Introduction
Autoimmune polyendocrine syndrome type 1 (APS-1;
OMIM 240300), also known as autoimmune
polyendocrinopathy-candidiasis-ectodermal dystrophy, is a rare
disease, characterized by chronic mucocutaneous candidiasis (CMC),
primary adrenocortical insufficiency (PAI) and hypoparathyroidism
(HP) (1). APS-1 may present with
other abnormities, such as primary ovarian insufficiency, vitiligo,
alopecia, type 1 diabetes mellitus (T1D), chronic intestinal
dysfunction, enamel hypoplasia, and autoimmune hepatitis (2,3).
Pure red cell aplasia (PRCA), metaphyseal dysplasia, retinitis and
polyarthritis have also been reported in patients with APS-1
(4,5).
APS-1 is primarily caused by variants in the
autoimmune regulator gene (AIRE), which is located on
chromosome 21q22.3 (6).
AIRE contains 14 exons and encodes a 57-kDa transcription
regulator of 545 amino acids (7,8).
AIRE is primarily expressed in medullary thymic epithelial cells,
which serve key roles in maintaining central immunological
tolerance via promoting the negative selection of autoreactive
thymocytes and building the thymic microarchitecture (6). AIRE is also distributed in the
periphery of antigen-presenting cells (9). If the functions of AIRE are impaired,
autoreactive T cells can escape into the periphery, resulting in a
number of autoimmune abnormities such as autoimmune attack against
parathyroid and adrenal glands in APS-1 (6).
APS-1 is relatively prevalent in Iranian Jews and
Sardinians, with a prevalence of 1:145,000 to 1:9,000 (10–12).
Over 100 different variants in AIRE gene have been found in
patients with APS-1, including substitutions, insertions, deletions
and splice site variants [BIOBASE Human Gene Mutation Database
(HGMD) professional 2019.1; hgmd.cf.ac.uk/ac/index.php]. To the
best of our knowledge, however, there are very few reports of APS-1
in Chinese patients, and its pathological mechanism has not been
fully elucidated.
The present study investigated the phenotype and
genotype of a Chinese woman with non-classical APS-1.
Characteristics of previously reported Chinese patients with APS-1
induced by AIRE gene variants were also summarized.
Materials and methods
Subjects
A 32-year-old woman of Chinese Han origin from a
non-consanguineous family was recruited following admission to the
Department of Endocrinology, Peking Union Medical College Hospital
(PUMCH; Beijing, China) in 2018, with suspected APS-1 based on the
clinical features of syncope, tetany, primary amenorrhea,
intermittent diarrhea and general fatigue.
Phenotypic evaluation
The functions of multiple endocrine glands and
examined levels of numerous autoantibodies were evaluated. Serum
levels of calcium, phosphorus, creatinine (Cr) and alanine
aminotransferase (ALT), and 24-h urinary concentrations of calcium
(24hUCa) and phosphorus (24hUP) were assessed via an AU5800 Beckman
Automatic Biochemical Analyzer (Beckman Coulter, Inc.). Serum
levels of parathyroid hormone (PTH), follicle-stimulating hormone
(FSH), luteinizing hormone (LH) and estradiol, as well as
serum-free thyroxine (FT4), free triiodothyronine (FT3),
thyroid-stimulating hormone (TSH) and autoantibodies to thyroid
peroxidase antigen (TPOAb) and thyroglobulin (TGAb) were measured
using an automated electrochemiluminescence system (Roche
Diagnostics). Blood samples measured for serum cortisol and plasma
adrenocorticotropic hormone (ACTH) were collected at 8 AM and
measured via fluorescent immunoassay (Diagnostic Products). Serum
autoantibodies to islet cells antibodies (ICA) and glutamic acid
decarboxylase antibodies (GADA) were measured using
radioimmunoassays kits (Cisbio; Perkin Elmer, Inc.). Serum
autoantibodies to antinuclear antibody (ANA), 52 kDa Ro/SS-A
molecule (RO-52), as well as anti-neutrophil cytoplasmic antibodies
(ANCA) were tested using an indirect immunofluorescence kit (ANA
screen 11; cat. no. EA 1590-9601-11 G; Euroimmun AG). The blood
count assays were measured using a Sysmex XE-5000 hematology
analyzer (Sysmex Corporation). Serum levels of erythropoietin (EPO;
cat. no. DEP00) and antibodies to IFN-ω, IFN-α (cat. no. 21100-1),
IL-17A (cat. no. MAB317-100), IL-17F (cat. no. MAB13353-100) and
IL-22 (cat. no. MAB7822-100) were detected using ELISA kits
(R&D Systems Inc.).
Morphology of thyroid, abdomen, uterus and ovary
were assessed via standard ultrasound scanning (Siemens
Healthineers). Calcification in the central nervous system was
evaluated via CT scan.
The diagnosis of hypoparathyroidism was determined
by hypocalcemia, hyperphosphatemia and decreased intact PTH level.
The diagnosis of primary ovarian insufficiency was based on
amenorrhea, absent pubertal development and elevated serum levels
of FSH or LH. The diagnosis of PRCA was based on normal marrow in
the absence of erythroblasts (<1% erythroblasts on the
differential count of bone marrow cells).
Identification of pathogenic
variants
DNA was harvested from peripheral leucocytes using
the DNA Extraction kit (QIAamp DNA; Qiagen GmbH) according to the
manufacturer's instructions. Targeted next-generation sequencing
(NGS) was used to identify the pathogenic variants. A panel
containing >700 genes of bone disease was created to capture all
exons sequences of the candidate genes of APS-1 and
hypoparathyroidism (such as AIRE, T-box transcription factor
1, tubulin-specific chaperone E and family with sequence similarity
111 member A), as previously described (13). Bioinformatic analysis was performed
as previously described (13).
Fragments from the patient, her parents and 500
unrelated healthy controls covering the variant sites in
AIRE identified by NGS were amplified via PCR. Primer
sequences were as follows: Exon 3: Forward
5′-ACCCTACCCCTGGAGAAAAC-3′ and reverse 5′-TGGTCCAGTGTGTGGGTCTA-3′;
and exon 5: forward, 5′-GCCTGCTTCTGGCATAGAGT-3′ and reverse,
5′-AACAGTCCGGTCTGCTGTG-3′.
PCR was performed using TaqMix DNA polymerase
(Biomed) under the following conditions: 95°C for 3 min, followed
by 35 cycles of 95°C for 30 sec, annealing at 60°C for 30 sec, and
72°C for 1 min, followed by a final extension at 72°C for 10 min.
Products of PCR were purified and sequenced on the ABI377 DNA
automated sequencer using Big Dye Terminators Cycle Sequencing
Ready Reaction kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions.
In order to predict the potential deleterious
effects of the variants, PolyPhen-2 (Polymorphism Phenotyping v2;
genetics.bwh.harvard.edu/pph2/), SIFT
(sift.jcvi.org/) and MutationTaster (mutationtaster.org/) were utilized. The conservation
of the amino acid substitution position among species was analyzed
via UniProt software (uniprot.org).
A literature review of reported Chinese patients
with APS-1 exhibiting AIRE variants was performed and
clinical and genetic characteristics were summarized (7,14–17).
Three-dimensional modeling of
AIRE
In order to investigate the change in protein
induced by the pathogenic variant in AIRE, the
three-dimensional structure of the AIRE protein was constructed via
SWISS-MODEL (swissmodel.expasy.org). Subsequently, the
disease-associated p.Gly208Val substitution of AIRE protein was
built using the mutagenesis module of PyMoL software (v2.3.1;
pymol.org/).
The present study was approved by the Scientific
Ethics Committee of PUMCH (approval no. JS-2081). A total of 500
unrelated healthy individuals were recruited as controls for
genetic study. Written informed consent was provided by all
subjects prior to participation. The pedigree of the family of the
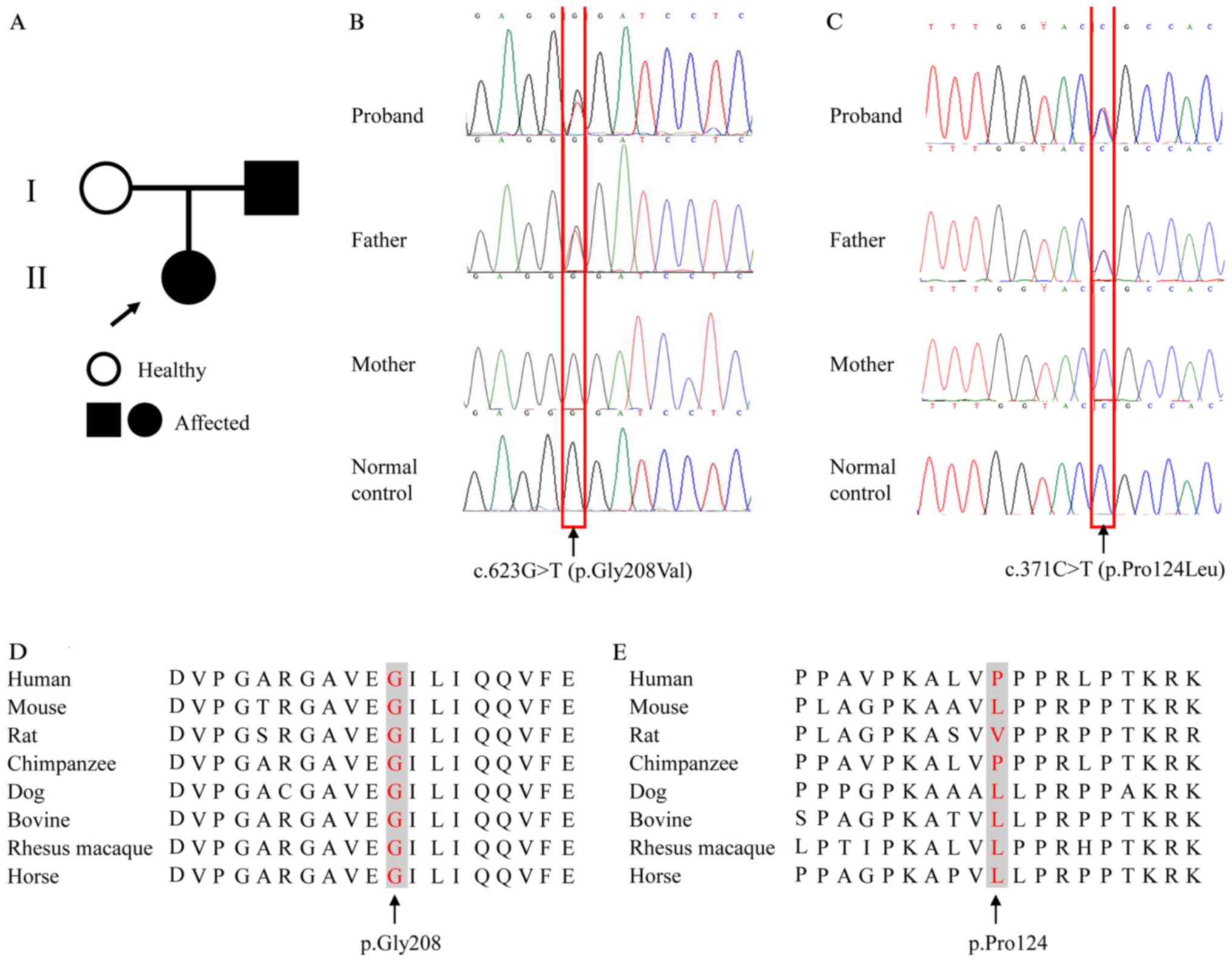
patient is shown in Fig. 1A.
Results
Phenotypes of the patient
The patient, a 32-year-old woman (height, 158.0 cm;
weight, 52.5 kg; body mass index, 21.0 kg/m2), came from
a Chinese non-consanguineous family with no history of autoimmune
disease. At 7 years of age, she presented with tetany and
hypocalcemia. She was supplemented with Calcichew D (500 mg
elemental calcium, 200 IU vitamin D per tablet; GE Pharmaceutical
Shanghai Co., Ltd.) three times daily and serum calcium levels were
not monitored. At 17 years of age, she suffered from syncope and a
generalized tonic-clonic seizure and was admitted to PUMCH for the
first time. She was diagnosed with hypoparathyroidism based on PTH
deficiency (PTH<1.0 pg/ml), hypocalcemia (serum total calcium,
1.35 mmol/l) and hyperphosphatemia (serum phosphorus, 1.91 mmol/l).
CT scans revealed bilateral calcifications in the basal ganglia of
the brain. One year later, she was admitted to the Department of
Gynecology with primary amenorrhea and was diagnosed as having
primary ovarian insufficiency, according to low estradiol (6.74
pg/ml), increased LH (33.7 mIU/ml) and FSH (82.6 mIU/ml) levels, as
well as uterine hypoplasia in ultrasonography. At 31 years of age,
she experienced intermittent diarrhea and general fatigue. Whole
blood cell count revealed a notably decreased hemoglobin level (6.1
g/dl), with normal platelet count (PLT, 306,000/µl) and white blood
cells (WBCs, 874,000/µl). The bone marrow aspirate showed
normocellularity, a lack of erythroid precursors (1.0% of bone
marrow nucleated cells), normal granulocyte precursors and
megakaryocytes with lymphocytosis (30.5%). In addition, serum level
of erythropoietin was notably elevated (>776.00 mIU/ml). The
patient was diagnosed with PRCA. Gastrointestinal endoscopy and
histological analysis of the biopsies revealed chronic atrophic
gastritis and gastric ulcers, with normal appearance of the
duodenal mucosa. Helicobacter pylori was not present in the
gastric mucosa. The abdominal ultrasound was normal. Serum
cortisol, plasma ACTH, serum FT4, FT3 and TSH levels were within
the normal range. For autoantibodies, RO-52, GADA and antibodies to
IFN-ω and IFN-α were positive, whereas ANA, ANCA, TGAb, TPOAb, ICA
and antibodies to IL-17A, IL-17F and IL-22 were negative.
Combined with clinical manifestations, the patient
was suspected of having APS-1. The clinical manifestations of the
patient are listed in Table I.
 | Table I.Clinical presentation and laboratory
results of a patient with APS-1. |
Table I.
Clinical presentation and laboratory
results of a patient with APS-1.
| Age, years | Clinical
manifestation | Laboratory
results | Reference
range |
|---|
| 7 | Tetany | NA | / |
| 17 | Syncope | Serum total
calcium, 1.350 mmol/l | 2.13–2.70
mmol/l |
|
| Generalized
tonic-clonic seizure | Serum phosphorus,
1.910 mmol/l | 0.81–1.45
mmol/l |
|
|
| Serum parathyroid
hormone, <1.000 pg/ml | 7.00–53.00
pg/ml |
| 18 | Amenorrhea | Serum estradiol,
6.740 pg/ml | 50.00–154.40
pg/ml |
|
|
| Serum luteinizing
hormone, 33.700 mIU/ml | 4.40–7.10
mIU/ml |
|
|
| Serum
follicle-stimulating hormone, 82.600 mIU/ml | 5.10–7.00
mIU/ml |
|
|
| Ultrasonography,
uterine hypoplasia |
|
| 31 | Intermittent
diarrhea | Hemoglobin, 6.100
g/dl | 11.00–15.00
g/dl |
|
| General
fatigue | Hematocrit,
19.000% | 35.00–50.00% |
|
|
| Platelets,
306,000.000/µl |
100,000.00–350,000.00/µl |
|
|
| White blood cells,
874,000.000/µl |
350,000.00–950,000.00/µl |
|
|
| Plasma
adrenocorticotropic hormone, 27.000 pg/ml | 0.00–46.00
pg/ml |
|
|
| Serum cortisol,
19.430 µg/dl | 4.00–22.30
µg/dl |
|
|
| Serum free
thyroxine, 1.100 ng/dl | 0.81–1.89
ng/dl |
|
|
| Serum free
triiodothyronine, 3.370 pg/ml | 1.80–4.10 g/ml |
|
|
| Serum
thyroid-stimulating hormone, 2.306 µIU/ml | 0.38–4.34
µIU/ml |
|
|
| Thyroglobulin
antibodies, 12.270 IU/ml | <115.00
IU/ml |
|
|
| Thyroperoxidase
antibodies, 10.910 IU/ml | <34.00
IU/ml |
|
|
| Erythropoietin,
>776.000 mIU/ml | 4.5.0–31.88
mIU/ml |
|
|
| Bone marrow
aspirate, normocelluiarity, lack of erythroid precursors |
|
|
|
| IFN-ω Abs/IFN-α
Abs/RO-52/GADA, Positive | GADA, <10.00
IU/ml |
|
|
| IL-17A Abs/IL-17F
Abs/IL-22 | IFN-ω
Abs/IFN-α |
|
|
|
Abs/ICA/IA-2/ANA/ANCA, Negative |
Abs/RO-52/IL-17A |
|
|
|
| Abs/IL-17F
Abs/IL-22 |
|
|
|
|
Abs/ICA/IA-2/ANA/ANCA, |
|
|
|
| Negative |
The parents did not display symptoms or signs of
APS-1; therefore, they were reluctant to receive further
biochemical and autoantibody profile examinations.
Treatment and follow-up
The patient was prescribed 3 tablets of calcium
carbonate (200 mg elemental calcium per tablet) and one capsule of
calcitriol (0.25 µg per capsule) three times daily to manage
hypoparathyroidism. For primary ovarian insufficiency, she received
a 21-day sequential therapy with Climen (2 mg estradiol valerate,
days 1–21 with 1 mg cyproterone acetate, days 12–21; Bayer, AG),
followed by a 7-day treatment-free interval. The patient received a
transfusion of packed red blood cells, and then 75 mg cyclosporine
A was given twice daily. As the serum ALT level increased to 304
U/l one month later, she switched to methylprednisolone (4 mg per
tablet). The therapeutic dosage was adjusted according to serum
levels of calcium, phosphate, 24-h urinary calcium excretion and
level of hemoglobin.
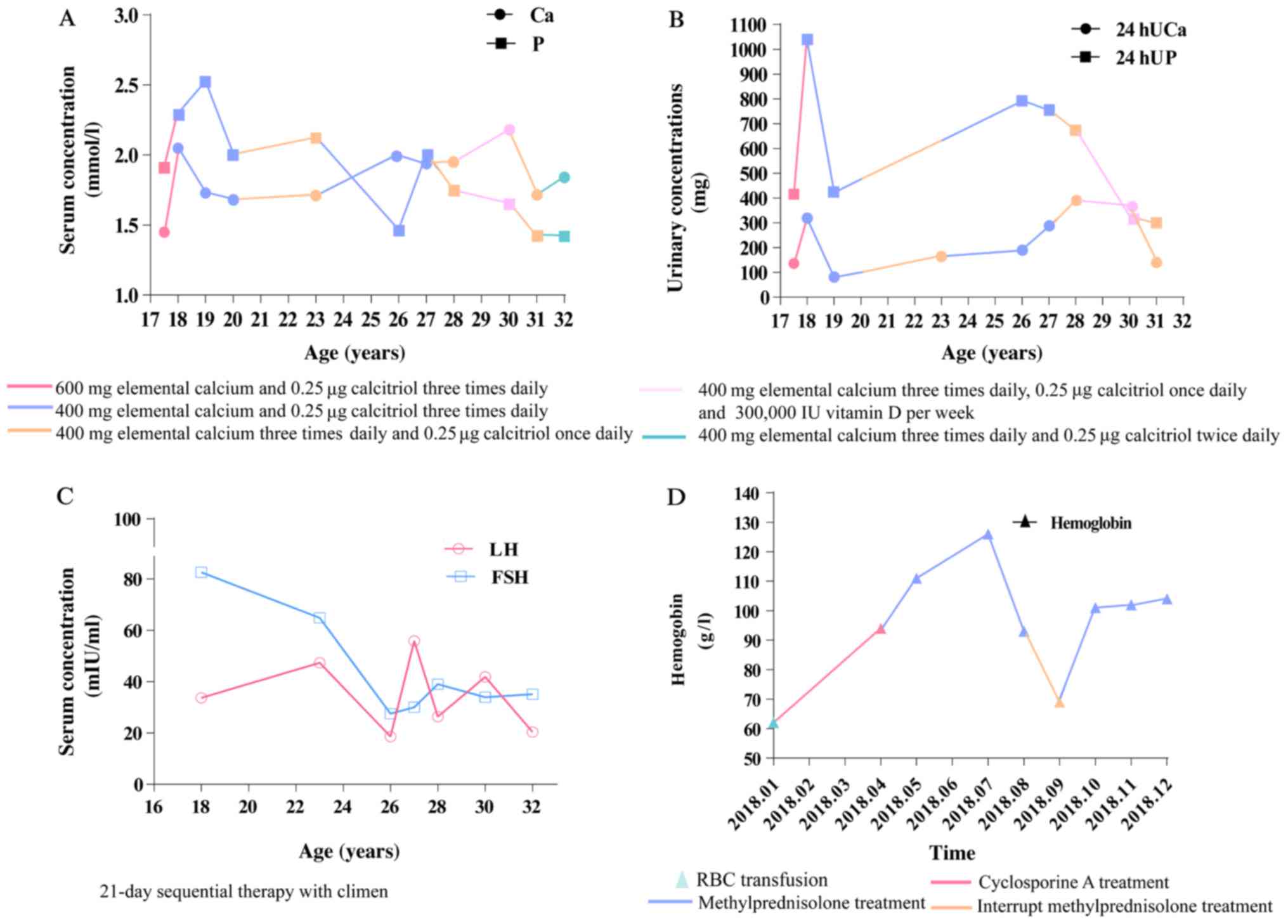
During the treatment, the frequency of tetany was
notably decreased, and the patient experienced regular
menstruation. On the final examination, serum calcium level
increased to 1.84 mmol/l and inorganic phosphate decreased to 1.42
mmol/l (Fig. 2A). Urinary calcium
and phosphate levels were monitored (Fig. 2B). Decreased LH and FSH were also
observed (Fig. 2C). The hemoglobin
level increased to 10.2 g/dl (Fig.
2D).
Variants in AIRE
A heterozygous single-base-pair variation in exon 5
(NM_000383.2: c.623G>T) of AIRE was identified, causing
an amino acid change of glycine to valine at position 208
(NP_000374.1: p.Gly208Val) (Fig.
1B). This variant was predicted to be pathogenic, with scores
of 1.00 by PolyPhen-2 and 0.00 by SIFT. The present study also
identified a sequence alteration (NM_000383.2: c.371C>T) in exon
3 of AIRE, which caused substitution of proline to leucine
at position 124 (NP_000374.1: p.Pro124Leu) (Fig. 1C). This variant was predicted to be
benign, with scores of 0.00 by PolyPhen-2 and 1.00 by SIFT.
Moreover, the affected amino acid residue, p.Gly208, was conserved
among different species, indicating the functional importance of
this amino acid residue throughout evolution (Fig. 1D). The 124th amino acid of the AIRE
protein varied between species, indicating that this residue may be
less important for the function and structure of the protein
(Fig. 1E).
Both sequence alterations were present in the
patient's father but were absent in the mother and the 500
unaffected controls (Fig. 1B and
C). Searching the HGMD (hgmd.cf.ac.uk/ac/index.php), Leiden
Open Variation Database 3.0 (lovd.nl/3.0/home), PubMed database,
the 1000 Genomes Project (browser.1000genomes.org) and Exome Aggregation
Consortium database (exac.broadinstitute.org/) indicated that c.623G>T
was a novel variant.
The change in the 3D structure of AIRE protein
induced by the pathogenic variant present in the patient is
presented in Fig. 3A. The glycine
to valine substitution at position 208 changes the structure of
Sp100, AIRE-1, NucP41/75, DEAF-1 (SAND).
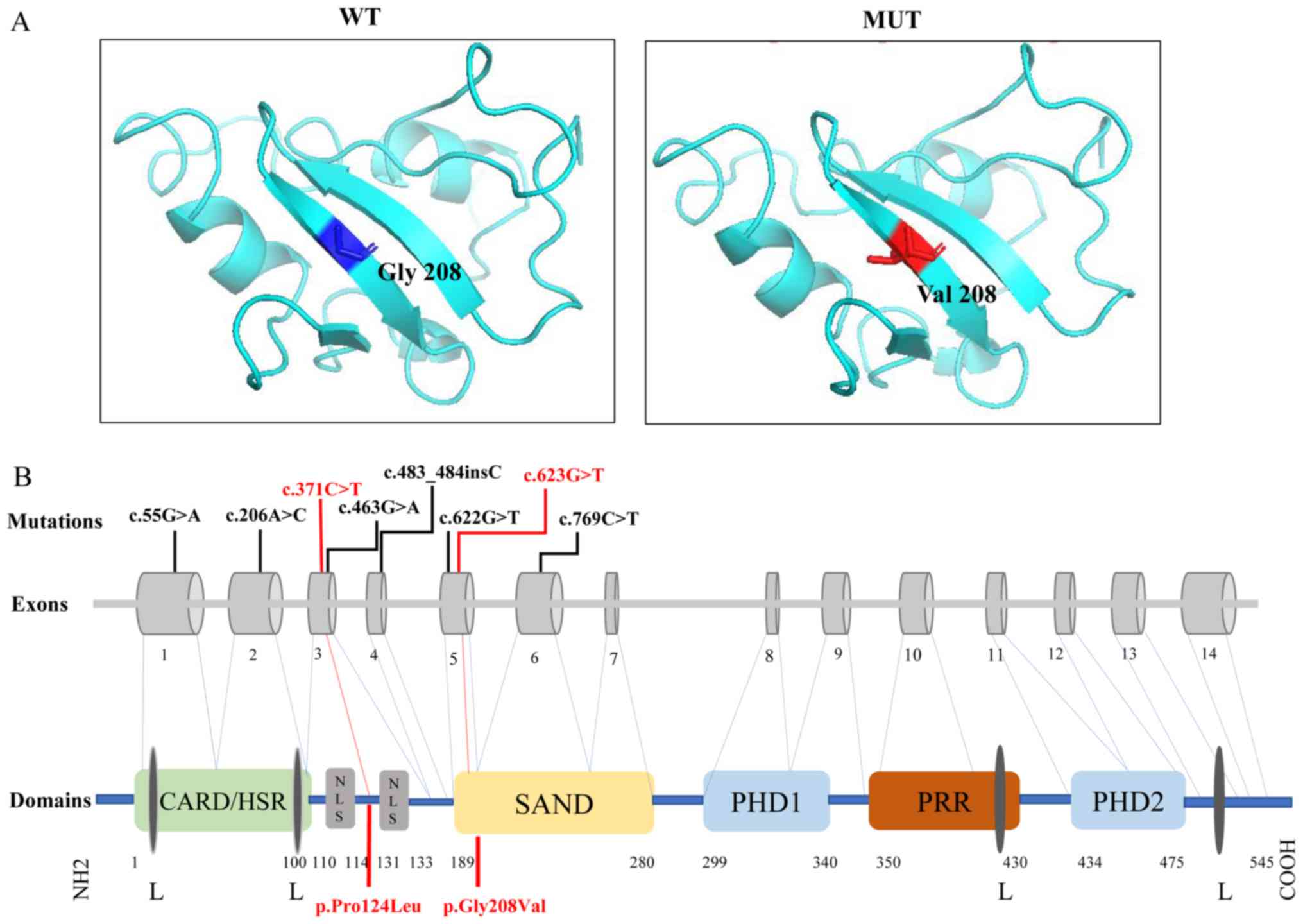 | Figure 3.Three-dimensional structural model
and schematic representation in AIRE protein. (A) Three-dimensional
structural model of the pathogenic-mutated position in AIRE. WT:
Position 208 was a glycine in normal population (blue). MUT: A
structural change caused by p.Gly208Val in SAND domain (red). (B)
Schematic representation of human AIRE variants and AIRE
protein. Boxes 1–14 represent exons. AIRE protein contained
CARD/HSR, NLS, SAND domain, PRR, two PHD fingers and four LXXLL
motifs. Variants are highlighted in red. AIRE, autoimmune
regulator; WT, wild type; MUT, mutant type; SAND, Sp100, AIRE-1,
NucP41/75, DEAF-1; CARD/HSR, caspase recruitment
domain/homogeneously staining region; NLS, nuclear localization
signal; PPR, proline-rich region; PHD, plant homeodomain; L,
leucine; X, any amino acid. |
Discussion
The diagnosis of APS-1 is primarily based on
clinical manifestations such as hypoparathyroidism, primary
adrenocortical insufficiency and chronic mucocutaneous candidiasis.
More recent clinical studies have demonstrated that APS-1 can be
classified into classical and non-classical type (18,19).
Classical APS-1 is characterized by autosomal recessive inheritance
and the presence of at least two of the three main features of
hypoparathyroidism, primary adrenocortical insufficiency and
chronic mucocutaneous candidiasis. Non-classical APS-1 follows an
autosomal dominant inheritance pattern and presents with varying
late-onset autoimmune phenotypes (18–21).
To the best of our knowledge, the present study is the first to
report a first Chinese patient with non-classical APS-1 caused by a
novel variant in AIRE in an autosomal dominant fashion. At
present, there are 15 known variants of AIRE that follow an
autosomal dominant inheritance pattern (including those identified
in the present study) (18,19).
The majority of variants are clustered within the first plant
homeodomain (PHD1) zinc finger domain (18). While two variants (p.Gly208Val and
p.Gly228Trp) were in the SAND domain, one variant (p.Cys446Gly) was
in the second plant homeodomain (PHD2) zinc finger domain (18,19,20).
The majority of patients with a dominant inheritance often did not
match the classical diagnostic criteria of APS-1. These patients
exhibited more variable phenotypes, ranging from no autoimmunity to
late-onset classical APS-1, and hypoparathyroidism was the most
prevalent phenotype (18,19). The present patient suffered from
tetany and syncope, primary amenorrhea, intermittent diarrhea and
general fatigue. Upon laboratory examination, she was diagnosed as
having hypoparathyroidism, primary ovarian insufficiency and PRCA.
A novel heterozygous variant of c.623G>T in exon 5 was
identified and regarded as a pathogenic variant of non-classical
APS-1. Another reported variant c.371C>T in exon 3 of
AIRE was also identified.
The AIRE gene encodes a 57-kDa protein that
promotes T-cell tolerance to self-antigens by upregulating the
expression levels of tissue-specific self-antigens in medullary
thymic epithelial cells (22).
Impaired functional AIRE leads to the escape of self-reactive
T-lymphocytes to the periphery and induces pathogenic autoimmune
reactions (23). Proper
subcellular location is important for the function of AIRE and its
ability to maintain macromolecular interactions. Previous reports
have shown that non-mutated AIRE localizes to the nucleus, whereas
mutant AIRE is retained in the cytoplasm, where it is unable to
regulate the transcription of a number of genes, such as major
histocompatibility complex class I and class II gene products, and
causes abnormal immune responses such as impaired negative
selection of autoreactive thymocytes (16,23–25).
AIRE contains a number of domains, including a
highly conserved caspase recruitment domain/N-terminal homogenously
staining region (CARD/HSR) domain, a conserved bipartite nuclear
localization signal (NLS), a SAND domain, two plant PHD zinc-finger
motifs separated by a proline-rich region, and four LXXLL (L,
leucine; X, any amino acid) nuclear receptor-binding motifs
(26). The SAND domain is
important for mediating the nuclear localization of AIRE (27). The variant c.623G>T in exon 5
may change the structure of the SAND domain, resulting in
aggregation of AIRE in the cytoplasm and impaired DNA binding
(Fig. 3A). However, p.Pro124Leu
caused by c.371C>T was between bipartite of NLS, which
constituted amino acids 110–114 and 131–33 (28), which may explain why this variant
was not pathogenic (Fig. 3B).
A previous study demonstrated that AIRE
variants in patients with hypoparathyroidism may induce
autoantibodies to NACHT leucine-rich-repeat protein 5, which is
selectively expressed in chief cells of the parathyroid glands
(29). An autoimmune reaction
targeting the calcium-sensing receptor was also found in
parathyroid cells of patients with APS-1 (30). In addition, a loss of T-lymphocyte
infiltration was observed surrounding follicles in female
AIRE-knockout mice (22).
These studies helped to elucidate the potential pathogenesis of the
AIRE variant underlying APS-1.
APS-1 is more frequent in European countries, with a
prevalence of 1:25,000 in Finland, 1: 14,000 in Sardinia, 1:43,000
in Slovenia and 1:500,000 in France (4,6,24).
p.R257X, located in the SAND domain, is the most common variant in
Finland, Russia and Eastern Europe (4,6,24).
R139X is most frequent in patients from Sardinia and results in
more severe phenotypes of APS-1 (10). The present study adds the clinical
and genetic characteristics of the patient to those of six
previously reported Chinese patients with APS-1 (7,14–17).
A total of 8 variants (7 missense variants and 1 insertion variant)
were detected in seven patients with APS-1 from six unrelated
families (Table II). Previous
research has revealed that the most common variants of the
AIRE gene exist in exons 1, 2, 6, 8 and 10 (24,31),
whereas the variants of the AIRE gene in Chinese patients
with APS-1occurred in exons 1–6 (c.55G>A, c.206A>C,
c.371C>T, c.463G>A, c.483_484insC, c.623G>T and
c.769C>T), without obvious hotspot variants. These variants
primarily affected the CARD, NLS and SAND domains (Fig. 3B). Variants in the CARD domain may
influence the multimerization of the AIRE protein and suppress its
transcriptional activation capacity (32). A number of gross deletions and
splice site variants (such as c.483_484insC in patient 2) have been
identified as associated with APS-1 because AIRE protein
translation is affected (33,34).
| Table II.Summary of clinical and genetic
features of 7 Chinese patients with APS-1 and AIRE
mutations. |
Table II.
Summary of clinical and genetic
features of 7 Chinese patients with APS-1 and AIRE
mutations.
|
| Gender/onset | Manifestations |
|
|
|
|
|---|
|
|
|
|
|
|
|
|
|---|
| Patient no. | age, years | Major
components | Other
components | AIRE
mutation/inheritance | Protein change | Domain | (Refs.) |
|---|
| 1 | F/4 | CMC; HP; PAI | None | Exon 1:
c.55G>A/AR | p.Ala19Thr | CARD | (14) |
|
|
|
|
| Exon 6:
c.769C>T/AR | p.Arg257X | SAND |
|
| 2 | F/10 | CMC; PAI | Autoimmune
thyroiditis | Exon 4:
c.483_484insC/AR | Truncated
protein | CARD | (16) |
| 3 | M/2 | HP; PAI | Enamel dystrophy;
binocular cataract; chronic intestinal dysfunction | Exon 2:
c.206A>C/AR | p.Gln69Pro | SAND | (7) |
| 4a | M/8 | HP; PAI | Epilepsy;
pernicious anemia; chronic/tension headaches; keratopathy; type 1
diabetes mellitus | Exon 3:
c.463G>A/AR | p.Gly155Ser | NA | (15) |
| 5a | F/1 | CMC; HP; PAI | Epilepsy;
pernicious anemia | Exon 3:
c.463G>A/AR | p.Gly155Ser | NA | (15) |
| 6 | F/12 | CMC; HP | Anemia | Exon 5:
c.622G>T/NA | p.Gly208Trp | SAND | (17) |
| 7 | F/7 | HP | Primary ovarian
insufficiency; | Exon 3:
c.371C>T/AD | p.Pro124Leu | Not domain | This |
|
|
|
| Pure red cell
aplasia; chronic atrophic gastritis | Exon 5:
c.623G>T/AD | p.Gly208Val | SAND | study |
Regarding the clinical phenotypes of Chinese
patients, the classic triad of CMC, PAI and HP were observed in two
patients, with four patients exhibiting two of the three major
manifestations. Generally, CMC is the most common, with an
occurrence of 77–100% in European patients with APS-1 (3,35–37),
but it is rare in Iranian Jews (17%) (11). CMC was only detected in four
Chinese patients with APS-1 and hypoparathyroidism was the most
common manifestation (six cases).
In patients with APS-1, the same pathogenic gene can
lead to different clinical phenotypes. A Chinese patient with APS-1
was diagnosed as having APS-1 without CMC, but his sister exhibited
this symptom (15). Cetani et
al (19), reported a missense
(p.Gly228Trp) mutation of the AIRE gene in a family with
APS-1. The p.Gly228Trp mutation was detected in the proband as well
as her mother, son and sister, and acted in a dominant manner.
However, only the proband and her sister exhibited the typical
components of APS-1. In another study, a proband and four of her
children carried the p.Cys311Tyr variant. The proband and three of
her children exhibited varying components and severity of APS-1,
whereas a son was diagnosed with autoantibodies against tyrosine
hydroxylase without any autoimmune manifestations (18). The patient's father exhibited the
same AIRE variants, but he did not present with symptoms of
APS-1. The mechanism governing the difference in penetration rates
is not clear in patients with APS-1.
The diagnosis of APS-1 is often delayed (2). The present patient presented with
major manifestations of hypoparathyroidism and other late-onset
autoimmune phenotypes, which was inconsistent with the phenotypes
of classic APS-1. However, APS-1 was confirmed by the detection of
AIRE variants. Thus, AIRE sequencing should be
performed at an early stage when patients are suspected of having
APS-1.
There is currently a lack of studies concerning the
effects of immunomodulatory and gene-targeted therapy on APS-1. The
primary therapeutic strategy for APS-1 is hormone replacement
therapy to correct endocrine function. For hypoparathyroidism,
supplementation with calcium and calcitriol is given to achieve
low-normal serum calcium and normal urine calcium levels, relieve
the symptoms of hypocalcemia and decrease long-term complications
(38). Hormone replacement therapy
is an alternative treatment used to manage primary ovarian
insufficiency. Cyclosporine A, with or without concurrent
corticosteroids, has been suggested to be an effective
immunotherapy for pure red aplastic anemia (39). Using these approaches, the clinical
symptoms of the present patient notably improved.
There were a number of limitations in in the present
study. First, the genotype-phenotype association was not well
established due to the small sample size. Second, the lack of
functional studies made it difficult to elucidate the detailed
mechanism underlying how variants in AIRE lead to APS-1.
Third, certain autoantibodies, such as 21-hydroxylase and
steroid-producing cells autoantibodies, were not investigated.
Last, detailed information on the patient's father was not
available.
In conclusion, a novel variant of c.623G>T
(p.Gly208Val) of the AIRE gene is associated with an APS-1
clinical syndrome of hypoparathyroidism, primary ovarian
insufficiency and PRCA. The novel missense variant of c.623G>T
in AIRE may impair its binding and regulation of the
macromolecular DNA binding complex, resulting in a clinical picture
of APS-1. The results of the present study expand the known genetic
and phenotypic spectra of APS-1.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81570802 and
81873668, respectively), Chinese Academy of Medical Sciences
Innovative Fund for Medical Sciences (grant no. 2016-I2M-3-003) and
the National Key Research and Development Program of China (grant
no. 2016YFC0901501).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WBZ performed molecular genetic studies,
participated in sequence alignment, analyzed data and wrote the
manuscript. LJL and DCZ collected, analyzed and interpreted the
data.. OW, YJ and WBX provided important medical decisions in the
clinical diagnose and treatment of patient, contributed the
analysis of clinical data and reviewed the manuscript. ML
contributed to the conception and design of the research,
acquisition and interpretation of data and revised the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Scientific Ethics
Committee of Peking Union Medical College Hospital (approval no.
JS-2081). Written informed consent was obtained from all subjects
prior to participation.
Patient consent for publication
The patient, her parents and the healthy controls
provided informed consent for publication of their data.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CMC
|
chronic mucocutaneous candidiasis
|
|
PAI
|
primary adrenocortical
insufficiency
|
|
HP
|
hypoparathyroidism
|
|
PRCA
|
pure red cell aplasia
|
|
Cr
|
creatine
|
|
ALT
|
alanine aminotransferase
|
|
PTH
|
parathyroid hormone
|
|
FSH
|
follicle-stimulating hormone
|
|
LH
|
luteinizing hormone
|
|
FT4
|
free thyroxine
|
|
FT3
|
free triiodothyronine
|
|
TSH
|
thyroid-stimulating hormone
|
|
TPOAb
|
autoantibodies to thyroid peroxidase
antigen
|
|
TGAb
|
autoantibodies to thyroglobulin
|
|
ACTH
|
adrenocorticotropic hormone
|
|
ICA
|
islet cell antibodies
|
|
GADA
|
glutamic acid decarboxylase
antibodies
|
|
ANA
|
autoantibodies to antinuclear
antibody
|
|
RO-52
|
52 kDa Ro/SS-A molecule
|
|
ANCA
|
anti-neutrophil cytoplasmic
antibodies
|
References
|
1
|
Sepe V, Velluzzi F and Songini M:
Autoimmune polyendocrine syndromes. N Engl J Med.
378:25432018.PubMed/NCBI
|
|
2
|
Husebye ES, Anderson MS and Kämpe O:
Autoimmune polyendocrine syndromes. N Engl J Med. 378:1132–1141.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bruserud Ø, Oftedal BE, Landegren N,
Erichsen MM, Bratland E, Lima K, Jørgensen AP, Myhre AG, Svartberg
J, Fougner KJ, et al: A longitudinal follow-up of autoimmune
polyendocrine syndrome type 1. J Clin Endocrinol Metab.
101:2975–2983. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Orlova EM, Sozaeva LS, Kareva MA, Oftedal
BE, Wolff ASB, Breivik L, Zakharova EY, Ivanova ON, Kämpe O, Dedov
II, et al: Expanding the phenotypic and genotypic landscape of
autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab.
102:3546–3556. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gutierrez MJ, Gilson J, Zacharias J,
Ishmael F and Bingham CA: Childhood polyarthritis as early
manifestation of autoimmune polyendocrinopathy with candidiasis and
ectodermal dystrophy syndrome. Front Immunol. 8:3772017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bruserud Ø, Oftedal BE, Wolff AB and
Husebye ES: AIRE-mutations and autoimmune disease. Curr Opin
Immunol. 43:8–15. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu W, Hu Z, Liao X, Chen X, Huang W,
Zhong Y and Zeng Z: A new mutation site in the AIRE gene causes
autoimmune polyendocrine syndrome type 1. Immunogenetics.
69:643–651. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nagamine K, Peterson P, Scott HS, Kudoh J,
Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis
SE, et al: Positional cloning of the APECED gene. Nat Genet.
17:393–398. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao B, Chang L, Fu H, Sun G and Yang W:
The role of autoimmune regulator (AIRE) in peripheral tolerance. J
Immunol Res. 2018:39307502018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meloni A, Willcox N, Meager A, Atzeni M,
Wolff AS, Husebye ES, Furcas M, Rosatelli MC, Cao A and Congia M:
Autoimmune polyendocrine syndrome type 1: An extensive longitudinal
study in Sardinian patients. J Clin Endocrinol Metab. 97:1114–1124.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zlotogora J and Shapiro MS: Polyglandular
autoimmune syndrome type I among Iranian Jews. J Med Genet.
29:824–826. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rosatelli MC, Meloni A, Meloni A, Devoto
M, Cao A, Scott HS, Peterson P, Heino M, Krohn KJ, Nagamine K, et
al: A common mutation in Sardinian autoimmune
polyendocrinopathy-candidiasis-ectodermal dystrophy patients. Hum
Genet. 103:428–434. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Y, Asan, Ma D, Lv F, Xu X, Wang J, Xia
W, Jiang Y, Wang O, Xing X, et al: Gene mutation spectrum and
genotype-phenotype correlation in a cohort of Chinese osteogenesis
imperfecta patients revealed by targeted next generation
sequencing. Osteoporos Int. 28:2985–2995. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu C, Shi Y, Yin H, Li H, Fan S, Wu S and
Yuan P: Autoimmune regulator gene mutations in a Chinese family
with autoimmune polyendocrinopathy syndrome type I. Zhonghua Yi Xue
Yi Chuan Xue Za Zhi. 27:18–22. 2010.(In Chinese). PubMed/NCBI
|
|
15
|
Zhang J, Liu H, Liu Z, Liao Y, Guo L, Wang
H, He L, Zhang X and Xing Q: A functional alternative splicing
mutation in AIRE gene causes autoimmune polyendocrine syndrome type
1. PLoS One. 8:e539812013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jin P, Zhang Q, Dong CS, Zhao SL and Mo
ZH: A novel mutation in autoimmune regulator gene causes autoimmune
polyendocrinopathy-candidiasis-ectodermal dystrophy. J Endocrinol
Invest. 37:941–948. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun YX, He YF and Li XL: Clinical analysis
and autoimmune regulator gene mutation of autoimmune
polyendocrinopathy syndrome type I in a family: A report of one
case. Zhongguo Dang Dai Er Ke Za Zhi. 18:147–151. 2016.(In
Chinese). PubMed/NCBI
|
|
18
|
Oftedal BE, Hellesen A, Erichsen MM,
Bratland E, Vardi A, Perheentupa J, Kemp EH, Fiskerstrand T, Viken
MK, Weetman AP, et al: Dominant mutations in the autoimmune
regulator AIRE are associated with common organ-specific autoimmune
diseases. Immunity. 42:1185–1196. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cetani F, Barbesino G, Borsari S, Pardi E,
Cianferotti L, Pinchera A and Marcocci C: A novel mutation of the
autoimmune regulator gene in an Italian kindred with autoimmune
polyendocrinopathy-candidiasis-ectodermal dystrophy, acting in a
dominant fashion and strongly cosegregating with hypothyroid
autoimmune thyroiditis. J Clin Endocrinol Metab. 86:4747–4752.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ilmarinen T, Eskelin P, Halonen M, Ruppell
T, Kilpikari R, Torres GD, Kangas H and Ulmanen I: Functional
analysis of SAND mutations in AIRE supports dominant inheritance of
the G228W mutation. Hum Mutat. 26:322–331. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Su MA, Giang K, Zumer K, Jiang H, Oven I,
Rinn JL, Devoss JJ, Johannes KP, Lu W, Gardner J, et al: Mechanisms
of an autoimmunity syndrome in mice caused by a dominant mutation
in Aire. J Clin Invest. 118:1712–1726. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Anderson MS, Venanzi ES, Klein L, Chen Z,
Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist
C and Mathis D: Projection of an immunological self shadow within
the thymus by the aire protein. Science. 298:1395–1401. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kisand K and Peterson P: Autoimmune
polyendocrinopathy candidiasis ectodermal dystrophy: Known and
novel aspects of the syndrome. Ann N Y Acad Sci. 1246:77–91. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Björses P, Halonen M, Palvimo JJ, Kolmer
M, Aaltonen J, Ellonen P, Perheentupa J, Ulmanen I and Peltonen L:
Mutations in the AIRE gene: Effects on subcellular location and
transactivation function of the autoimmune
polyendocrinopathy-candidiasis-ectodermal dystrophy protein. Am J
Hum Genet. 66:378–392. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Johnnidis JB, Venanzi ES, Taxman DJ, Ting
JP, Benoist CO and Mathis DJ: Chromosomal clustering of genes
controlled by the aire transcription factor. Proc Natl Acad Sci
USA. 102:7233–7238. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Peterson P, Org T and Rebane A:
Transcriptional regulation by AIRE: Molecular mechanisms of central
tolerance. Nat Rev Immunol. 8:948–957. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ramsey C, Bukrinsky A and Peltonen L:
Systematic mutagenesis of the functional domains of AIRE reveals
their role in intracellular targeting. Hum Mol Genet. 11:3299–3308.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ilmarinen T, Melen K, Kangas H, Julkunen
I, Ulmanen I and Eskelin P: The monopartite nuclear localization
signal of autoimmune regulator mediates its nuclear import and
interaction with multiple importin alpha molecules. FEBS J.
273:315–324. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alimohammadi M, Björklund P, Hallgren A,
Pöntynen N, Szinnai G, Shikama N, Keller MP, Ekwall O, Kinkel SA,
Husebye ES, et al: Autoimmune polyendocrine syndrome type 1 and
NALP5, a parathyroid autoantigen. N Engl J Med. 358:1018–1028.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Habibullah M, Porter JA, Kluger N, Ranki
A, Krohn KJE, Brandi ML, Brown EM, Weetman AP and Kemp EH:
Calcium-sensing receptor autoantibodies in patients with autoimmune
polyendocrine syndrome type 1: Epitopes, specificity, functional
affinity, IgG subclass, and effects on receptor activity. J
Immunol. 201:3175–3183. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Meloni A, Perniola R, Faà V, Corvaglia E,
Cao A and Rosatelli MC: Delineation of the molecular defects in the
AIRE gene in autoimmune polyendocrinopathy-candidiasis-ectodermal
dystrophy patients from Southern Italy. J Clin Endocrinol Metab.
87:841–846. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pitkänen J, Doucas V, Sternsdorf T,
Nakajima T, Aratani S, Jensen K, Will H, Vähämurto P, Ollila J,
Scott HS, et al: The autoimmune regulator protein has
transcriptional transactivating properties and interacts with the
common coactivator CREB-binding protein. J Biol Chem.
275:16802–16809. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cihakova D, Trebusak K, Heino M, Fadeyev
V, Tiulpakov A, Battelino T, Tar A, Halasz Z, Blümel P, Tawfik S,
et al: Novel AIRE mutations and P450 cytochrome autoantibodies in
Central and Eastern European patients with APECED. Hum Mutat.
18:225–232. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bøe Wolff AS, Oftedal B, Johansson S,
Bruland O, Lovas K, Meager A, Pedersen C, Husebye ES and Knappskog
PM: AIRE variations in Addison's disease and autoimmune
polyendocrine syndromes (APS): Partial gene deletions contribute to
APS I. Genes Immun. 9:130–136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wolff AS, Erichsen MM, Meager A, Magitta
NF, Myhre AG, Bollerslev J, Fougner KJ, Lima K, Knappskog PM and
Husebye ES: Autoimmune polyendocrine syndrome type 1 in Norway:
Phenotypic variation, autoantibodies, and novel mutations in the
autoimmune regulator gene. J Clin Endocrinol Metab. 92:595–603.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Perheentupa J: Autoimmune
polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin
Endocrinol Metab. 91:2843–2850. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ahonen P, Myllärniemi S, Sipilä I and
Perheentupa J: Clinical variation of autoimmune
polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a
series of 68 patients. N Engl J Med. 322:1829–1836. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brandi ML, Bilezikian JP, Shoback D,
Bouillon R, Clarke BL, Thakker RV, Khan AA and Potts JT Jr:
Management of hypoparathyroidism: Summary statement and guidelines.
J Clin Endocrinol Metab. 101:2273–2283. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Means RT Jr: Pure red cell aplasia. Blood.
128:2504–2509. 2016. View Article : Google Scholar : PubMed/NCBI
|