Introduction
Homocysteine (Hcy) is a known risk factor for
various cardiovascular diseases (CVDs) (1). It is well known that endothelial
dysfunction plays a crucial role in CVDs (2,3).
Notably, Hcy is a modifiable risk factor for endothelial
dysfunction. Evidence has demonstrated that hyperhomocysteinemia
(HHcy), as defined as plasma total Hcy levels ≥15 µmol/l (1), is associated with impaired
endothelium-dependent vascular dilation (4,5).
Previous studies reported that Hcy induced endoplasmic reticulum
(ER) stress and apoptosis of human umbilical vein endothelial cells
(HUVECs), indicating the involvement of ER stress in Hcy-induced
endothelial injury (6–8). However, the precise mechanism of
Hcy-induced endothelial dysfunction is not completely
understood.
Hcy is formed during the conversion of methionine to
cysteine (9). Folate plays an
important role in Hcy catabolism via the remethylation pathways.
Polymorphisms in the methylenetetrahydrofolate reductase gene or
inadequate folate intake are associated with high Hcy levels and
worse CVD outcomes (10–13). Moreover, human folate receptors
(hFRs), particularly hFRα, have a high affinity for folate and have
a pivotal role in folate uptake (14). Previous studies have detected the
expression levels of hFRs in healthy tissues, with higher levels of
protein expression in human lung and kidney (15,16).
However, it remains unclear whether hFRs are expressed on HUVECs.
Furthermore, little is known about the potential role of
endothelial hFRs in Hcy-induced endothelial injury.
The present study investigated the role of hFRs in
Hcy-induced HUVECs injury. Furthermore, the effect of hFRα
inhibition through RNA interference (RNAi) on ER stress marker
expression in HUVECs was studied.
Materials and methods
Materials
HUVECs (cat. no. KG419; http://www.keygentec.com.cn/index.php) were purchased
from the Nanjing KeyGen Biotech Co., Ltd. Hcy was purchased from
Sigma-Aldrich (Merck KGaA). Dulbecco's modified Eagle's medium
(DMEM), fetal bovine serum (FBS) and penicillin/streptomycin were
purchased from Gibco (Thermo Fisher Scientific, Inc.).
TransLipid® HL Transfection Reagent and Cell Counting
Kit-8 (CCK-8) were purchased from Beijing TransGen Biotech Co.,
Ltd. The bicinchoninic acid (BCA) protein assay kit was purchased
from Wuhan Boster Biological Technology Ltd. Dimethyl sulfoxide,
which was used for freezing and storing HUVECs, and
radioimmunoprecipitation assay (RIPA) buffer were purchased from
Applygen Technologies, Inc. The AnnexinV-fluorescein isothiocyanate
(FITC) Apoptosis Detection kit was purchased from BD Pharmingen.
The antibody against hFRα (cat. no. ab3361) was purchased from
Abcam. The antibody against β-tubulin (cat. no. sc-5274) was
purchased from Santa Cruz Biotechnology, Inc. Antibodies against
activating transcription factor 4 (ATF4; cat. no. 60035-1-lg) and
caspase 12 (cat. no. 55238-1-AP) were purchased from Wuhan Sanying
Biotechnology. Antibodies against protein kinase RNA-like ER kinase
(PERK; cat. no. 5683), phosphorylated (p)-PERK (cat. no. 3179),
p-eukaryotic translation initiation factor 2α (p-eIF2α; cat. no.
3398) and C/EBP homologous protein (CHOP; cat. no. 2895) were
purchased from Cell Signaling Technology, Inc. The antibody against
eIF2α (cat. no. AF6087) was purchased from Affinity Biosciences,
Inc. The antibody against β-actin (cat. no. TA-09) was purchased
from OriGene Technologies, Inc. The western blotting detection
reagents were purchased from Sigma-Aldrich (Merck KGaA). The small
interfering RNA (siRNA) targeting hFRα and control siRNA were
purchased from Novobio Co., Ltd. The siRNA targeting PERK and
corresponding control siRNA were purchased from Shanghai GenePharma
Co., Ltd.
Cell culture and treatment
HUVECs were cultured in DMEM with high sugar,
containing 10% FBS and 1% penicillin/streptomycin at 37°C in a 5%
CO2 humidified atmosphere. The medium was changed every
48 h, the cells were passaged every 2–3 days. For Hcy treatment,
HUVECs were incubated with mild-to-moderate concentrations of Hcy
(0, 50, 100 and 200 µM). For knocking down hFRα, HUVECs were
transfected with siRNA targeting hFRα. The sequences of siRNAs were
as follows: FRα-siRNA-1 sense, 5′-GGACUGAGCUUCUCAAUGUTT-3′ and
anti-sense, 5′-ACAUUGAGAAGCUCAGUCCTT-3′; FRα-siRNA-2 sense,
5′-GAUGUUUCCUACCUAUAUATT-3′ and anti-sense,
5′-UAUAUAGGUAGGAAACAUCTT-3′; FRα-siRNA-3 sense,
5′-CCACUGUUCUGUGCAAUGATT-3′ and anti-sense,
5′-UCAUUGCACAGAACAGUGGTT-3′; and negative control siRNA sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and anti-sense,
5′-ACGUGACACGUUCGGAGAATT-3′. For knocking down PERK, HUVECs were
transfected with siRNAs targeting PERK. The sequences were as
follows: PERK- siRNA-1 sense, 5′-ACCTCCAAGACCAACCACTTT-3′ and
anti-sense, 5′-AAAGTGGTTGGTCTTGGAGGT-3′; PERK-siRNA-2 sense,
5′-GUAGCUGGAAUGACAUAAATT-3′ and anti-sense,
5′-UUUAUGUCAUUCCAGCUACTT-3′; PERK-siRNA-3 sense,
5′-GUGGAAAGGUGAGGUAUAUTT-3′ and anti-sense,
5′-AUAUACCUCACCUUUCCACTT-3′; and negative control siRNA sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and anti-sense 5′-ACGUGACACGUUCGGGATT
−3′.
For cell transfection, HUVECs were plated in a
6-well plate, at a density of ~50%. The cells were transfected with
20 µM corresponding siRNA or negative control siRNA using
TransLipid® HL Transfection reagent (TransGen Biotech
Co., Ltd.) and incubated at room temperature for 20 min, according
to the manufacture's protocol. After 48 h, the effect of target
gene knockdown was confirmed by western blotting.
Cell morphology, viability and
apoptosis
For morphological observation, HUVECs were seeded
into 6-well plates (5×105 cells/well) and incubated with
different concentrations of Hcy (0, 50, 100 and 200 µM) for 24 h at
37°C. Cell morphology was examined with an inverted light
microscope Leica DMi1 (magnification, ×50; Leica Microsystems
GmbH).
CCK-8 assay was used to measure cell viability
according to the manufacturer's protocol. Cells (1×105
cells/ml; 100 µl/well) were seeded in a 96-well culture plate and
incubated for 24 h. After pretreatment with Hcy at different
concentrations (0, 50, 100 and 200 µM) for 24 h, CCK-8 (10 µl/100
µl fresh culture medium) was added to each well and incubated for 1
h at 37°C. A microplate reader (Thermo Fisher Scientific Inc.) was
used to measure the absorbance at a wavelength of 490 nm. Cell
viability = (ODtreatment -
ODblank)/(ODcontrol - ODblank) ×
100%, where OD refers to optical density.
Annexin V-FITC/propidium iodide (PI) double staining
was performed to measure cell apoptosis. After incubation with
siRNA targeting hFRα for 24 h, the HUVECs were collected and
centrifuged at 300 × g for 10 min at 4°C, washed three times with
cold PBS and then resuspended in binding buffer (1×106
cells/ml). Subsequently, cells were incubated with Annexin V-FITC
for 15 min and then PI for 5 min at room temperature in the dark.
The results were measured using a Backman CytoFLEX LX flow
cytometer (Beckman Coulter, Inc.) and analyzed with CytExpert
software (v2.1; Beckman Coulter, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The mRNA expression levels of hFRα, hFRβ and hFRγ
were determined by RT-qPCR after 24 h of Hcy treatment. mRNA
expression levels of solute carrier family 46 member 1 (SLC46A1)
and solute carrier family 19 member 1 (SLC19A1), which are the main
folate transporters in mammals (17,18)
were also determined. Briefly, total RNA was extracted from HUVECs
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc) and reverse transcription was performed with a
EasyScript® One-Step RT-PCR SuperMix kit (Beijing
Transgen Biotech Co., Ltd.). For the synthesis of the first-strand
cDNA, a total of 20 µl reaction solution was incubated for 30 min
at 45°C. qPCR was performed using an ABI 7900 real-time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) with
TransStart® Tip Green qPCR SuperMix kit (Beijing
Transgen Biotech Co., Ltd.). For PCR amplification, a total of 20
µl reaction solution, which included 2 µl cDNA, was first incubated
for 30 sec at 94°C, followed by 42 amplification cycles
(denaturation at 94°C for 5 sec, annealing at 55°C for 15 sec and
extension at 72°C for 10 sec). Fold changes in target gene
expression were determined using the 2−ΔΔCq method
(19) and relative levels of mRNA
were normalized to mRNA levels of β-actin for each sample. The
following primers for RT-qPCR were used: hFRα sense,
5′-GAATGCCTGCTGTTCTACCA-3′ and antisense, 5′-TGCGACAATCTTCCCACC-3′;
hFRβ sense, 5′-ATGCCACTTCTGCTGCTTCT-3′ and antisense,
5′-AGTGACTCCAGAGGCCTTCA-3′; hFRγ sense,
5′-TCAATGTCTGCATGAACGCCAAGC-3′ and antisense,
5′-TAAAGTTGTACAGGCGGGAGGTGT-3′; SLC46A1 sense,
5′-CTGGACCCTCTACATGAACG-3′ and antisense,
5′-GGTAGAGTGAGTTGAAGATG-3′; SLC19A1 sense,
5′-CCTCGTGTGCTACCTTTGCTT-3′ and antisense,
5′-TGATCTCGTTCGTGACCTGCT-3′; and β-actin sense,
5′-AGCGAGCATCCCCCAAAGTT-3′ and antisense,
5′-GGGCACGAAGGCTCATCATT-3′.
Western blot analysis
For western blotting, HUVECs were collected and
total proteins were lysed using RIPA buffer on ice; proteins were
quantified by the BCA protein assay kit. Equal amounts of total
protein (50 µg) were separated by sodium-dodecyl sulfate
polyacrylamide gel electrophoresis on a 10% gel, and transferred to
polyvinylidene difluoride membranes. The membranes were blocked
with 5% skimmed milk for 2 h at room temperature and then incubated
overnight at 4°C with primary antibody against hFRα (1:800
dilution), PERK (1:1,000 dilution), p-PERK (1:1,000 dilution), ATF4
(1:1,000 dilution), caspase 12 (1:1,000 dilution), CHOP (1:1,000
dilution), eIF2α (1:400 dilution), p-eIF2α (1:1,000 dilution),
β-actin (1:2,000 dilution) or β-tubulin (1:1,000 dilution),
followed by incubation with the respective HRP-conjugated secondary
antibodies (cat. nos. HS101 and HS201; 1:5,000 dilution; Beijing
TransGen Biotech Co., Ltd.) for 2 h at room temperature. Finally,
the target bands were visualized using an enhanced
chemiluminescence detection reagent (cat. no. DW101; Beijing
Transgen Biotech Co., Ltd.) and images were captured by Quantity
One 1-D analysis software (Bio-Rad Laboratories, Inc.). Intensity
of the bands were assessed with ImageJ (v1.46, National Institutes
of Health) and normalized to the intensity of loading controls
β-actin or β-tubulin.
Statistical analysis
The experiments were repeated three times and the
data are expressed as mean ± standard error. Statistical analysis
was performed with the SPSS statistics program (v22.0; IBM Corp.).
One-way ANOVA followed by Tukey post hoc test or Kruskal-Wallis
followed by Dunn-Bonferroni post hoc test were applied as
appropriate. A two-sided P<0.05 was considered to indicate a
statistically significant difference.
Results
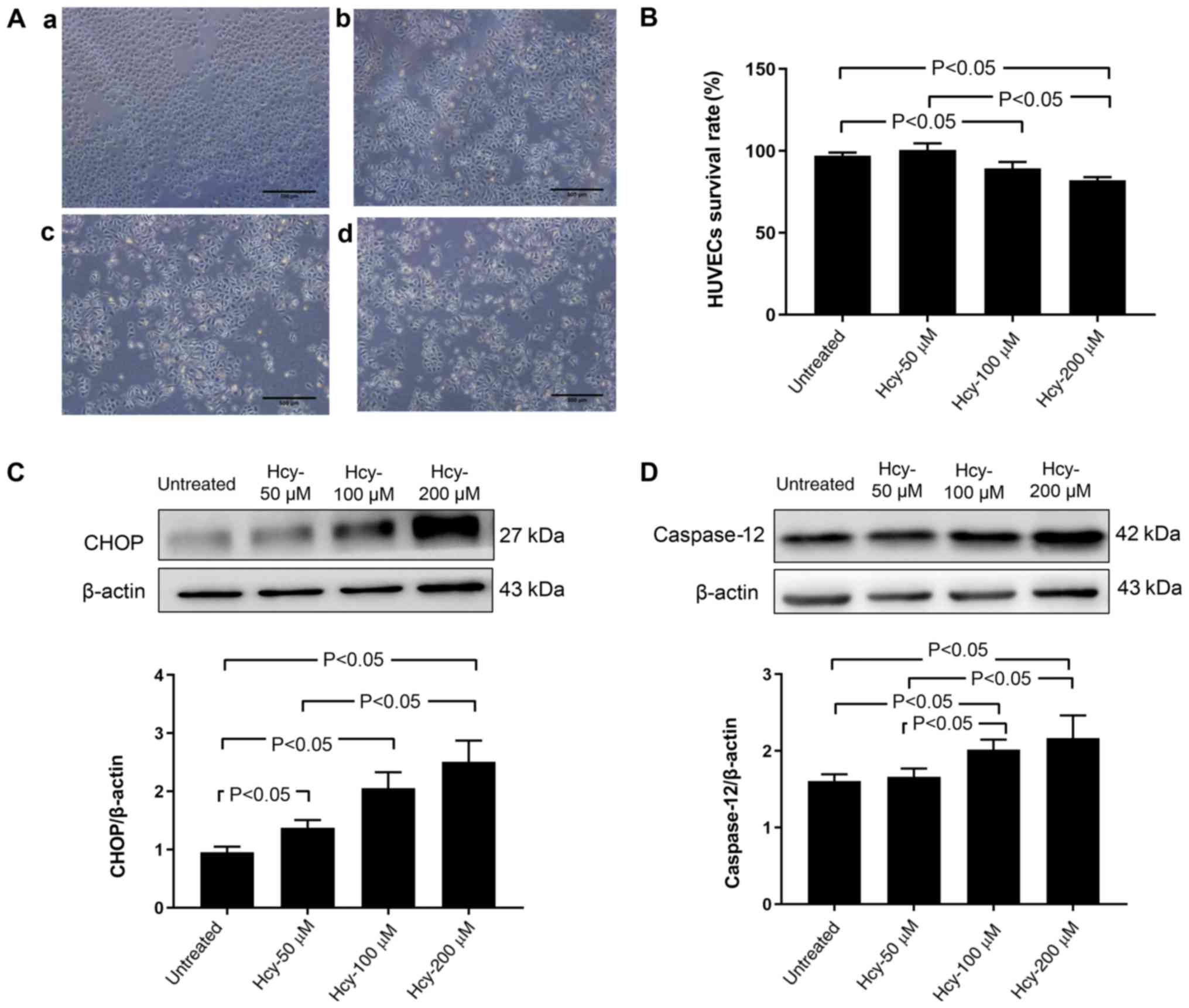
Hcy induces morphological changes and
decreases viability of HUVECs
As shown in Fig.
1A, HUVECs treated with 0 and 50 µM Hcy were smooth and plump,
arranged in a tight and neat conformation. Conversely, exposure of
HUVECs to higher concentrations of Hcy (100 and 200 µM) induced
marked changes. There were fewer adherent cells, and an increase in
cell shedding (Fig. 1A).
As shown in Fig.
1B, no significant differences in cell viability were found
between untreated cells and cells treated with 50 µM Hcy
(P>0.05). Hcy at a concentration of 100 and 200 µM significantly
reduced the percentage of viable cells compared with untreated
cells (P<0.05). Cells treated with 200 µM Hcy were also
significantly less viable than cells treated with 50 µM Hcy
(P<0.05; Fig. 1B).
As shown in Fig.
1C, Hcy at 50, 100 and 200 µM significantly increased the
protein expression of CHOP, relative to β-actin, compared with the
untreated cells (P<0.05). As shown in Fig. 1D, treatment with Hcy at 100 and 200
µM significantly increased the protein expression of caspase 12,
relative to β-actin, compared with the untreated cells and cells
treated with 50 µM Hcy (P<0.05).
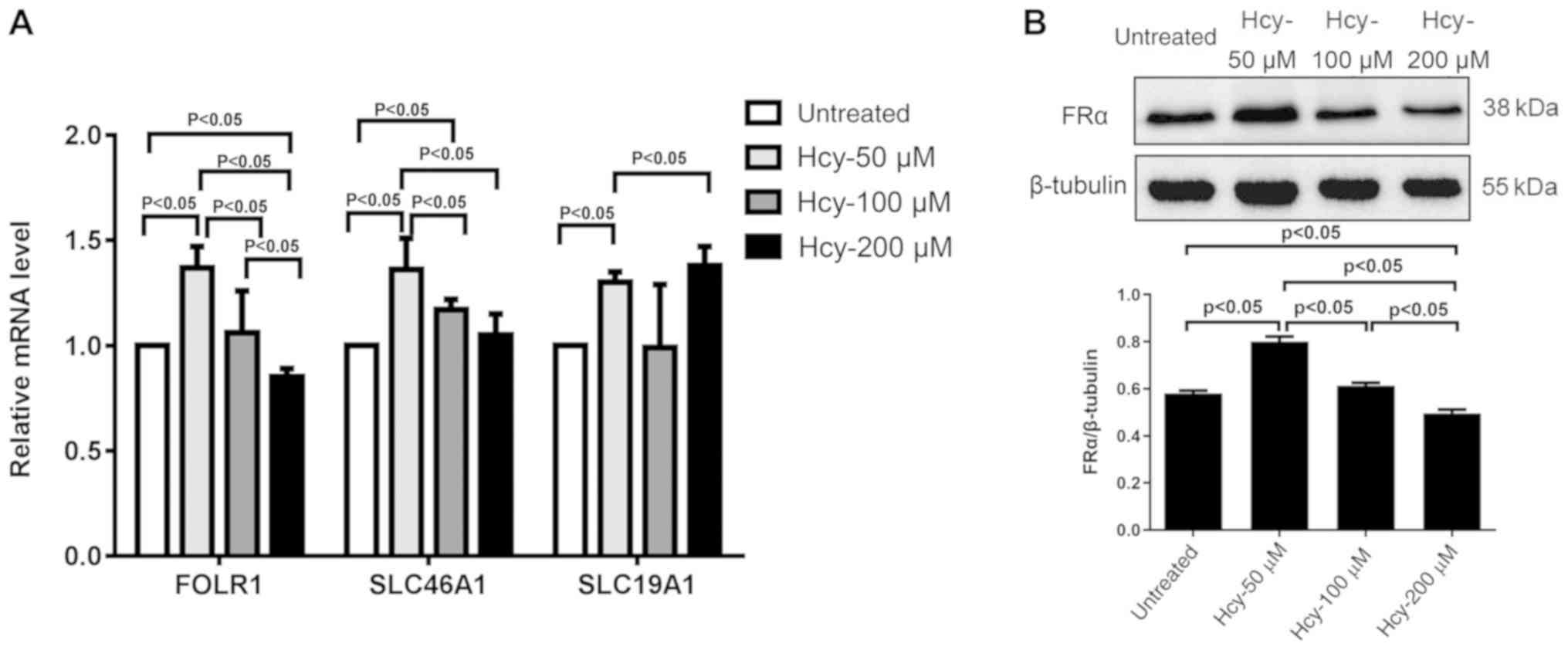
Hcy reduces expression of mRNA and
protein expression levels of hFRα
As shown in Fig.
2A, treatment with 50 µM Hcy for 24 h significantly increased
the mRNA expression of hFRα (FOLR1), SLC46A1 and SLC19A1
(P<0.05) compared with the untreated cells. The mRNA expression
of hFRα was significantly lower in cells treated with 200 µM Hcy
compared with untreated cells and cells treated with 50 and 100 µM
Hcy (P<0.05; Fig. 2A). Compared
with cells treated with 50 µM Hcy, the mRNA expression levels of
hFRα were significantly lower in cells treated with 100 and 200 µM
Hcy (P<0.05). Cells treated with 50 µM Hcy also expressed
significantly higher levels of SLC46A1 mRNA compared with cells
treated with 100 and 200 µM (P<0.05). Conversely, the mRNA
levels of SLC19A1 increased significantly in cells treated with 200
µM Hcy, compared with cells treated with 50 µM Hcy (P<0.05;
Fig. 2A). On the other hand, the
study failed to detect the mRNA levels of hFRβ and hFRγ (data not
shown).
The protein expression levels of hFRα in HUVECs were
also measured after Hcy treatment for 48 h. As shown in Fig. 2B, the protein expression of hFRα
was significantly increased in cells treated with 50 µM Hcy
compared with untreated cells (P<0.05). Compared with cells
treated with 50 µM Hcy, protein expression levels in cells treated
with 100 µM Hcy were significantly reduced (P<0.05). Compared
with the untreated cells, or cells treated with 50 or 100 µM Hcy,
cells treated with 200 µM Hcy also had a significantly lower
protein expression (P<0.05).
hFRα knockdown increases apoptosis and
induces PERK activation in HUVECs
To determine the role of hFRα in Hcy-induced HUVECs
injury, hFRα expression was inhibited using siRNA. Transfection was
confirmed by immunofluorescence staining with siRNAs that had an
immunofluorescent component (data not shown). hFRα expression was
significantly reduced in HUVECs transfected with hFRα siRNA
compared with HUVECs that were not transfected (untreated) or
transfected with a non-specific control siRNA (Fig. 3A and B). Among the three siRNAs,
hFRα-siRNA-1 generated the most significant knockdown results and
was therefore chosen for further experiments. Flow cytometric
analysis was conducted 12 h post-siRNA transfection. As shown in
Fig. 3C and D, the apoptotic rate
of HUVECs transfected with hFRα siRNA was significantly higher
compared with the untreated and the control siRNA groups
(P<0.05).
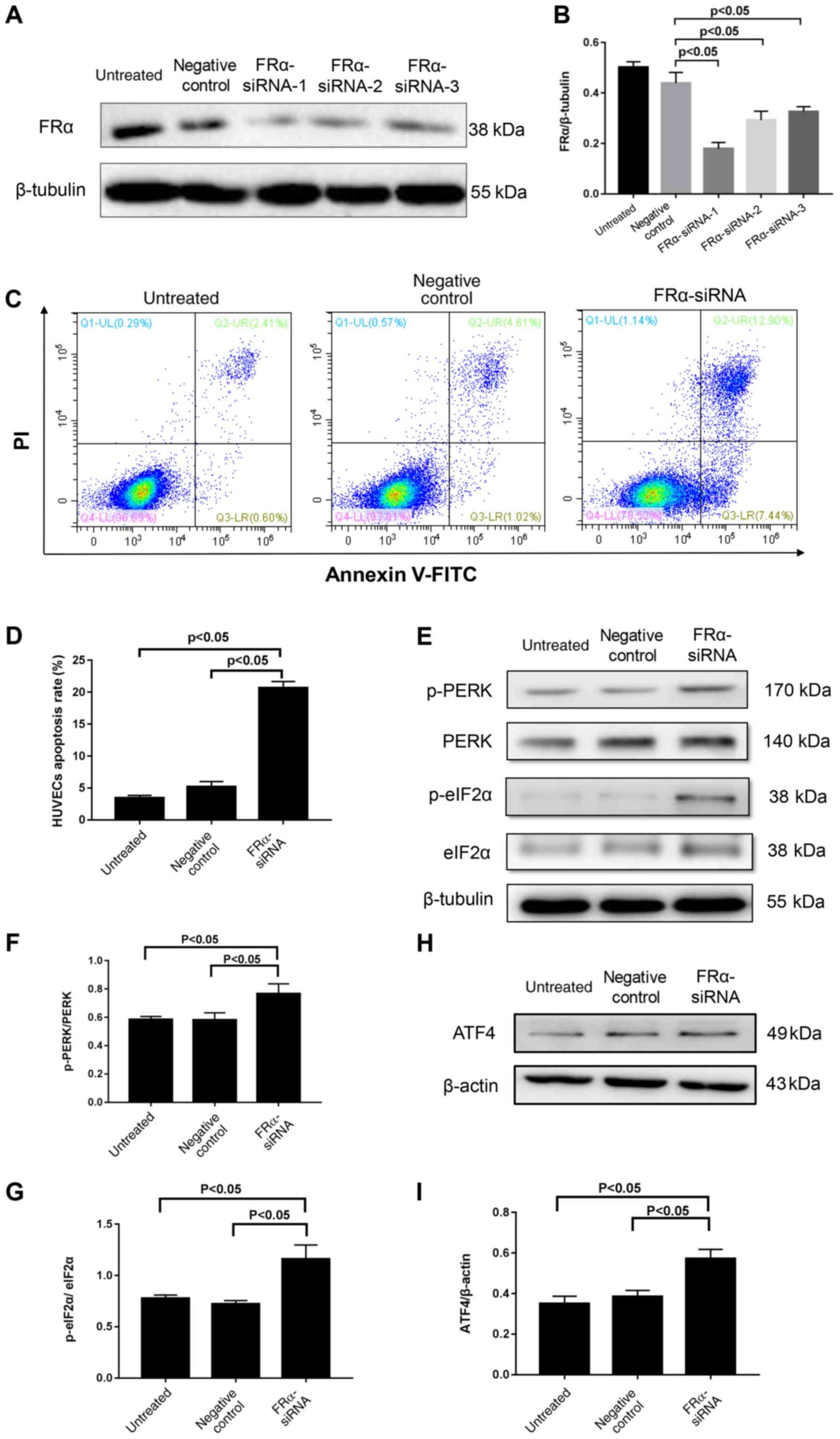 | Figure 3.Effect of hFRα inhibition on HUVECs
apoptosis and PERK activation. (A) Representative western blot of
FRα following knockdown of FRα. (B) Semi-quantification of
FRα/β-tubulin. (C) Flow cytometric analysis of HUVECs stained with
Annexin V/PI. (D) Quantitative analysis of percentage of apoptotic
cells based on flow cytometry of Annexin V/PI double-staining. (E)
Representative western blot of PERK, p-PERK, eIF2α and p-eIF2α. (F)
Semi-quantification of p-PERK/PERK. (G) Semi-quantification of
p-eIF2α/eIF2α. (H) Representative western blot of ATF4 and (I)
semi-quantification of ATF4/β-actin. Data are presented as mean ±
standard error. (B, D, F and G) Statistical analysis was conducted
by one-way ANOVA followed by Tukey's multiple comparison post hoc
test. (I) Statistical analysis was conducted by the Kruskal-Wallis
test followed by Dunn-Bonferroni post-hoc test between each group.
hFRα, human folate receptor α; HUVECs, human umbilical vein
endothelial cells; PERK, protein kinase RNA-like endoplasmic
reticulum kinase; p, phosphorylated; siRNA, small interfering RNA;
ATF4, activating transcription factor 4; FITC, fluorescein
isothiocyanate; PI, propidium iodide; eIF2α, eukaryotic translation
initiation factor 2α. |
The PERK signaling pathway, which is a sensor of the
unfolded protein response in ER stress (20), was also analyzed. As shown in
Fig. 3E and F, the p-PERK/PERK
ratio was significantly higher in cells transfected with hFRα siRNA
than the untreated and control siRNA groups (P<0.05). In
addition, as shown in Fig. 3E, G, H
and I, hFRα siRNA transfection caused a significant increase in
the expression of ATF4 mRNA (P<0.05) and p-eIF2α (P<0.05) in
comparison with the untreated and control siRNA groups.
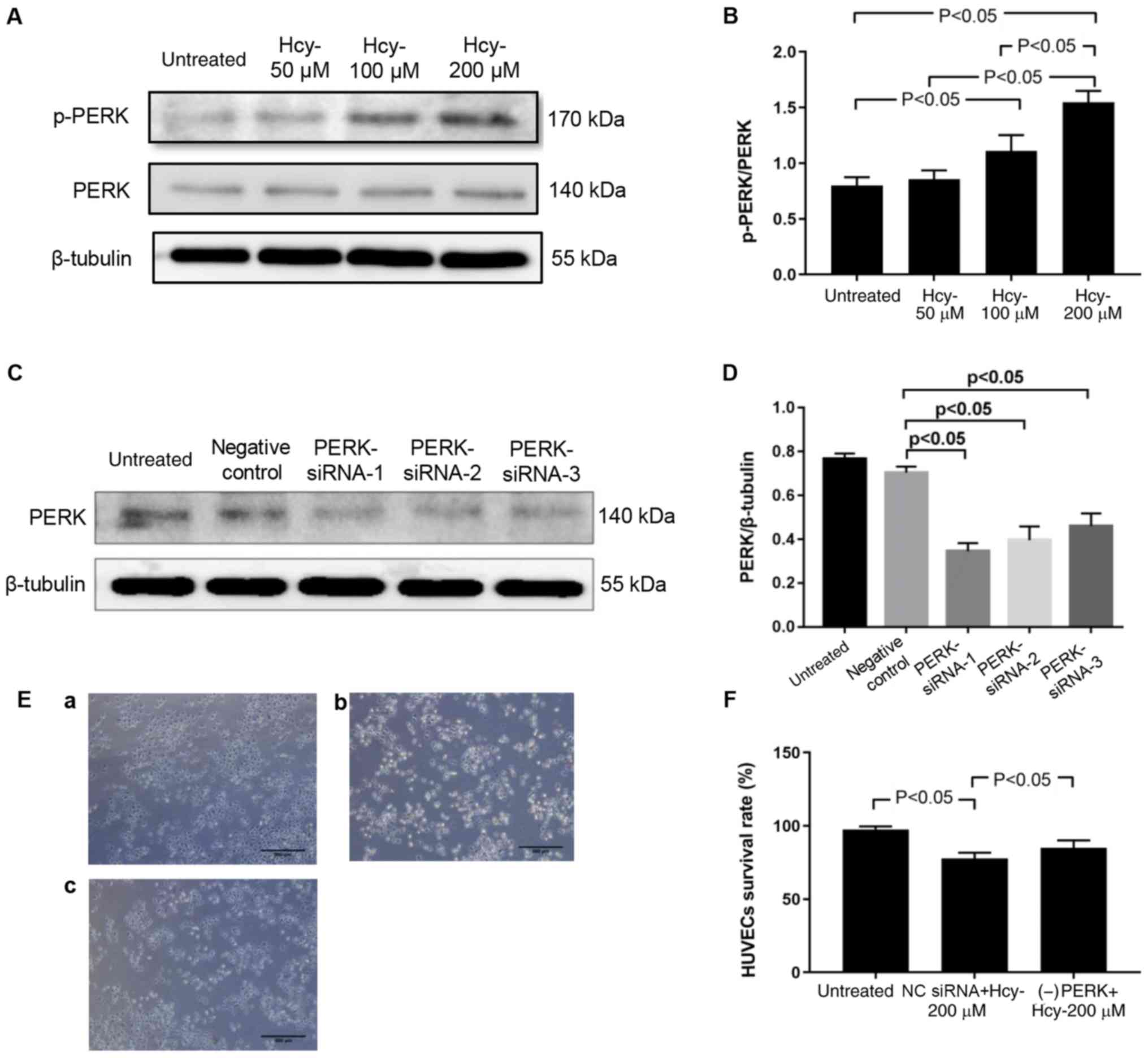
Knockdown of PERK attenuates
Hcy-induced cell injury in HUVECs
As shown in Fig. 4A and
B, in cells treated with 100 and 200 µM Hcy for 48 h the
p-PERK/PERK ratio was significantly increased compared with
untreated cells (P<0.05). In order to determine the role of PERK
in Hcy-induced injury, PERK mRNA expression was knocked down using
siRNA (Fig. 4C and D). Among the
three siRNAs, PERK-siRNA-1 generated the most significant knockdown
results and was therefore chosen for further experiments. As shown
in Fig. 4E and F, PERK siRNA
transfection ameliorated Hcy-induced morphological changes and
decreased HUVECs viability (P<0.05).
Discussion
Previous studies have demonstrated the detrimental
effect of Hcy on HUVECs, which was significantly altered by the
addition of folic acid (8,21,22).
The present study showed that hFRα expression was regulated by Hcy
and depletion of hFRα mimicked Hcy-induced ER stress and cell
injury in HUVECs. The principle findings of the present study were
as follows: i) Hcy dose-dependently decreased the expression of
hFRα in HUVECs and ii) inhibition of hFRα expression resulted in
increased apoptosis and activation of the PERK signaling pathway of
ER stress. Collectively, the present study highlighted the critical
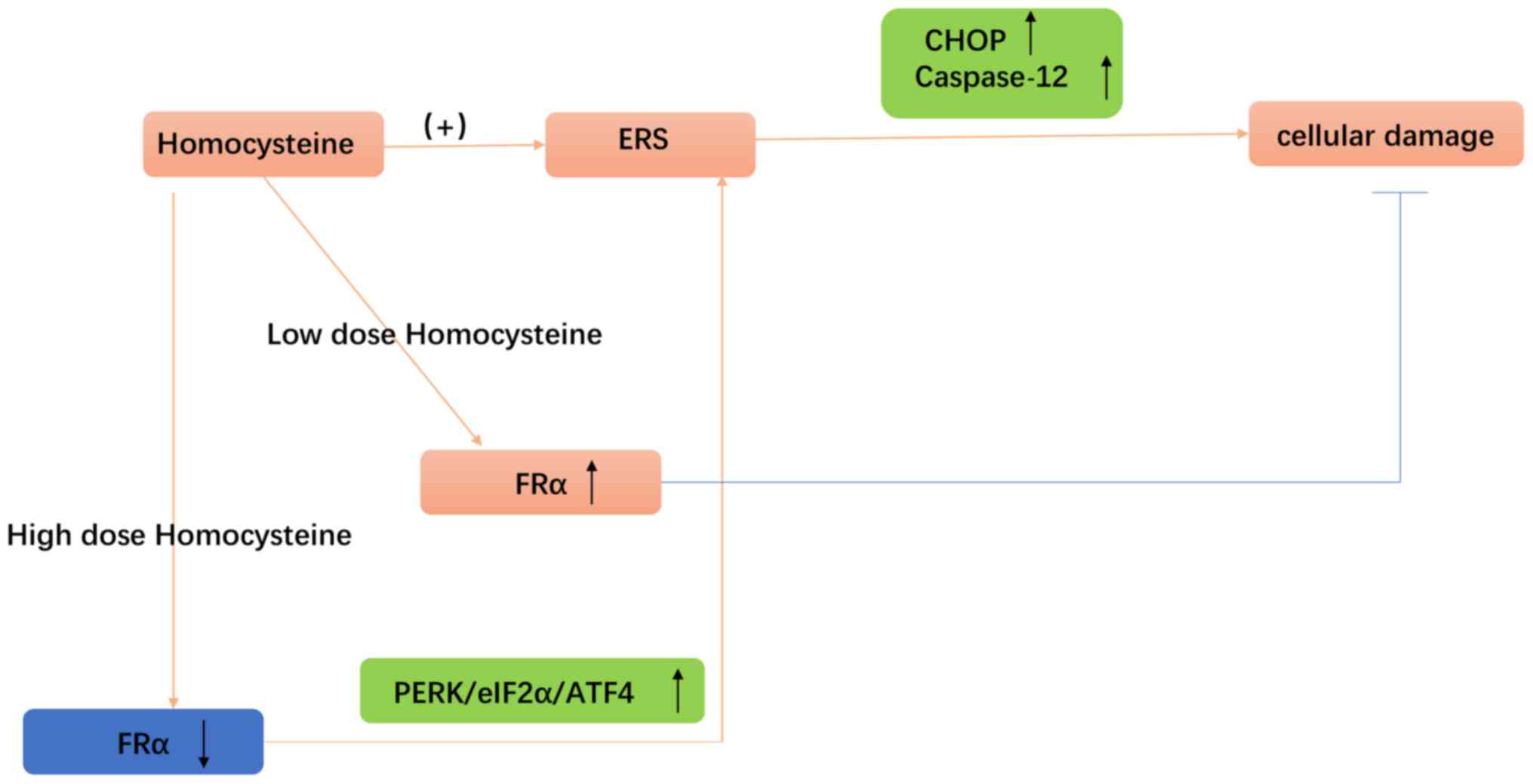
role of hFRα in protecting against Hcy-induced endothelial injury.
The schematic illustration of the proposed model is presented in
Fig. 5.
The present study investigated the acute toxicity of
mild-to-moderate doses of Hcy in HUVECs, because mild and moderate
HHcy is more common in the general population (9). The dose of Hcy used in this study is
a similar concentration to what is used in HUVECs in previous
reports (8,23,24).
Furthermore, 2,000 µM Hcy was reported as a moderate HHcy
concentration (8). Therefore, the
doses of Hcy used in this study should be considered as
low-to-moderate concentrations. Previous studies have demonstrated
that mild and moderate HHcy induced apoptosis and injury in HUVECs
in a dose-dependent manner (8,23,24).
Consistently, this study found that Hcy induced morphological
injury and reduced viability of HUVECs, particularly at higher
doses. In addition, a previous clinical study reported that in
patients at risk for atherosclerosis, 17% of men with higher Hcy
concentrations (21.27±5.37 µM) died during follow up (25). However, the finding in the present
study that there were no significant morphological changes to the
endothelial cells following treatment with 50 µM Hcy is
inconsistent with this report (25). This discrepancy might be caused by
the different experimental conditions, such as different
experimental subject (HUVEC cells vs. a human male), or different
study duration (24 h vs. 5 years).
It has been reported that Hcy treatment may reduce
HUVEC folate levels and folate supplements can decrease Hcy levels
and improve cell viability (21).
Since hFRs play a key role in cell folate uptake, it is important
to investigate whether Hcy modulates hFR levels in HUVECs. The
present study found that hFRs, mainly hFRα, exist in HUVECs.
Moreover, low-dose Hcy treatment induced an increase in hFRα mRNA
and protein levels. Notably, higher dose Hcy induced a decrease in
hFRα expression. To the best of our knowledge, this is the first
study to demonstrate that hFRα exists in HUVECs and can be
regulated by Hcy treatment. It has been suggested that
intracellular Hcy can stimulate the mRNA expression of FRs, which
may represent a mechanism underlying the effect of low-dose Hcy on
hFRα levels (26). However, the
present study did not allow us to elucidate the precise mechanism
of Hcy in the regulation of hFRs. Further study is needed to
determine the precise mechanism of Hcy-induced hFRα expression.
It has been reported that ER stress induced by Hcy
is one of the main mechanisms for endothelial injury (8,20,22).
ER stress is initiated by the accumulation of unfolded proteins,
and chronic or severe ER stress in endothelial cells can result in
cell death (27). Activation of
the PERK pathway, represented by p-PERK expression, is the most
definitive marker of ER stress. It has been demonstrated that Hcy
can activate the PERK pathway (8,24).
However, few studies have investigated the role of hFRα in ER
stress (8,20). This study demonstrated that hFRα
inhibition was associated with PERK signal pathway activation.
Additionally, hFRα downregulation was also associated with an
increased apoptotic rate in HUVECs. In agreement with previous
studies (8,20), these results showed that PERK was
strongly involved in Hcy-mediated HUVEC injury, since depletion of
PERK significantly reduced Hcy-induced cell injury. Considering
these results, it is possible that hFRα may serve a protective role
against Hcy-induced ER stress and cell injury in HUVECs.
In conclusion, this study revealed that hFRα was
present in HUVECs, and may negatively regulate ER stress, apoptosis
and reduced cell viability induced by Hcy treatment in HUVECs. The
present results indicated that hFRα may be an important target for
Hcy treatment.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81560079), Major Projects of
the Jiangxi Province Natural Science Foundation of China (grant no.
20152ACB20022) and the Education Department of Jiangxi Province
(grant no. GJJ150265).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
The work presented here was carried out in
collaboration between each author. JC, CC, HH, LQW and XH performed
the experiments. JC, CC and PL wrote the main manuscript. HZ and
YJY statistically analyzed the data. PL and JC designed the
experiments. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Han L, Wu Q, Wang C, Hao Y, Zhao J, Zhang
L, Fan R, Liu Y, Li R, Chen Z, et al: Homocysteine, ischemic
stroke, and coronary heart disease in hypertensive patients: a
population-based, prospective cohort study. Stroke. 46:1777–1786.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bonetti PO, Lerman LO and Lerman A:
Endothelial dysfunction: A marker of atherosclerotic risk.
Arterioscler Thromb Vasc Biol. 23:168–175. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vita JA: Endothelial function.
Circulation. 124:e906–e912. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stamler JS, Osborne JA, Jaraki O, Rabbani
LE, Mullins M, Singel D and Loscalzo J: Adverse vascular effects of
homocysteine are modulated by endothelium-derived relaxing factor
and related oxides of nitrogen. J Clin Invest. 91:308–318. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Woo KS, Chook P, Lolin YI, Cheung AS, Chan
LT, Sun YY, Sanderson JE, Metreweli C and Celermajer DS:
Hyperhomocyst(e)inemia is a risk factor for arterial endothelial
dysfunction in humans. Circulation. 96:2542–2544. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cimellaro A, Perticone M, Fiorentino TV,
Sciacqua A and Hribal ML: Role of endoplasmic reticulum stress in
endothelial dysfunction. Nutr Metab Cardiovasc Dis. 26:863–871.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang XC, Sun WT, Yu CM, Pun SH, Underwood
MJ, He GW and Yang Q: ER stress mediates homocysteine-induced
endothelial dysfunction: Modulation of IKCa and SKCa channels.
Atherosclerosis. 242:191–198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Z, Wei C, Zhou Y, Yan T, Wang Z, Li
W and Zhao L: Homocysteine induces apoptosis of human umbilical
vein endothelial cells via mitochondrial dysfunction and
endoplasmic reticulum stress. Oxid Med Cell Longev.
2017:57365062017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
De Bree A, Verschuren WM, Kromhout D,
Kluijtmans LA and Blom HJ: Homocysteine determinants and the
evidence to what extent homocysteine determines the risk of
coronary heart disease. Pharmacol Rev. 54:599–618. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Casas JP, Bautista LE, Smeeth L, Sharma P
and Hingorani AD: Homocysteine and stroke: Evidence on a causal
link from mendelian randomisation. Lancet. 365:224–232. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huo Y, Li J, Qin X, Huang Y, Wang X,
Gottesman RF, Tang G, Wang B, Chen D, He M, et al CSPPT
Investigators, : Efficacy of folic acid therapy in primary
prevention of stroke among adults with hypertension in China: The
CSPPT randomized clinical trial. JAMA. 313:1325–1335. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Klerk M, Verhoef P, Clarke R, Blom HJ, Kok
FJ and Schouten EG; MTHFR Studies Collaboration Group, : MTHFR
677C-->T polymorphism and risk of coronary heart disease: A
meta-analysis. JAMA. 288:2023–2031. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mark SD, Wang W, Fraumeni JF Jr, Li JY,
Taylor PR, Wang GQ, Guo W, Dawsey SM, Li B and Blot WJ: Lowered
risks of hypertension and cerebrovascular disease after
vitamin/mineral supplementation: The Linxian Nutrition Intervention
Trial. Am J Epidemiol. 143:658–664. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen C, Ke J, Zhou XE, Yi W, Brunzelle JS,
Li J, Yong EL, Xu HE and Melcher K: Structural basis for molecular
recognition of folic acid by folate receptors. Nature. 500:486–489.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parker N, Turk MJ, Westrick E, Lewis JD,
Low PS and Leamon CP: Folate receptor expression in carcinomas and
normal tissues determined by a quantitative radioligand binding
assay. Anal Biochem. 338:284–293. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weitman SD, Weinberg AG, Coney LR,
Zurawski VR, Jennings DS and Kamen BA: Cellular localization of the
folate receptor: Potential role in drug toxicity and folate
homeostasis. Cancer Res. 52:6708–6711. 1992.PubMed/NCBI
|
|
17
|
Hou Z and Matherly LH: Biology of the
major facilitative folate transporters SLC19A1 and SLC46A1. Curr
Top Membr. 73:175–204. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qiu A, Jansen M, Sakaris A, Min SH,
Chattopadhyay S, Tsai E, Sandoval C, Zhao R, Akabas MH and Goldman
ID: Identification of an intestinal folate transporter and the
molecular basis for hereditary folate malabsorption. Cell.
127:917–928. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Harding HP, Zhang Y, Bertolotti A, Zeng H
and Ron D: Perk is essential for translational regulation and cell
survival during the unfolded protein response. Mol Cell. 5:897–904.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cui S, Li W, Wang P, Lv X, Gao Y and Huang
G: Folic acid inhibits homocysteine-induced cell apoptosis in human
umbilical vein endothelial cells. Mol Cell Biochem. 444:77–86.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu X, Zhang L, Miao Y, Yang J, Wang X,
Wang CC, Feng J and Wang L: Homocysteine causes vascular
endothelial dysfunction by disrupting endoplasmic reticulum redox
homeostasis. Redox Biol. 20:46–59. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bao XM, Wu CF and Lu GP: Atorvastatin
attenuates homocysteine-induced apoptosis in human umbilical vein
endothelial cells via inhibiting NADPH oxidase-related oxidative
stress-triggered p38MAPK signaling. Acta Pharmacol Sin.
30:1392–1398. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu L, Jia F, Wei J, Yu Y, Yu T, Wang Y,
Sun J and Luo G: Salidroside protects against homocysteine-induced
injury in human umbilical vein endothelial cells via the regulation
of endoplasmic reticulum stress. Cardiovasc Ther. 35:33–39. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Blum A, Hijazi I, Eizenberg MM and Blum N:
Homocysteine (Hcy) follow-up study. Clin Invest Med. 30:21–25.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Perna AF, Lanza D, Sepe I, Conzo G,
Altucci L and Ingrosso D: Altered folate receptor 2 expression in
uraemic patients on haemodialysis: Implications for folate
resistance. Nephrol Dial Transplant. 28:1214–1224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hotamisligil GS: Endoplasmic reticulum
stress and the inflammatory basis of metabolic disease. Cell.
140:900–917. 2010. View Article : Google Scholar : PubMed/NCBI
|