Introduction
Spontaneous miscarriage (SM) is a major cause of
pregnancy failure. It is estimated that ~10-15% of all clinically
recognized pregnancies terminate in SM (1,2). In
addition, >50% of all SMs have chromosomal abnormalities (CAs)
(3–5), including mosaicism, structural
abnormalities and numerical chromosomal defects, such as trisomy,
monosomy, polyploidy and monosomy X (6,7).
Furthermore, SM increases the risk of pregnancy loss and
complications. Therefore, analysis of CAs in aborted tissues would
provide insight into the etiology of pregnancy termination, as well
as improved management of subsequent pregnancies in patients with
SM (8,9). Previous studies suggested that
patient follow-up is more cost-effective when CA analyses are
performed in patients who had experienced miscarriage (10,11).
Conventional methods used to detect CAs and
determine the cause of pregnancy loss include karyotyping,
fluorescence in situ hybridization, quantitative fluorescent-PCR
(QF-PCR) and multiplex ligation-dependent probe amplification.
However, these methods have inherent limitations (10,12).
Following the rapid development of molecular biology technologies,
array comparative genomic hybridization (CGH) and single nucleotide
polymorphism (SNP) microarray (13,14)
have become the standard methods used to investigate possible
chromosomal causes of miscarriage because of their ability to
analyze the whole genome at high resolution. However, microarray
assays have numerous limitations such as high cost, low throughput
and requirement of a large amount of high-quality DNA. With the
development of next-generation sequencing (NGS) and reduced
sequencing costs, low-coverage NGS assays have been widely used for
noninvasive pre-natal testing in China, which is also gradually
expanding to the detection of CAs in SM (1,9,15,16).
The aim of the present study was to develop a method
based on low-coverage NGS to detect CAs in SM through a
retrospective, case-controlled approach. The clinical performance
of the developed method was then assessed in a prospective study.
The performance of copy number variant (CNV) analysis based on
low-coverage NGS technology is dependent on the sequencing coverage
(15,17). Increasing the coverage may increase
the sensitivity of the CNV analysis method, while simultaneously
increase the sequencing cost (17). The present study used low-coverage
NGS CNV analysis, which yielded >3.5 million sequencing reads
with CNVs >1 Mb in length. Overall, the sequencing coverage was
~0.13X, with an average fragment length ~110 bp.
Materials and methods
Study design
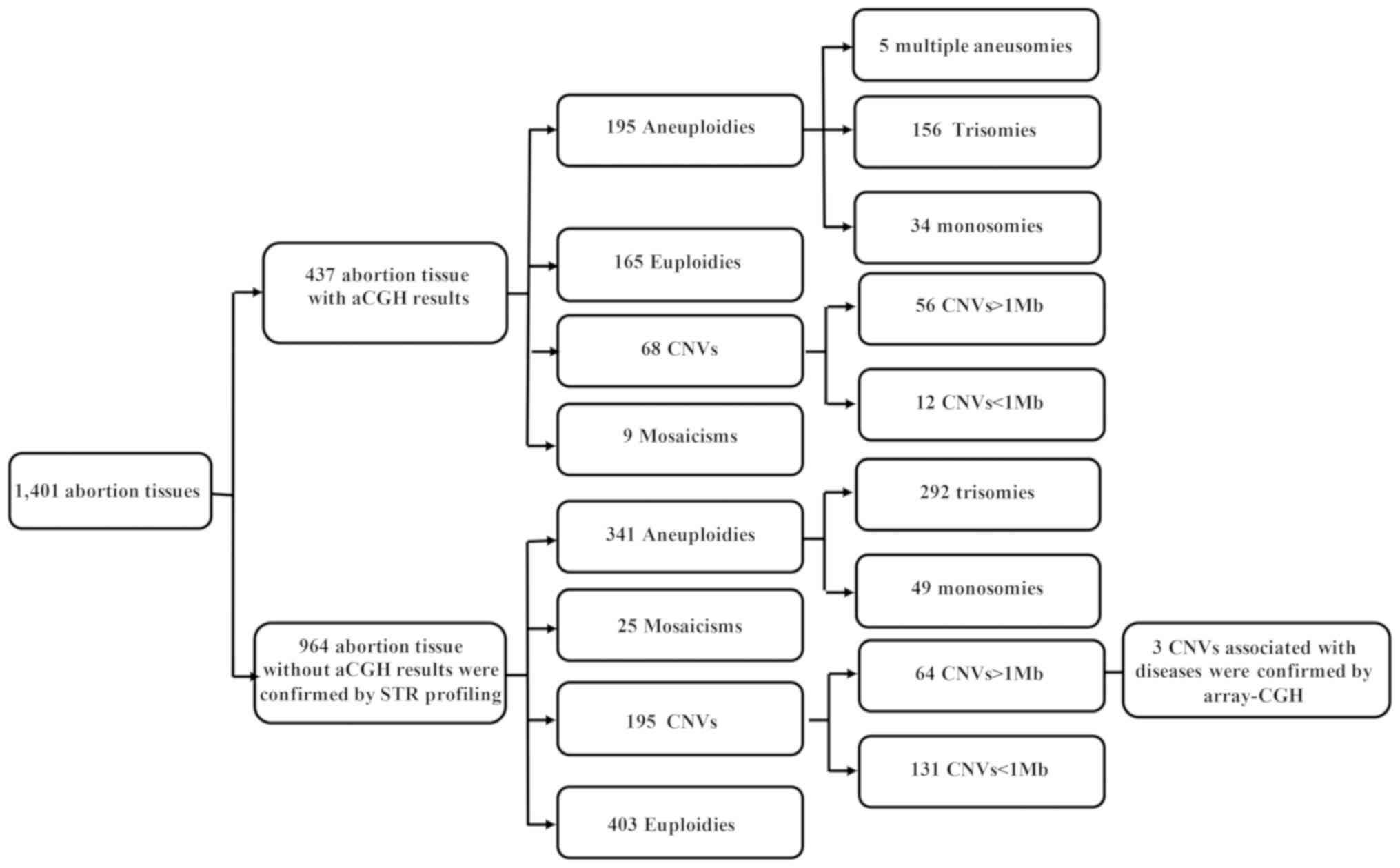
In total, 1,401 patients with SM were enrolled in
the present study and divided into two groups. Group I included 437
samples previously validated by array CGH. Samples in group I were
used to establish a method to detect CAs by semiconductor
sequencing, using a retrospective, case-controlled study design.
Group II, which lacked verified results, comprised 964 samples
tested for clinical significance via a prospective design. Finally,
CNVs with clear clinical significance in group II were verified by
array CGH. The CNV-positive and euploid samples were subjected to
short tandem repeat (STR) profiling to identify polyploidies.
Samples and clinical materials
All samples were obtained under Institutional Review
Board approved protocols with informed consent from all
participants for research use at Nanfang Hospital, Southern Medical
University (approval no. NFEC-2017-050). In total, 437 SM samples
within 20 weeks of gestation with array CGH results, and 964 SM
samples within 20 weeks of gestation but without array CGH results,
were collected between August 2017 and February 2018. The maternal
age range was 18–47 years, with a mean of 30 years. Gestational age
ranged from 5 to 20 weeks, with a mean of 9 weeks and 2 days.
Following collection, SM samples (chorionic or dermal tissue of SM)
were rinsed three times in PBS and then stored in 15 ml centrifuge
tubes (Corning Inc.) at −20°C until use.
DNA extraction and fragmentation
Genomic DNA was extracted from SM samples using the
DNEasy Blood and Tissue kit (Qiagen, GmbH) following the
manufacturer's instructions, and stored at −80°C until use. DNA
quality was evaluated using a NanoDrop™ spectrophotometer (Thermo
Fisher Scientific, Inc.). Genomic DNA was sheared using the M220
instrument (Covaris) and DNA fragments 150–200 bp in length were
purified using Agencourt AMPure XP beads (Beckman Coulter, Inc.),
quantified using the Qubit® 3.0 fluorometer (Thermo
Fisher Scientific, Inc.), and stored at −80°C until use.
DNA library construction and
sequencing
Fragmented DNA samples served as input DNA to
construct a DNA library for sequencing, using an Ion Plus Fragment
Library kit (Thermo Fisher Scientific, Inc.). Agencourt AMPure XP
beads (Beckman Coulter, Inc.) were used for purification during
library construction. The DNA libraries were quantified using
Qubit® 3.0 (Thermo Fisher Scientific, Inc.) and their
size distributions were verified using the Agilent High Sensitivity
DNA kit on a 2100 Bioanalyzer (Agilent Technologies, Inc.). In
total, 15 libraries were pooled and amplified by emulsion PCR using
the Ion OneTouch™ 2 system (Thermo Fisher Scientific, Inc.).
Template-positive ion sphere particles were enriched using the Ion
OneTouch™ ES instrument (Thermo Fisher Scientific, Inc.). The ion
sphere particles were immediately loaded onto the ion semiconductor
chip, which was placed in an Ion Proton instrument (Thermo Fisher
Scientific, Inc.) for sequencing, according to the manufacturer's
instructions (300-cycle workflow).
Data analysis
In total, ~5 million raw reads were obtained per
sample. The mean length of sequencing reads was ~150 bp. The raw
data were aligned to The National Center for Biotechnology
Information GRCh37 human reference genome (https://ftp.ncbi.nlm.nih.gov/genomes/refseq/vertebrate_mammalian/Homo_sapiens/all_assembly_versions),
and duplicates were identified using the Ion Torrent Server V5.4.11
(Torrent Mapping Alignment Program; Thermo Fisher Scientific,
Inc.). Reads that mapped to multiple locations, had duplicate PCR
products, or had a mapping quality score <30 were discarded from
analysis. Overall, ~75% (3.5 million) of the total reads were
unique reads, which were retained. The genome was then partitioned
into 50-kb non-overlapping bins, and raw counts were obtained for
each bin. After binning, regions with high variability or low
‘mappability’ were excluded. To normalize the raw bin count, GC
biases were corrected using Loess regression and principal
component analysis (PCA) to remove higher-order artefacts, then
divided by the total autosomal sequence length count to obtain
genomic representation (GR) values (18). The normalization process was based
on previously reported studies (19,20)
and was as follows: i) Calibrate clean data to 10 million reads and
normalize the read counts in each bin; ii) organize the normalized
read counts and the baseline from 200 normal samples (in-house
database) into a matrix and carry out PCA; iii) construct linear
model by the top 20% principal components and normalized reads
count; and iv) changing the residual error to reduce the effect of
the data variance on the GR value of the test sample.
The GR values of normal samples were determined
using a reference set, which was obtained from 200 healthy men (46,
XY) and women (46, XX) (in-house database). To detect microdeletion
and microduplication syndromes, the GR value of the test samples
was divided by that of the reference set and the ratio was
normalized to that of 10 million reads. The circular binary
segmentation algorithm of the DNAcopy package in R (version 1.36.0;
R Development Core Team) was used to distinguish copy number
regions. The z score of each region was calculated using the
formula: Z score = (region representation - median population)/MAD
population, where region representation corresponds to the GRs of
different copy number regions, the median population is the median
region representation of all samples, and MAD population is the
median absolute deviation of region representations within the
reference set. A negative result was defined as a z score <10
and a positive result as a z score ≥10.
Array CGH validation
High-quality (A260/A280 ratio, 1.8:2.0; A260/A230
ratio, >1.0) DNA was labeled and hybridized to the SurePrint G3
Human CGH Microarray 8×60K, consisting of 60,000 oligonucleotides.
The whole genome was assayed at a backbone resolution of 200 kb.
Slides were then scanned using the Agilent SureScan Microarray
scanner. The images were analyzed using Agilent Genomic Workbench
V7.0 (all from Agilent Technologies, Inc.).
STR profiling validation by
QF-PCR
First, 10–50 ng genomic DNA was amplified using
QF-PCR using a thermal cycler. The thermocycling conditions were:
95°C for 5 min, 95°C for 30 sec, followed by 35 cycles at 58°C for
40 sec, 72°C for 50 sec, and finally 10 min at 72°C in a reaction
volume of 25 µl. The resulting PCR products were subjected to
capillary electrophoretic separation using the ABI3500 Genetic
Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Finally, the data were analyzed using GeneMapper®
Analysis V4.1 software (Applied Biosystems; Thermo Fisher
Scientific; Inc.).
Results
Method development for the
retrospective study
In the retrospective study, a method was developed
based on low-coverage NGS to detect CAs in 437 abortion samples.
The results obtained using this method presented high concordance
with the array CGH results. In total, >1 Mb CNV sequences were
detected, and 3.5 million unique sequencing reads were obtained, at
a lower cost. Of the 437 samples, 272 (62.2%) had abnormal
chromosome numbers, including 195 (44.6%) aneuploidies, of which
156 (80.0%) were trisomies and 34 (17.4%) monosomies. In total, 68
(15.6%) samples were CNVs (size range, 204 kb-147 Mb), among which
56 were >1 Mb and 12 were 0.2–1 Mb in length. In addition, 9
(2.0%) samples were mosaicisms, including five (46, XX/45, X)
cases; one (46, XX/47, XX, +21) case, 46, XX/47, XX, +7 (with 50 Mb
loss), one (47, XXY/46, XY) case and one (46, XY/47, XY, +8) case.
There were 165 (37.8%) euploidy cases (Fig. 1). Furthermore, the most common CA
detected among the SM samples was trisomy. In addition, four double
aneusomies [(48, XY, +12, +15); (48, XY, +9, +22); (48, XX, +3,
+5); (48, XX, +8, +10)] and one case of multiple aneusomy (49, XX,
+13, +14, +21) were detected. Table
I summarizes the diagnostic performance of the present method
for detecting CAs in SM.
 | Table I.Diagnostic performance of NGS and
identification of chromosomal abnormalities in 437 spontaneous
miscarriage samples. |
Table I.
Diagnostic performance of NGS and
identification of chromosomal abnormalities in 437 spontaneous
miscarriage samples.
|
| Sample size, n |
|
|---|
|
|
|
|
|---|
| Abnormality | NGS | Array CGH | CF, % |
|---|
| Trisomy | 156 | 156 | 100.0 |
| Monosomy | 34 | 34 | 100.0 |
| Multiple
aneusomies | 5 | 5 | 100.0 |
| Mosaicism | 9 | 9 | 100.0 |
| CNVs | 68 | 74 |
91.9 |
| Euploidy | 165 | 159 | / |
| Total | 437 | 437 | / |
All semiconductor sequencing platform (SSP) results
agreed with those of array CGH, except for six CNVs <1 Mb, which
did not present CAs according to the SSP results. We searched for
the 12 CNVs 0.2–1 Mb in size detected by array CGH within the
Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home), DECIPHER database
(https://decipher.sanger.ac.uk/index),
and International Standard Cytogenomic Array database (http://dbsearch.clinicalgenome.org/search). All CNVs
were found to be variants of uncertain significance (VOUS, referred
to as CNV without further sub-classification).
Prospective study
For the prospective study, abortion samples were
obtained from 964 patients with SM. Using the present NGS method,
561 (58.2%) abnormal samples were detected, including 341 (35.4%)
aneuploidies, 195 (20.2%) CNVs, 25 (2.6%) mosaicisms, and 403
(41.8%) euploidies (Fig. 1). Of
the 341 aneuploid samples, T16 was the most common abnormality,
followed by T22, T15, T21 and T13. Aneuploidy of chromosomes 1 and
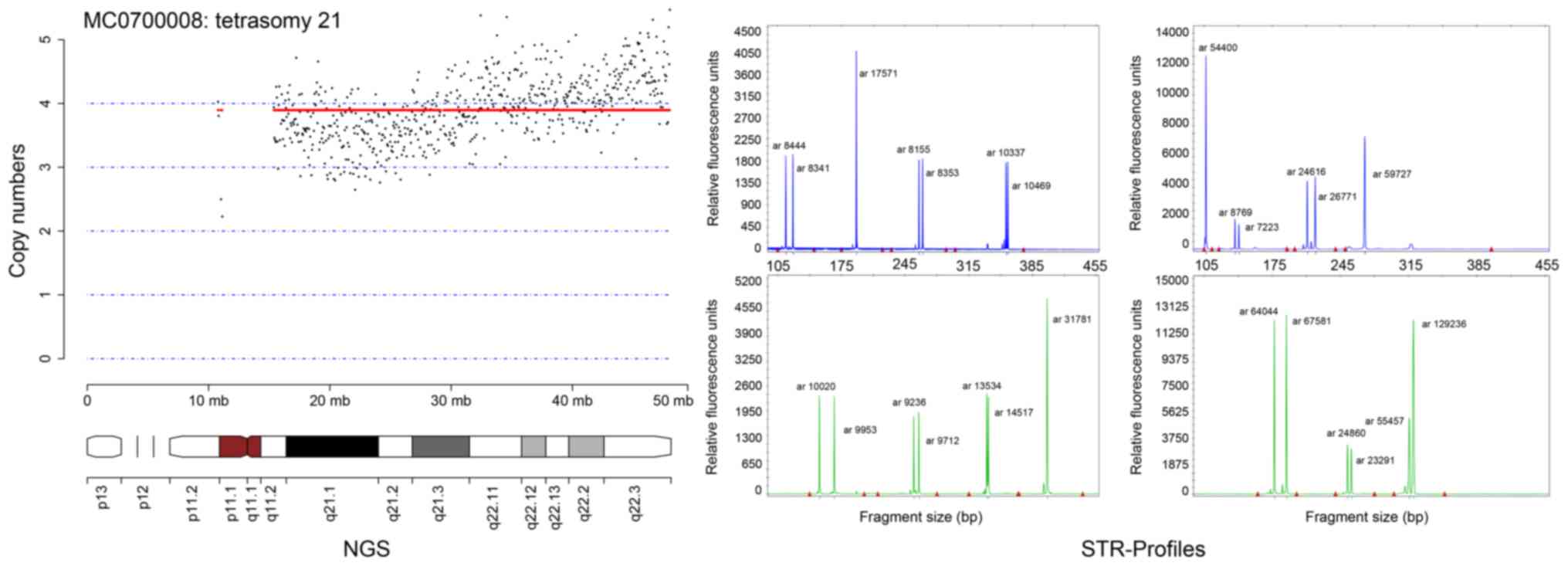
19 was not seen (data not shown). However, one tetrasomy of
chromosome 21 was observed using the NGS method and validated by
STR profiling with QF-PCR (Fig.
2).
Among the 195 CNV cases (size range, 200 kb-96.75
Mb), 64 were >1 Mb and 131 were <1 Mb. Overall, 192 were not
associated with any pathology, thus classified as VOUS. The
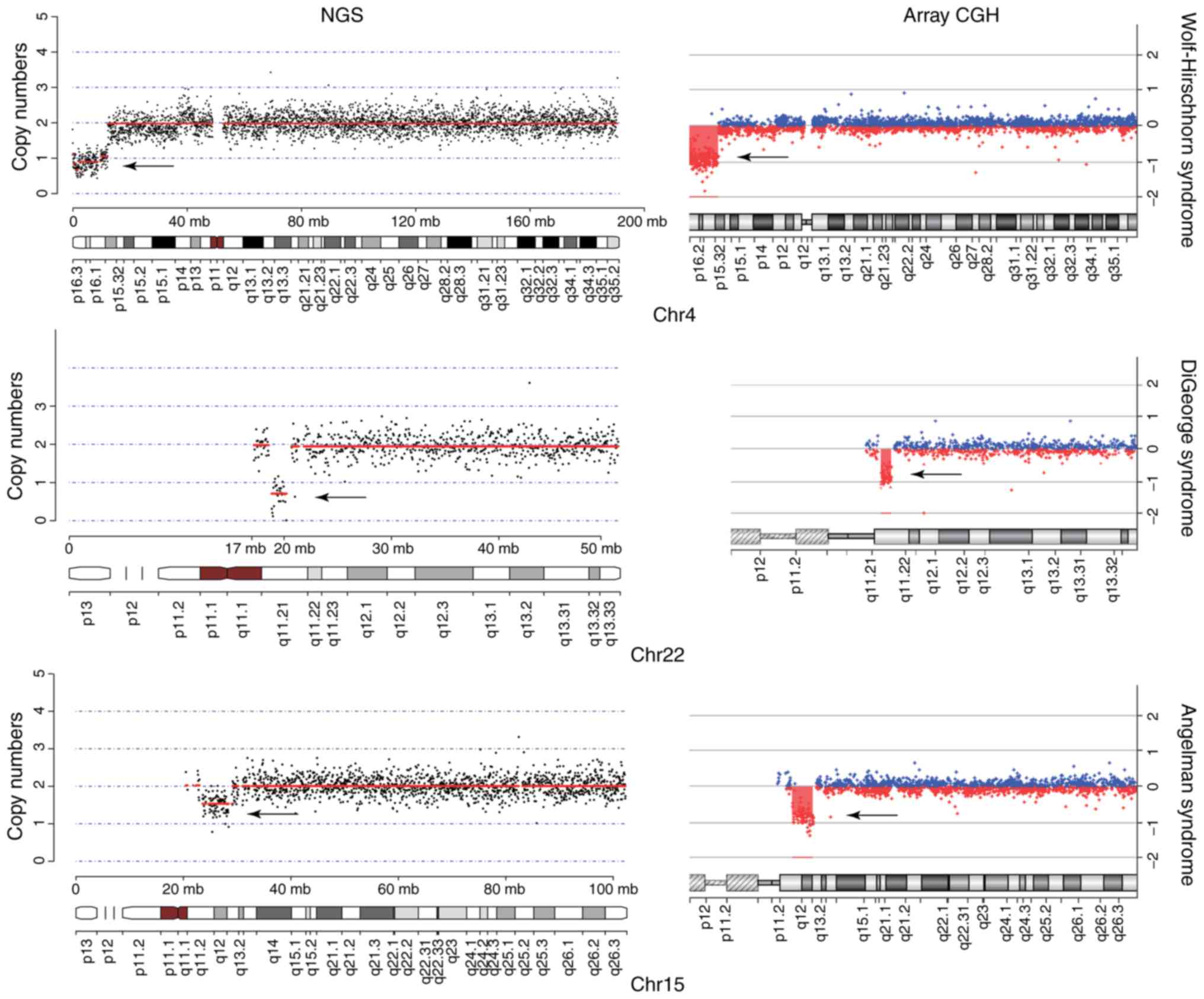
remaining three CNVs comprised a contiguous gene deletion at
4p16.3-p15.33 (9.25 Mb) associated with Wolf-Hirschhorn syndrome
(WHS), a microdeletion in chromosome 22q11.21 (1.35 Mb) associated
with DiGeorge syndrome (DGS), and a loss-of-function gene located
at 15q11.2-q13.1 (5.56 Mb) associated with both Prader-Willi (PW)
and Angelman syndrome (AS). These three CNVs were pathogenic and
confirmed by array CGH (Fig.
3).
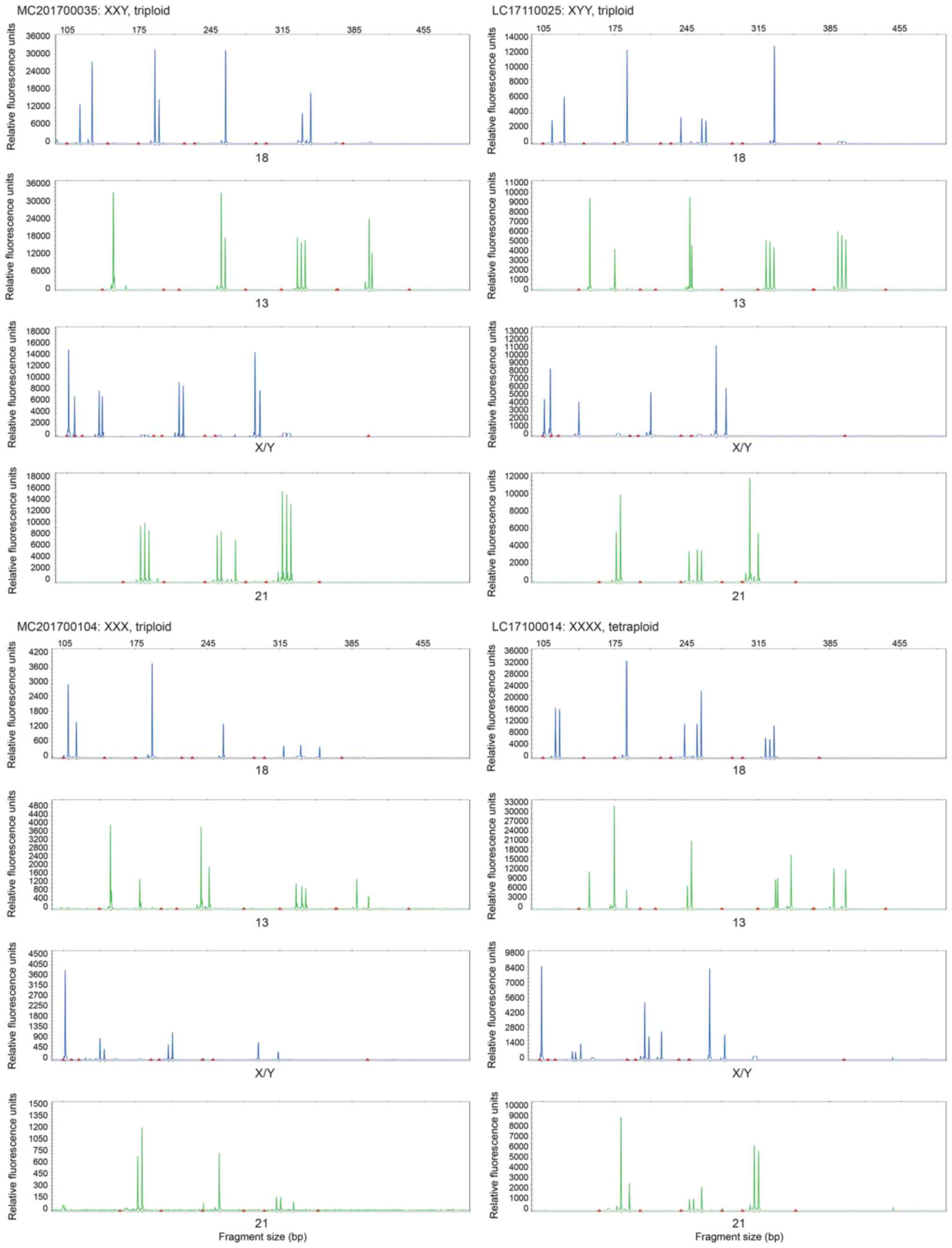
Polyploidy is often observed in SM (21). Since both array CGH and SSPs have a
limited ability to detect polyploidies (17), STR profiling using QF-PCR was
performed in all samples to identify polyploidies. Overall, nine
polyploidies were detected in female euploidy and sex chromosomal
abnormalities, including five cases of (69, XXY) triploidy, two
cases of (69, XXX) triploidy, one case of (69, XYY) triploidy and
one of (92, XXXX) tetraploidy (Fig.
4). These were determined as (47, XXY), (46, XX), (47, XYY) and
(46, XX), respectively, using the CNV analysis method based on
low-coverage NGS and array CGH (data not shown).
Distribution of CAs among all samples. The 1,401
abortion samples carrying a CA comprised 536 (38.3%) aneuploidies,
263 (18.8%) CNVs, 34 (2.4%) mosaicisms and 568 (40.5%) euploidies
(Fig. 1). The most common
aneuploidies were T16 (n=101), (45, X) (n=81), T22 (n=66), T15
(n=36), T21 (n=29) and T13 (n=34) (data not shown). Furthermore,
two monosomy 21 cases, 27 double aneusomies, two multiple
aneusomies, and one tetrasomy 21 case were found.
Discussion
DNA sequencing is widely used in medical research,
and many biological problems can only be solved by sequencing
technologies (22). Due to its
simplicity and rapidity, as well as its high throughput and
resolution, NGS also has numerous applications in the clinical
setting. For instance, NGS is considered to have clinical utility
for the prevention of infectious diseases, noninvasive prenatal
diagnosis, identification and diagnosis of genetic disorders, early
diagnosis and treatment of cancer and pre-implantation (23–26).
In the present study, a novel NGS method was developed for the
detection of CAs in SM samples using an SSP, which could reliably
and accurately diagnose genetic anomalies commonly associated with
CAs. The results suggested that the performance of the present
method was equivalent to that of array-based techniques.
Over the last 10 years, array-based methods, such as
array CGH and SNP microarray, have become the gold standard for
detection of aneuploidies, microdeletions and duplications,
allowing high-resolution (0.1 Mb) chromosomal analysis (27). However, array-based methods require
large amounts of high-quality DNA and are too expensive for
clinical testing of CAs. Compared with these technologies, NGS
possesses clear advantages, including a lower requirement for
nucleic acids, lower cost and high-throughput capability. More
importantly, NGS technology can identify poorly represented DNA
sequences missed by array-based methods. Advances in NGS technology
have led to its application in CNV analysis. Several comparative
studies demonstrated that NGS-based methods were a viable
alternative technology to karyotyping and arrays for CA detection
(1,12,15,28,29).
Previous prospective studies also suggested that NGS-based
approaches are sensitive, reliable and accurate when detecting CAs
in either SM or pre-natal samples (27,30).
Chromosomal abnormalities are the main genetic
causes for SM (3,4). Aneuploidies, polyploidies and CNVs
are the most common type of chromosomal abnormalities (6,7). The
resolution for CNVs detection is different in pre-natal diagnosis
and spontaneous abortion analysis (0.2 and 1 Mb, respectively)
(17,27). In the present study, the resolution
for CNV detection was set at 1 Mb, because: i) It is generally
thought that CNVs >10 Mb are directly associated with SM, 10 Mb
> CNVs > 2 Mb recommend to reference the CNV database
(31,32); ii) the majority of copy-number
polymorphisms are <50 kb (33);
and iii) the purpose of present method was to define cost-effective
approach for clinical settings in the spontaneous abortions
analysis ($100 per sample; 0.25X coverage). A total 437 SM samples
were initially screened in a retrospective study, and the detection
rates were in accordance with those of array CGH, with the
exception of six CNVs <1 Mb that were nonpathogenic repeats. In
addition, the present method unambiguously detected aneuploidies,
CNVs, and mosaicisms in SM samples, as well as CAs involving
several chromosomes. Among the 964 samples analyzed prospectively,
341 aneuploidies were detected, the most common being T16, T22,
T15, T21 and T13. Monosomy X was the most prevalent, which was in
agreement with the findings of previous studies (34,35).
Previous studies indicated that T16, T22, and T15 trisomy in SM
samples were associated with high probabilities of fetal death and
anomalies, preterm delivery, and intrauterine growth retardation
(36). T21, T13 and monosomy X are
often seen in SM during the first trimester (37). Only a small portion of such fetuses
survive to metaphase and advanced-stage pregnancy. If born,
congenital malformations, such as Down's syndrome, Edward's
syndrome and Turner's syndrome, may manifest (30). Aneuploidy of chromosomes 1 and 19
is relatively rare (3,38,39).
In the present study, one case of tetrasomy 21 was detected.
According to the small number of reported cases, tetrasomy 21 is
extremely rare in constitutionally normal patients but is seen
frequently in patients with acute megakaryoblastic leukemia
(40). It was reported that
tetrasomy 21 has an association with physical features consistent
with Down's syndrome (42).
However, of the 195 cases of CNVs in the present study, most were
not associated with any pathological disease, with the exception of
one case associated with WHS, one with DGS, and another associated
with both PW and AS. WHS, DGS and PW or AS can affect some
pregnancies, ultimately causing neonatal defects, such as
developmental delays, skeletal anomalies, as well as cardiac,
neurological and endocrinal abnormalities (43–45).
Neither array CGH nor low-coverage NGS can detect
polyploidy. Therefore, supplementary STR profiling was performed,
which allowed the identification of nine polyploidy cases in female
euploidy and sex CA confirmatory tests. Although rare, polyploidy
can cause miscarriage (45,46).
Altogether, these validation results suggested that comprehensive
experimental results could be achieved by combining STR profiling
with the NGS method described in the present study method, as a
reliable and accurate approach for the diagnosis CAs associated
with miscarriage.
In conclusion, CAs are the most common causes of
abortion in SM, with trisomy being the most frequent, followed by
CNVs and mosaicisms. In the present study, 565 samples were normal
diploids. Early studies have reported that if the cytogenetics were
normal, there was an increased risk of the next pregnancy failing
(47,48). There are many other causes of SM
aside from genetic factors, such as maternal thrombophilic
disorders, immune dysfunction and various endocrine dysregulation
(49,50). Due to the complexity of SM, studies
with larger sample numbers and a variety of detection methods are
needed to improve the diagnostic accuracy of spontaneous
abortions.
Acknowledgements
Not applicable.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant no. 81871177), The
Science and Technology Program of Guangzhou (grant nos.
201604020104, 201803040009 and 201500000004-4), and The Science and
Technology Program of Guangdong (grant nos. 2019B020208009 and
2018A030313286).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XXY designed and supervised the study. FXL and MJX
analyzed and interpreted the data and prepared the manuscript. SFQ
and YSW designed the study, provided technical support and analyzed
the NGS data. FY designed the study and provided samples. DH, LW
and ZKL performed experiments, and collected and analyzed the data.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
All samples were obtained under protocols approved
by the Institutional Review Board at Nanfang Hospital, Southern
Medical University (approval no. NFEC-2017-050). Samples were
collected for research use with informed consent from all
participants.
Patient consent for publication
Not applicable.
Competing interests
The present low-coverage next-generation sequencing
assay for the detection of chromosomal abnormalities in spontaneous
miscarriage has been patented in China (patent no. ZL
201611028711.5; registration date, 18/11/2016; approval date,
14/06/2019).
References
|
1
|
Liu S, Song L, Cram DS, Xiong L, Wang K,
Wu R, Liu J, Deng K, Jia B, Zhong M, et al: Traditional karyotyping
vs copy number variation sequencing for detection of chromosomal
abnormalities associated with spontaneous miscarriage. Ultrasound
Obstet Gynecol. 46:472–477. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van den Berg MM, van Maarle MC, van Wely M
and Goddijn M: Genetics of early miscarriage. Biochim Biophys Acta.
1822:1951–1959. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Daniely M, Aviram-Goldring A, Barkai G and
Goldman B: Detection of chromosomal aberration in fetuses arising
from recurrent spontaneous abortion by comparative genomic
hybridization. Hum Reprod. 13:805–809. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim JW, Lee WS, Yoon TK, Seok HH, Cho JH,
Kim YS, Lyu SW and Shim SH: Chromosomal abnormalities in
spontaneous abortion after assisted reproductive treatment. BMC Med
Genet. 11:1532010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lomax B, Tang S, Separovic E, Phillips D,
Hillard E, Thomson T and Kalousek DK: Comparative genomic
hybridization in combination with flow cytometry improves results
of cytogenetic analysis of spontaneous abortions. Am J Hum Genet.
66:1516–1521. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lathi RB, Gustin SL, Keller J,
Maisenbacher MK, Sigurjonsson S, Tao R and Demko Z: Reliability of
46,XX results on miscarriage specimens: A review of 1,222
first-trimester miscarriage specimens. Fertil Steril. 101:178–182.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schaeffer AJ, Chung J, Heretis K, Wong A,
Ledbetter DH and Lese Martin C: Comparative genomic
hybridization-array analysis enhances the detection of aneuploidies
and submicroscopic imbalances in spontaneous miscarriages. Am J Hum
Genet. 74:1168–1174. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kimberly L, Case A, Cheung AP, Sierra S,
AlAsiri S, Carranza-Mamane B, Case A, Dwyer C, Graham J, Havelock
J, et al: Advanced reproductive age and fertility: No. 269,
November 2011. Int J Gynaecol Obstet. 117:95–102. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shen J, Wu W, Gao C, Ochin H, Qu D, Xie J,
Gao L, Zhou Y, Cui Y and Liu J: Chromosomal copy number analysis on
chorionic villus samples from early spontaneous miscarriages by
high throughput genetic technology. Mol Cytogenet. 9:72016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bagheri H, Mercier E, Qiao Y, Stephenson
MD and Rajcan-Separovic E: Genomic characteristics of miscarriage
copy number variants. Mol Hum Reprod. 21:655–661. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Foyouzi N, Cedars MI and Huddleston HG:
Cost-effectiveness of cytogenetic evaluation of products of
conception in the patient with a second pregnancy loss. Fertil
Steril. 98:151–155. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liang D, Peng Y, Lv W, Deng L, Zhang Y, Li
H, Yang P, Zhang J, Song Z, Xu G, et al: Copy number variation
sequencing for comprehensive diagnosis of chromosome disease
syndromes. J Mol Diagn. 16:519–526. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kearney HM, South ST, Wolff DJ, Lamb A,
Hamosh A and Rao KW; Working Group of the American College of
Medical Genetics, : American College of Medical Genetics
recommendations for the design and performance expectations for
clinical genomic copy number microarrays intended for use in the
postnatal setting for detection of constitutional abnormalities.
Genet Med. 13:676–679. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shaffer LG and Rosenfeld JA:
Microarray-based prenatal diagnosis for the identification of fetal
chromosome abnormalities. Expert Rev Mol Diagn. 13:601–611. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang MZ, Lin FQ, Li M, He D, Yu QH, Yang
XX and Wu YS: Semiconductor Sequencing Analysis of Chromosomal Copy
Number Variations in Spontaneous Miscarriage. Med Sci Monit.
23:5550–5557. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wen J, Hanna CW, Martell S, Leung PC,
Lewis SM, Robinson WP, Stephenson MD and Rajcan-Separovic E:
Functional consequences of copy number variants in miscarriage. Mol
Cytogenet. 8:62015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang H, Dong Z, Zhang R, Chau MHK, Yang Z,
Tsang KYC, Wong HK, Gui B, Meng Z, Xiao K, et al: Low-pass genome
sequencing versus chromosomal microarray analysis: Implementation
in prenatal diagnosis. Genet Med. 22:500–510. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chiu RW, Chan KC, Gao Y, Lau VY, Zheng W,
Leung TY, Foo CH, Xie B, Tsui NB, Lun FM, et al: Noninvasive
prenatal diagnosis of fetal chromosomal aneuploidy by massively
parallel genomic sequencing of DNA in maternal plasma. Proc Natl
Acad Sci USA. 105:20458–20463. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li X, Chen S, Xie W, Vogel I, Choy KW,
Chen F, Christensen R, Zhang C, Ge H, Jiang H, et al: PSCC:
Sensitive and reliable population-scale copy number variation
detection method based on low coverage sequencing. PLoS One.
9:e850962014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Szatkiewicz JP, Wang W, Sullivan PF, Wang
W and Sun W: Improving detection of copy-number variation by
simultaneous bias correction and read-depth segmentation. Nucleic
Acids Res. 41:1519–1532. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Menasha J, Levy B, Hirschhorn K and Kardon
NB: Incidence and spectrum of chromosome abnormalities in
spontaneous abortions: New insights from a 12-year study. Genet
Med. 7:251–263. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Levy SE and Myers RM: Advancements in
Next-Generation Sequencing. Annu Rev Genomics Hum Genet. 17:95–115.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gu W, Miller S and Chiu CY: Clinical
Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu
Rev Pathol. 14:319–338. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bianchi DW, Parker RL, Wentworth J,
Madankumar R, Saffer C, Das AF, Craig JA, Chudova DI, Devers PL,
Jones KW, et al CARE Study Group, : DNA sequencing versus standard
prenatal aneuploidy screening. N Engl J Med. 370:799–808. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang J, Li J, Saucier JB, Feng Y, Jiang
Y, Sinson J, McCombs AK, Schmitt ES, Peacock S, Chen S, et al:
Non-invasive prenatal sequencing for multiple Mendelian monogenic
disorders using circulating cell-free fetal DNA. Nat Med.
25:439–447. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen M and Zhao H: Next-generation
sequencing in liquid biopsy: Cancer screening and early detection.
Hum Genomics. 13:342019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dong Z, Zhang J, Hu P, Chen H, Xu J, Tian
Q, Meng L, Ye Y, Wang J, Zhang M, et al: Low-pass whole-genome
sequencing in clinical cytogenetics: A validated approach. Genet
Med. 18:940–948. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Duan J, Zhang JG, Deng HW and Wang YP:
Comparative studies of copy number variation detection methods for
next-generation sequencing technologies. PLoS One. 8:e591282013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luthra R, Chen H, Roy-Chowdhuri S and
Singh RR: Next-Generation Sequencing in Clinical Molecular
Diagnostics of Cancer: Advantages and Challenges. Cancers (Basel).
7:2023–2036. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang J, Chen L, Zhou C, Wang L, Xie H,
Xiao Y, Zhu H, Hu T, Zhang Z, Zhu Q, et al: Prospective chromosome
analysis of 3429 amniocentesis samples in China using copy number
variation sequencing. Am J Obstet Gynecol. 219:287.e1–287.e18.
2018. View Article : Google Scholar
|
|
31
|
South ST, Lee C, Lamb AN, Higgins AW and
Kearney HM; Working Group for the American College of Medical
Genetics and Genomics Laboratory Quality Assurance Committee, :
ACMG Standards and Guidelines for constitutional cytogenomic
microarray analysis, including postnatal and prenatal applications:
Revision 2013. Genet Med. 15:901–909. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kearney HM, Thorland EC, Brown KK,
Quintero-Rivera F and South ST; Working Group of the American
College of Medical Genetics Laboratory Quality Assurance Committee,
: American College of Medical Genetics standards and guidelines for
interpretation and reporting of postnatal constitutional copy
number variants. Genet Med. 13:680–685. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
33. Brothman AR, Dolan MM, Goodman BK,
Park JP, Persons DL, Saxe DF, Tepperberg JH, Tsuchiya KD, Van Dyke
DL, Wilson KS, et al: College of American Pathologists/American
College of Medical Genetics proficiency testing for constitutional
cytogenomic microarray analysis. Genet Med. 13:765–769. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Du Y, Chen L, Lin J, Zhu J, Zhang N, Qiu
X, Li D and Wang L: Chromosomal karyotype in chorionic villi of
recurrent spontaneous abortion patients. Biosci Trends. 12:32–39.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yuan SM, Liao C, Li DZ, Huang JZ, Hu SY,
Ke M, Zhong HZ and Yi CX: Chorionic villus cell culture and
karyotype analysis in 1,983 cases of spontaneous miscarriage.
Zhonghua Fu Chan Ke Za Zhi. 52:461–466. 2017.(In Chinese).
PubMed/NCBI
|
|
36
|
Benn P: Trisomy 16 and trisomy 16
Mosaicism: A review. Am J Med Genet. 79:121–133. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wapner RJ, Martin CL, Levy B, Ballif BC,
Eng CM, Zachary JM, Savage M, Platt LD, Saltzman D, Grobman WA, et
al: Chromosomal microarray versus karyotyping for prenatal
diagnosis. N Engl J Med. 367:2175–2184. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hanna JS, Shires P and Matile G: Trisomy 1
in a clinically recognized pregnancy. Am J Med Genet. 68:981997.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dunn TM, Grunfeld L and Kardon NB: Trisomy
1 in a clinically recognized IVF pregnancy. Am J Med Genet.
99:152–153. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ohsaka A, Hisa T, Watanabe N, Kojima H and
Nagasawa T: Tetrasomy 21 as a sole chromosome abnormality in acute
myeloid leukemia. fluorescence in situ hybridization and spectral
karyotyping analyses. Cancer Genet Cytogenet. 134:60–64. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Slavotinek AM, Chen XN, Jackson A, Gaunt
L, Campbell A, Clayton-Smith J and Korenberg JR: Partial tetrasomy
21 in a male infant. J Med Genet. 37:E302000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Alkan G, Emiroglu MK and Kartal A:
DiGeorge Syndrome with Sacral Myelomeningocele and Epilepsy. J
Pediatr Neurosci. 12:344–345. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Siew JX and Yap F: Growth trajectory and
pubertal tempo from birth till final height in a girl with
Wolf-Hirschhorn syndrome. Endocrinol Diabetes Metab Case Rep.
2018:18–0001. 2018.PubMed/NCBI
|
|
44
|
Warner ME, Martin DP, Warner MA, Gavrilova
RH, Sprung J and Weingarten TN: Anesthetic Considerations for
Angelman Syndrome: Case Series and Review of the Literature. Anesth
Pain Med. 7:e578262017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hyde KJ and Schust DJ: Genetic
considerations in recurrent pregnancy loss. Cold Spring Harb
Perspect Med. 5:a0231192015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rull K, Nagirnaja L and Laan M: Genetics
of recurrent miscarriage: Challenges, current knowledge, future
directions. Front Genet. 3:342012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ogasawara M, Aoki K, Okada S and Suzumori
K: Embryonic karyotype of abortuses in relation to the number of
previous miscarriages. Fertil Steril. 73:300–304. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sullivan AE, Silver RM, LaCoursiere DY,
Porter TF and Branch DW: Recurrent fetal aneuploidy and recurrent
miscarriage. Obstet Gynecol. 104:784–788. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Larsen EC, Christiansen OB, Kolte AM and
Macklon N: New insights into mechanisms behind miscarriage. BMC
Med. 11:1542013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sugiura-Ogasawara M, Ozaki Y, Katano K,
Suzumori N, Kitaori T and Mizutani E: Abnormal embryonic karyotype
is the most frequent cause of recurrent miscarriage. Hum Reprod.
27:2297–2303. 2012. View Article : Google Scholar : PubMed/NCBI
|