treated with UVB. Furthermore, in untreated HLECs, HN
pretreatment did not further reduce the apoptotic rate, but the
levels of Bcl-2 and Bax proteins were altered. Thus, it is
hypothesized that exogenous HN may inhibit apoptosis at the protein
level in untreated conditions, but it is difficult to further
reduce apoptosis due to its low basal rate. Gottardo et al
(49) revealed that in untreated
GH3 cells, HN was able to upregulate Bcl-2 expression, reduce Bax
expression and significantly increase the ratio of Bcl-2 to Bax,
which is a good index of antiapoptotic cell behavior. Therefore, HN
may be involved in the regulation of Bcl-2 family proteins and have
a complex relationship with apoptosis-related proteins involved in
the mechanism of preventing apoptosis; the binding between HN and
Bax has been previously reported (12). Furthermore, caspase-3 plays a key
role in the execution of apoptosis and the apoptotic pathway in the
development of cataracts (50–52),
and caspase activities may be activated by the release of
cytochrome c from mitochondria (48). Moreover, the present results
suggested that HN reduced the levels of cleaved caspase-3 in
UVB-treated HLECs.
In conclusion, the present results indicated that HN
may reduce the damage and apoptosis of HLECs under oxidative stress
by reducing the production of intracellular ROS and protecting the
function of mitochondria. To the best of our knowledge, the present
study was the first to demonstrate that exogenous HN may enhance
the occurrence of intracellular mitochondrial autophagy to remove
dysfunctional mitochondria, and remove harmful byproducts and
oxidants to help maintain intracellular environmental balance and
cell survival. Moreover, the expression levels of HN have been
reported to decrease with age, thus a deficiency in endogenous HN
may lead to insufficient oxidative resistance and may be involved
in the pathogenesis of age-related diseases (53). The present study investigated the
protective effect of HN on HLECs under in vitro oxidative
stress. The lack of in vivo studies or clinical data is the
main limitation of the present study. To examine the role of HN in
the process of cataracts, further in vivo study of HN is
required. Given the multiple protective effects of HN in HLECs
under oxidative stress, HN may be a valuable potential protective
molecule in the prevention and treatment of ARCs.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank the Dr Chenqi Luo of
the Eye Center of the Second Affiliated Hospital for excellent
technical assistance.
Funding
This work was supported by the National Natural
Science Foundation of China (grant no. 81670834), the National
Natural Science Foundation of China (grant no. 81970781), the
Natural Science Foundation of Zhejiang Province (gran no.
LY17H090004), the National Natural Science Foundation of China
(grant no. 81800807) and the National Natural Science Foundation of
China (grant no. 81800869).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HY wrote the manuscript. HY and YC performed the
experiments. HY and QY analyzed the data. YT performed the flow
cytometry analysis. JZ, XT, XY and XS designed the study and
interpreted the data. XS modified the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HLECs
|
human lens epithelial cells
|
|
ARCs
|
age-related cataracts
|
|
HN
|
humanin
|
|
MDP
|
mitochondrial-derived peptide
|
|
ROS
|
reactive oxygen species
|
|
T2DM
|
type 2 diabetes mellitus
|
|
AMD
|
age-related macular degeneration
|
|
CCK-8
|
Cell Counting Kit-8
|
|
mtDNA
|
mitochondrial DNA
|
|
Δψm
|
mitochondrial membrane potential
|
References
|
1
|
Skinner C and Miraldi Utz V:
Pharmacological approaches to restoring lens transparency: Real
world applications. Ophthalmic Genet. 38:201–205. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Beebe DC, Holekamp NM and Shui YB:
Oxidative damage and the prevention of age-related cataracts.
Ophthalmic Res. 44:155–165. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu D, Zhao J, Wu D and Zhang J:
Ultraviolet A exposure induces reversible disruption of gap
junction intercellular communication in lens epithelial cells. Int
J Mol Med. 28:239–245. 2011.PubMed/NCBI
|
|
4
|
Li WC and Spector A: Lens epithelial cell
apoptosis is an early event in the development of UVB-induced
cataract. Free Radic Biol Med. 20:301–311. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Löfgren S: Solar ultraviolet radiation
cataract. Exp Eye Res. 156:112–116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wolfle U, Esser PR, Simon-Haarhaus B,
Martin SF, Lademann J and Schempp CM: UVB-induced DNA damage,
generation of reactive oxygen species, and inflammation are
effectively attenuated by the flavonoid luteolin in vitro and in
vivo. Free Radic Biol Med. 50:1081–1093. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hightower KR: A review of the evidence
that ultraviolet irradiation is a risk factor in cataractogenesis.
Doc Ophthalmol. 88:205–220. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dillon J: Sunlight exposure and cataract.
JAMA. 28:2291999. View Article : Google Scholar
|
|
9
|
Ji Y, Cai L, Zheng T, Ye H, Rong X, Rao J
and Lu Y: The mechanism of UVB irradiation induced-apoptosis in
cataract. Mol Cell Biochem. 401:87–95. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Berthoud VM and Beyer EC: Oxidative
stress, lens gap junctions, and cataracts. Antioxid Redox Signal.
11:339–353. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gupta SK, Trivedi D, Srivastava S, Joshi
S, Halder N and Verma SD: Lycopene attenuates oxidative stress
induced experimental cataract development: An in vitro and in vivo
study. Nutrition. 19:794–799. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo B, Zhai D, Cabezas E, Welsh K,
Nouraini S, Satterthwait AC and Reed JC: Human in peptide
suppresses apoptosis by interfering with Bax activation. Nature.
423:456–461. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kin T, Sugie K, Hirano M, Goto YI, Nishino
I and Ueno S: Humanin expression in skeletal muscles of patients
with chronic progressive external ophthalmoplegia. J Hum Genet.
51:555–558. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang D, Li H, Yuan H, Zheng M, Bai C, Chen
L and Pei X: Human in delays apoptosis in K562 cells by
downregulation of P38 MAP kinase. Apoptosis. 10:963–971. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sreekumar PG, Ishikawa K, Spee C, Mehta
HH, Wan J, Yen K, Cohen P, Kannan R and Hinton DR: The
mitochondrial-derived peptide human in protects RPE cells from
oxidative stress, senescence, and mitochondrial dysfunction. Invest
Ophthalmol Vis Sci. 57:1238–1253. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Klein LE, Cui L, Gong Z, Su K and Muzumdar
R: A humanin analog decreases oxidative stress and preserves
mitochondrial integrity in cardiac myoblasts. Biochem Biophys Res
Commun. 440:197–203. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hashimoto Y, Niikura T, Tajima H, Yasukawa
T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, et al: A
rescue factor abolishing neuronal cell death by a wide spectrum of
familial Alzheimer's disease genes and A beta. Proc Natl Acad Sci
USA. 98:6336–6341. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kariya S, Takahashi N, Ooba N, Kawahara M,
Nakayama H and Ueno S: Humanin inhibits cell death of
serum-deprived PC12h cells. Neuroreport. 13:903–907. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bachar AR, Scheffer L, Schroeder AS,
Nakamura HK, Cobb LJ, Oh YK, Lerman LO, Pagano RE, Cohen P and
Lerman A: Humanin is expressed in human vascular walls and has a
cytoprotective effect against oxidized LDL-induced oxidative
stress. Cardiovasc Res. 88:360–366. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu Y, Jiang H, Li H, Song W and Xia X:
Alpha-A-crystallin protects lens epithelial cell-derived iPSC-like
cells against apoptosis induced by oxidative stress. Cell
Reprogram. 18:327–332. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rwei P, Alex Gong CS, Luo LJ, Lin MB, Lai
JY and Liu HL: In vitro investigation of ultrasound-induced
oxidative stress on human lens epithelial cells. Biochem Biophys
Res Commun. 482:954–960. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yen K, Lee C, Mehta H and Cohen P: The
emerging role of the mitochondrial-derived peptide humanin in
stress resistance. J Mol Endocrinol. 50:R11–R19. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
West SK, Longstreth JD, Munoz BE, Pitcher
HM and Duncan DD: Model of risk of cortical cataract in the US
population with exposure to increased ultraviolet radiation due to
stratospheric ozone depletion. Am J Epidemiol. 162:1080–1088. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yam JC and Kwok AK: Ultraviolet light and
ocular diseases. Int Ophthalmol. 34:383–400. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hua H, Yang T, Huang L, Chen R, Li M, Zou
Z, Wang N, Yang D and Liu Y: Protective effects of lanosterol
synthase up-regulation in UV-B-induced oxidative Stress. Front
Pharmacol. 10:9472019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bodzioch M, Lapicka-Bodzioch K, Zapala B,
Kamysz W, Kiec-Wilk B and Dembinska-Kiec A: Evidence for potential
functionality of Nuclearly-encoded humanin isoforms. Genomics.
94:247–256. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ramanjaneya M, Bettahi I, Jerobin J,
Chandra P, Abi Khalil C, Skarulis M, Atkin SL and Abou-Samra AB:
Mitochondrial-derived peptides are down regulated in diabetes
subjects. Front Endocrinol (Lausanne). 10:3312019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu L, Yu R, Shi Y, Dai Y, Zeng Z, Guo X,
Ji Q, Wang G and Zhong J: Transduced protein transduction domain
linked HSP27 protected LECs against UVB radiation-induced damage.
Exp Eye Res. 120:36–42. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Marchetti MA, Lee W, Cowell TL, Wells TM,
Weissbach H and Kantorow M: Silencing of the methionine sulfoxide
reductase A gene results in loss of mitochondrial membrane
potential and increased ROS production in human lens cells. Exp Eye
Res. 83:1281–1286. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ylä-Herttuala S: Oxidized LDL and
atherogenesis. Ann N Y Acad Sci. 874:134–137. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stadtman ER and Levine RL: Protein
oxidation. Ann N Y Acad Sci. 899:191–208. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu H, Lin L, Giblin F, Ho YS and Lou MF:
Glutaredoxin 2 knockout increases sensitivity to oxidative stress
in mouse lense pithelial cells. Free Radic Biol Med. 51:2108–2117.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brooks MM, Neelam S, Fudala R, Gryczynski
I and Cammarata PR: Lenticular mitoprotection. Part A: Monitoring
mitochondrial depolarization with JC-1 and artifactual fluorescence
by the glycogen synthase kinase-3β inhibitor, SB216763. Mol Vis.
19:1406–1412. 2013.PubMed/NCBI
|
|
36
|
Ristow M, Zarse K, Oberbach A, Klöting N,
Birringer M, Kiehntopf M, Stumvoll M, Kahn CR and Blüher M:
Antioxidants prevent health-promoting effects of physical exercise
in humans. Proc Natl Acad Sci USA. 106:8665–8660. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Campbell CT, Kolesar JE and Kaufman BA:
Mitochondrial transcription factor A regulates mitochondrial
transcriptioni nitiation, DNA packaging, and genome copy number.
Biochim Biophys Acta. 1819:921–929. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee C, Wu SB, Hong CH, Liao WT, Wu CY,
Chen GS, Wei YH and Yu HS: Aberrant cell proliferation by enhanced
mitochondrial biogenesis via mtTFA in arsenical skin cancers. Am J
Pathol. 178:2066–2076. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu Q, Guo D, Bi H, Wang D and Du Y: UVB
irradiation-induced dysregulation of plasma membrane calcium
ATPase1 and intracellular calcium homeostasis in human lens
epithelial cells. Mol Cell Biochem. 382:263–272. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shefa U, Jeong NY, Song IO, Chung HJ, Kim
D, Jung J and Huh Y: Mitophagy links oxidative stress conditions
and neurodegenerative diseases. Neural Regen Res. 14:749–756. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gong Z, Tasset I, Diaz A, Anguiano J, Tas
E, Cui L, Kuliawat R, Liu H, Kühn B, Cuervo AM and Muzumdar R:
Humanin is an endogenous activator of chaperone-mediated autophagy.
J Cell Biol. 217:635–647. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Han K, Jia N, Zhong Y and Shang X:
S14G-humanin alleviates insulin resistance and increases autophagy
in neurons of APP/PS1 transgenic mouse. J Cell Biochem.
119:3111–3117. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wride MA: Lens fibre cell differentiation
and organelle loss: Many paths lead to clarity. Philos Trans R Soc
Lond B Biol Sci. 366:1219–1233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Brennan LA, McGreal-Estrada R, Logan CM,
Cvekl A, Menko AS and Kantorow M: BNIP3L/NIX is required for
elimination of mitochondria, endoplasmic reticulum and Golgi
apparatus during eye lens organelle-free zone formation. Exp Eye
Res. 74:173–184. 2018. View Article : Google Scholar
|
|
45
|
Costello MJ, Brennan LA, Basu S, Chauss D,
Mohamed A, Gilliland KO, Johnsen S, Menko S and Kantorow M:
Autophagy and mitophagy participate in ocular lens organelle
degradation. Exp Eye Res. 116:141–150. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen J, Ma Z, Jiao X, Fariss R, Kantorow
WL, Kantorow M, Pras E, Frydman M, Pras E, Riazuddin S, et al:
Mutations in FYCO1 cause autosomal-recessive congenital cataracts.
Am J Hum Genet. 88:827–838. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Balaban RS, Nemoto S and Finkel T:
Mitochondria, oxidants, and aging. Cell. 120:483–495. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gottardo MF, Ayala MM, Ferraris J, Zárate
S, Pisera D, Candolfi M, Jaita G and Seilicovich A: Humanin
inhibits apoptosis in pituitary tumor cells through several
signaling pathways including NF-κB activation. J Cell Commun
Signal. 11:329–340. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Roy S: Caspases at the heart of the
apoptotic cell death pathway. Chemical Res Toxicol. 13:961–962.
2000. View Article : Google Scholar
|
|
51
|
Andersson M, Honarvar A, Sjöstrand J,
Peterson A and Karlsson JO: Decreased caspase-3 activity in human
lens epithelium from posterior sub capsular cataracts. Exp Eye Res.
76:175–182. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yao H, Tang X, Shao X, Feng L, Wu N and
Yao K: Parthenolide protects human lens epithelial cells from
oxidative stress-induced apoptosis via inhibition of activation of
caspase-3 and caspase-9. Cell Res. 17:565–571. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Muzumdar RH, Huffman DM, Atzmon G,
Buettner C, Cobb LJ, Fishman S, Budagov T, Cui L, Einstein FH,
Poduval A, et al: Humanin: A novel central regulator of peripheral
insulin action. PLoS One. 4:e63342009. View Article : Google Scholar : PubMed/NCBI
|














