Introduction
Asthma is a chronic inflammatory airway disease that
is considered an important public health concern. It usually
develops in response to the inhalation of allergens, inhalants and
air pollutants, and is characterized by pulmonary inflammation,
airway hyper-responsiveness and mucus overproduction (1). Asthma and allergic diseases are
considered to be complex genetic diseases caused by interactions
between various genes and environmental factors, which are very
important in the pathogenesis of asthma (2). These gene-by-environment interactions
can be explained by the phenomenon of epigenetic regulation, which
has provided mechanistic explanations linking molecular events and
early-life environmental exposure with subsequent disease
development (3). Epigenetic
markers have emerged as potential participants in the mechanism of
asthma, and have been demonstrated to modulate various immune cell
processes involved in the development of disease (4–7).
Furthermore, several previous epigenomic and transcriptomic studies
have identified DNA methylation as a biomarker involved in the
development of asthma (8,9). Numerous asthma-promoting
environmental factors can alter the epigenetic profiles of airway
cells, suggesting that environmental exposure may cause asthma or
contribute to phenotypic heterogeneity changes via DNA methylation
(10–12).
Experimental asthma models have been established
using a variety of stimuli, such as house dust mites, ovalbumin
(OVA), fungi, cockroach extracts, ragweed and latex (13). These animal models have focused on
the role of type 2 immune responses driven by allergic reactions in
the development of asthma (14).
An OVA-induced asthma model was developed to evaluate the efficacy
of therapeutic agents prior to clinical trials and to investigate
their mechanisms of action; it has been used to identify the
signaling pathways involved in the development of asthma (15). Additionally, the OVA-induced asthma
model has often been used to identify the epigenetic mechanisms of
therapeutic agents and the pathogenesis of asthma (16–18);
however, to the best of the authors' knowledge, no studies have
explained the association between DNA methylation and gene
expression in the development of OVA-induced asthma.
The present study aimed to establish an integrated
network of DNA methylation and RNA expression in an OVA-induced
asthma model and to investigate the involvement of
epigenetically-regulated genes related to the development of
asthma.
Materials and methods
Animals
A total of 6 female BALB/c mice (8 weeks old and
weight 20–25 g) were obtained from Daehan BioLink Co., Ltd., and
were housed under standard laboratory conditions (22±2°C, 60%
relative humidity, 12/12 h light/dark cycle, and food and water
ad libitum) for at least 1 week before the experiment began.
Ethical and scientific management procedures for all animals were
approved by The Institutional Animal Care and Use Committee of the
Korea Institute of Oriental Medicine (approval no. 17-073).
Induction of asthma using OVA
Mice were randomly divided into two groups:
Vehicle-treated (n=3) and OVA-treated (n=3). Asthma was induced
using OVA according to the previously described experimental
procedures (19). To boost immune
responses, mice were injected intraperitoneally with OVA (50 µg)
emulsified with 200 µl aluminum sulfate (InvivoGen) twice, 7 days
apart (on day 0 and 7). The mice were administered intranasally
with 50 µl OVA (25 µg) from day 12 to 15 under anesthesia with 2%
isoflurane (Piramal Critical Care, Ltd.).
Evidence of OVA-induced asthma
Pathophysiological evidence of OVA-induced asthma
was indicated by airway inflammation, eosinophil and interleukin
(IL)-4 production (Fig. S1). The
right lungs of the mice were fixed with 10% neutral buffered
formalin at 25°C for 24 h, embedded in paraffin, sectioned into
4-µm thick slices, and stained with hematoxylin for 5 min and eosin
3 min (Sigma-Aldrich; Merck KGaA) at 25°C to evaluate inflammatory
responses in lung tissues, as well as with periodic acid-Schiff
(IMEB Inc.) to evaluate mucus production. A tracheostomy was
performed in mice anesthetized with 85 mg/kg alfaxan
(Alfaxalone®; Jurox Pty Ltd.) to obtain bronchoalveolar
lavage fluid (BALF). The lungs were lavaged twice with cold PBS
(total volume 1.4 ml) and the obtained BALF was centrifuged at 380
× g for 5 min at 4°C. The supernatant was used for an ELISA assay
(cat. no. M400B; R&D Systems, Inc.) in order to analyze level
of IL-4, according to the manufacturer's protocol. The pellets of
BALF were used to count inflammatory cells in the BALF. The
eosinophil count was determined using a cell counting machine
(Countess II™ Automated Cell Counter, Thermo Fisher Scientific,
Inc.).
Statistical analysis
Statistical analysis was performed using Student's
t-test with Prism 8 software (GraphPad Software, Inc.). P<0.05
was considered to indicate a statistically significant
difference.
Microarray data
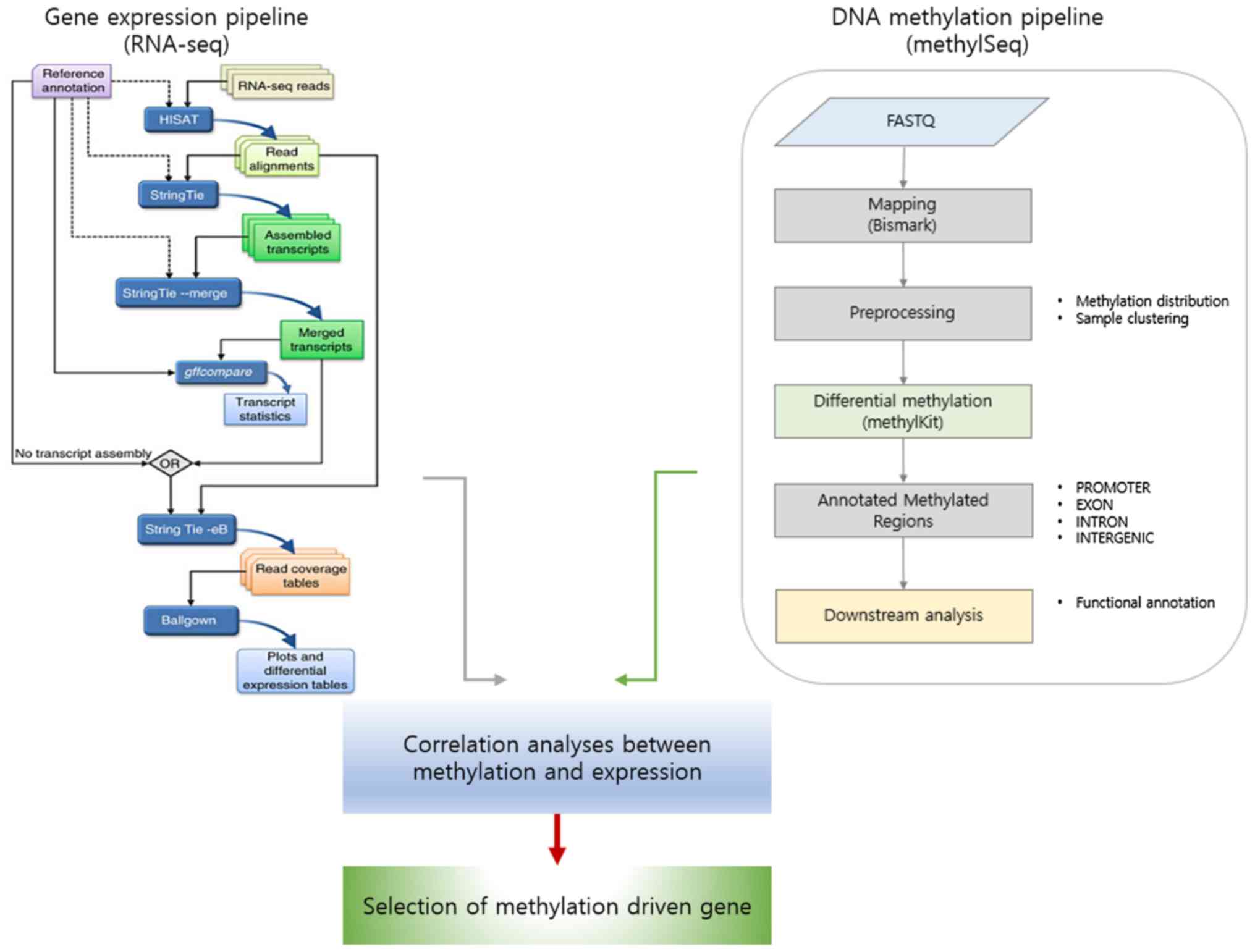
A schematic diagram for identifying target genes
involved in asthma development is presented in Fig. 1. Gene expression profile data was
used from a previous study by Baek et al (18), which were deposited in Gene
Expression Omnibus (GSE114587; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114587).
A total of six chips were available for further analysis, including
samples from three vehicle- and three OVA-treated animals. Detailed
information is available from the GEO database (18).
RNA-sequencing (RNA-seq) and data
analysis
Total RNA was extracted from the lung tissues of
saline-treated (normal) and OVA-induced mice. RNA isolation was
performed using the PureLink™ RNA Mini kit (cat. no. 12183018A;
Thermo Fisher Scientific, Inc.). RNA-seq libraries were prepared
using the TruSeq RNA Sample Prep kit (Illumina, Inc.) and sequenced
with a NextSeq® 500 System Whole-Genome Sequencing
Solution (Illumina, Inc.) using the 76-bp paired-end reads. The raw
FASTQ reads were trimmed to remove adapters and low-quality reads
(per-base quality <20) using cutadapt (v.1.10, http://cutadapt.readthedocs.io/en/stable/) (20), then the high-quality sequence reads
were aligned to the Mus musculus genome (mm10) using HISAT2
(v.2.1.0) and StringTie (v.1.3.4, http://ccb.jhu.edu/software/stringtie/) (21). Gene expression was quantified using
the ballgown package in R (v.2.6.0) (22). Differentially expressed genes
(DEGs) between the normal and OVA-induced mice groups were
identified using the edgeR package (v.3.16.5) (23) based on negative binomial models of
RNA-seq count data. Candidate DEGs were filtered using
log2(FC) ≥|1| and P<0.05 as thresholds, where FC
indicates the fold change. DAVID was used to functionally annotate
the genes (Tables SI and SII) (24,25).
Methyl-sequencing and data
analysis
DNA methylation profile data from the previous study
by Baek et al (18) was
used. DNA libraries were prepared according to the SureSelectXT
Methyl-Seq Target Enrichment System protocol (Agilent Technologies,
Inc.) and sequenced using an Illumina HiSeq 2500 platform
(Illumina, Inc.) to generate 101-bp paired-end reads. After
sequencing, the raw FASTQ reads were trimmed to remove adapters and
low-quality reads (per-base quality <20) using cutadapt (v.1.10)
and mapped onto the bisulfite converted Mus musculus genome
(mm10) using Bismark v.0.19.0 (https://www.bioinformatics.babraham.ac.uk/projects/bismark/).
The methylKit R package (v.1.6.0) (26) was used to analyze DNA methylation.
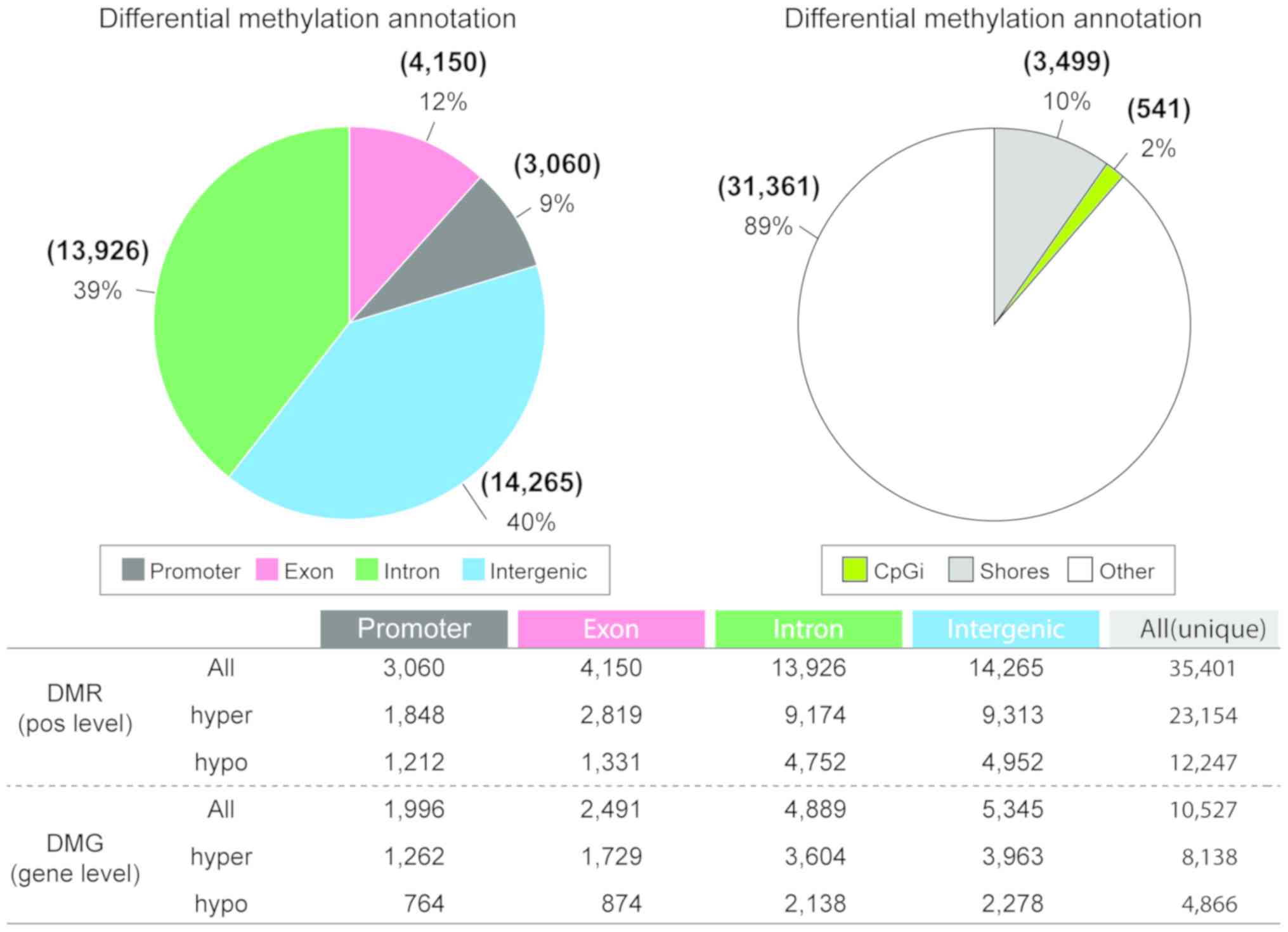
Differentially methylated regions (DMRs) are presented in Fig. 2. The base-pair methylation profile
was summarized over a tiling window (window size: 1,000 bp and
step-size: 500 bp). Statistically significant DMRs were selected
based on their Q-value and the percentage of methylation difference
(CpGs, mean methylation difference in groups >10%; Q<0.01).
The genomation R package (27) was
used to annotate the genomic features of DMRs, which were
classified into six types (intergenic, 5′UTR, 3′UTR, promoter, CDS
and intron) based on University of California Santa Cruz Genome
Browser (28).
Combined analysis of DNA methylation
and RNA expression
DNA methylation and gene expression are known to be
closely related. To identify the genes whose RNA expression levels
were affected by DNA methylation (i.e. inverse correlation between
RNA expression and DNA methylation) in asthmatic mice, the
expression level was adjusted to between 0 and 1 (as methylation β
values range between 0 and 1). Spearman's rank correlation
coefficient was used to evaluate the correlation between CpG
methylation and gene expression. To understand the biological
mechanisms and pathways related to these genes in asthma, Gene
Ontology (GO) enrichment analysis (29,30)
and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
(31) mapping were conducted using
DAVID (24).
Pathway analysis
Ingenuity Pathway Analysis (IPA; v.1.13, Qiagen,
Inc.) software was used to predict putative sub-networks containing
the four candidate genes, whose expression altered due to changes
in DNA methylation in asthma. Briefly, the Molecule Activity
Predictor tool in IPA was used to interrogate sub-networks using
the methylation-expression candidate genes, then integrated the
sub-network by overlaying the genes involved in asthma. To examine
how each gene affected the sub-network, the fold change of gene
expression and DNA methylation β values were included.
Data access
The data from the present study were deposited in
the GEO under accession number GSE114587 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114587;
secure token for reviewer: itytgkuoptclnej) (18).
Results
Methyl-sequencing exhibits DNA
methylation alterations between vehicle-treated mice and mice with
OVA-induced asthma
The DNA methylation of samples from the same animals
were profiled in order to identify DMRs between the vehicle and the
OVA-induced asthma samples (Fig.
2). After filtering the methylation profile, a total of 35,401
DMRs (base pair-level) were identified between the OVA-induced
asthma and the saline-treated mice. Of these, 3,060 were located in
promoter regions and 370 of the genes containing these DMRs
demonstrated an inverse correlation between methylation and gene
expression. Additionally, the number of hyper-methylated regions
was twice as high as the number of hypo-methylated regions.
Nevertheless, the proportion of functional regions, such as
promoters and exons, was similar between the hyper- and
hypo-methylated regions (hyper-methylated: 20.1%; hypo-methylated:
20.7%). The regional distribution of DMRs was assessed based on
their distance from the nearest CpG island (CpGi); shores were
defined as the regions flanking the CpGi (0–2 kb range). The
majority of DMRs identified in mice with OVA-induced asthma were
located in other regions (89% non-CpGi and non-shore); this was
followed by shores (10%) and CpGi (2%). The basepair-level DMRs
were combined with the differentially methylated genes to perform
an integrated analysis of methylation and gene expression. The
number of hyper-methylated genes was also higher than the number of
hypo-methylated genes. These DMRs, which were verified as important
genes, were used to infer the mechanisms underlying the development
of asthma. DAVID was used to functionally annotate the genes
(Tables I and II). KEGG pathway analysis identified
that 1,235 genes were more highly expressed in the OVA-induced
asthma samples, than in the vehicle samples, and were mainly
involved in ‘chemokine signalling pathway’, ‘cytokine-cytokine
receptor interaction’, ‘MAPK signalling pathway’, ‘neuroactive
ligand receptor interaction’, ‘olfactory transduction’ and
‘oxidative phosphorylation’ pathways (Table I). Conversely, 170 genes were
expressed at lower levels in the OVA-induced asthma samples than in
the vehicle samples, and were enriched in the ‘cytokine-cytokine
receptor interaction’, MAPK signalling pathway’, ‘neuroactive
ligand receptor interaction’ and ‘olfactory transduction’ pathways
(Table II).
 | Table I.Hyper-KEGG pathway functional
annotation of DNA methylation. |
Table I.
Hyper-KEGG pathway functional
annotation of DNA methylation.
| Term | Hit | Total | P-value |
|---|
| Olfactory
transduction | 13 | 389 |
3.80×10−141 |
| Cytokine-cytokine
receptor interaction | 89 | 267 |
1.22×10−43 |
| Neuroactive ligand
receptor interaction | 98 | 272 |
1.08×10−41 |
| Pathways in
cancer | 170 | 328 |
4.18×10−34 |
| Systemic lupus
erythematosus | 25 | 140 |
8.18×10−34 |
| Huntington's
disease | 60 | 185 |
4.45×10−31 |
| MAPK signalling
pathway | 129 | 267 |
2.51×10−30 |
| Oxidative
phosphorylation | 30 | 135 |
2.68×10−29 |
| Chemokine
signalling pathway | 70 | 190 |
1.13×10−28 |
| Alzheimer's
disease | 55 | 169 |
1.66×10−28 |
| Natural killer cell
mediated cytotoxicity | 36 | 137 |
5.31×10−27 |
| Parkinson's
disease | 34 | 133 |
1.01×10−26 |
| Antigen processing
and presentation | 10 | 89 |
5.96×10−26 |
| Spliceosome | 33 | 128 |
1.28×10−25 |
| Purine
metabolism | 55 | 159 |
2.00×10−25 |
| Regulation of
action cytoskeleton | 102 | 216 |
2.19×10−25 |
| Focal adhesion | 100 | 201 |
3.38×10−22 |
| Cell cycle | 41 | 128 |
4.23×10−22 |
| Lysosome | 36 | 121 |
5.19×10−22 |
| Endocytosis | 87 | 183 |
1.66×10−21 |
| Table II.Hypo-KEGG pathway functional
annotation of DNA methylation. |
Table II.
Hypo-KEGG pathway functional
annotation of DNA methylation.
| Term | Hit | Total | P-value |
|---|
| Olfactory
transduction | 7 | 389 |
5.65×10−102 |
| Neuroactive ligand
receptor interaction | 55 | 272 |
5.59×10−33 |
| Systemic lupus
erythematosus | 14 | 140 |
1.07×10−25 |
| Cytokine-cytokine
receptor interaction | 72 | 267 |
5.55×10−25 |
| Pathways in
cancer | 113 | 328 |
9.82×10−23 |
| Huntington's
disease | 38 | 185 |
1.69×10−22 |
| Alzheimer's
disease | 31 | 169 |
2.02×10−22 |
| Oxidative
phosphorylation | 20 | 135 |
1.04×10−20 |
| MAPK signalling
pathway | 88 | 267 |
1.00×10−19 |
| Parkinson's
disease | 22 | 133 |
5.98×10−19 |
| Spliceosome | 22 | 128 |
4.89×10−18 |
| Drug metabolism
cytochrome P450 | 3 | 72 |
2.35×10−17 |
| Regulation of
action cytoskeleton | 69 | 216 |
7.53×10−17 |
| Antigen processing
and presentation | 9 | 89 |
1.05×10−16 |
| Ubiquitin mediated
proteolysis | 30 | 138 |
2.40×10−16 |
| Purine
metabolism | 41 | 159 |
6.54×10−16 |
| Calcium signalling
pathway | 52 | 178 |
1.37×10−15 |
| Cell cycle | 27 | 128 |
1.63×10−15 |
| Endocytosis | 57 | 183 |
6.20×10−15 |
| Steroid hormone
biosynthesis | 1 | 55 |
7.97×10−15 |
Identification of
epigenetically-regulated genes in the development of asthma
To verify the genes modulated by DNA methylation in
the development of asthma, the RNA-seq data were analyzed for
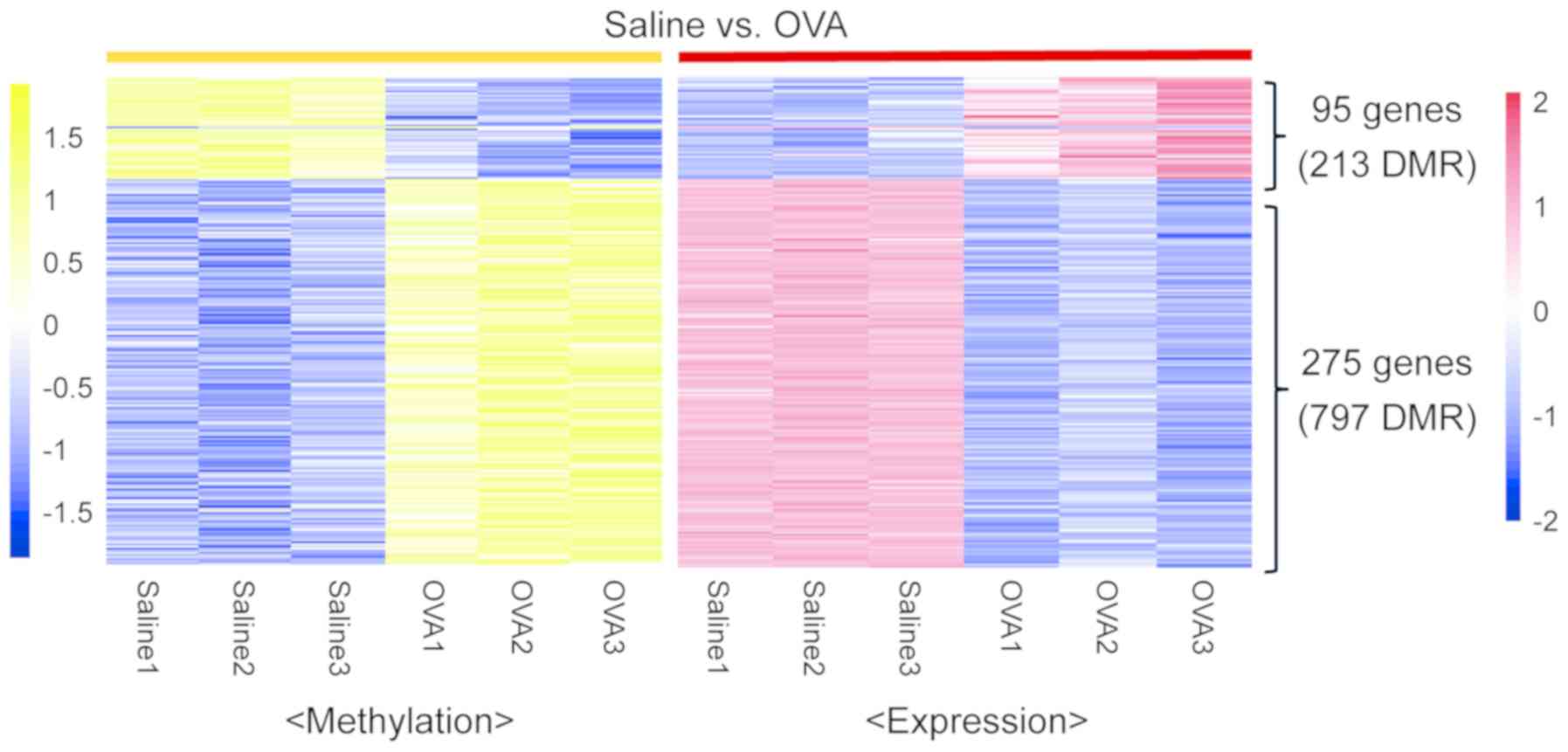
correlations between DNA methylation and gene expression (Fig. 3). A total of 95 genes were
upregulated, whilst 275 genes were downregulated in the OVA-induced
asthma samples. DAVID was used to functionally annotate the genes.
KEGG pathway analysis identified that 368 genes were upregulated or
downregulated in the OVA-induced asthma samples, and were mainly
involved in ‘acute myeloid leukemia’, ‘chemokine signalling
pathway’, ‘focal adhesion’, ‘leukocyte transendothelial migration’,
‘vascular smooth muscle contraction’ and ‘neurotrophin signalling
pathway’ (Table III).
| Table III.Kyoto Encyclopaedia of Genes and
Genomes pathway functional annotation of DNA methylation and gene
expression. |
Table III.
Kyoto Encyclopaedia of Genes and
Genomes pathway functional annotation of DNA methylation and gene
expression.
| Term | Count | % | P-value |
|---|
| mmu05221: Acute
myeloid leukemia | 7 | 2.07715134 | 0.00156622 |
| mmu04062: Chemokine
signalling pathway | 12 | 3.56083086 | 0.00232787 |
| mmu04510: Focal
adhesion | 12 | 3.56083086 | 0.00446246 |
| mmu04670: Leukocyte
transendothelial migration | 9 | 2.67062315 | 0.00486667 |
| mmu04270: Vascular
smooth muscle contraction | 9 | 2.67062315 | 0.00511869 |
| mmu04070:
Phosphatidylinositol signalling system | 7 | 2.07715134 | 0.00627478 |
| mmu04916:
Melanogenesis | 8 | 2.37388724 | 0.00669627 |
| mmu04722:
Neurotrophin signalling pathway | 9 | 2.67062315 | 0.00822449 |
| mmu05200: Pathways
in cancer | 15 | 4.45103858 | 0.01234863 |
| mmu04666: Fc γ
R-mediated phagocytosis | 7 | 2.07715134 | 0.02170314 |
| mmu05212:
Pancreatic cancer | 6 | 1.7841543 | 0.02172617 |
| mmu05213:
Endometrial cancer | 5 | 1.48367953 | 0.02805151 |
| mmu00562: Inositol
phosphate metabolism | 5 | 1.48367953 | 0.03168093 |
| mmu04512:
ECM-receptor interaction | 6 | 1.78041543 | 0.03721468 |
Identification of four genes
associated with asthma development using integrated network
analysis
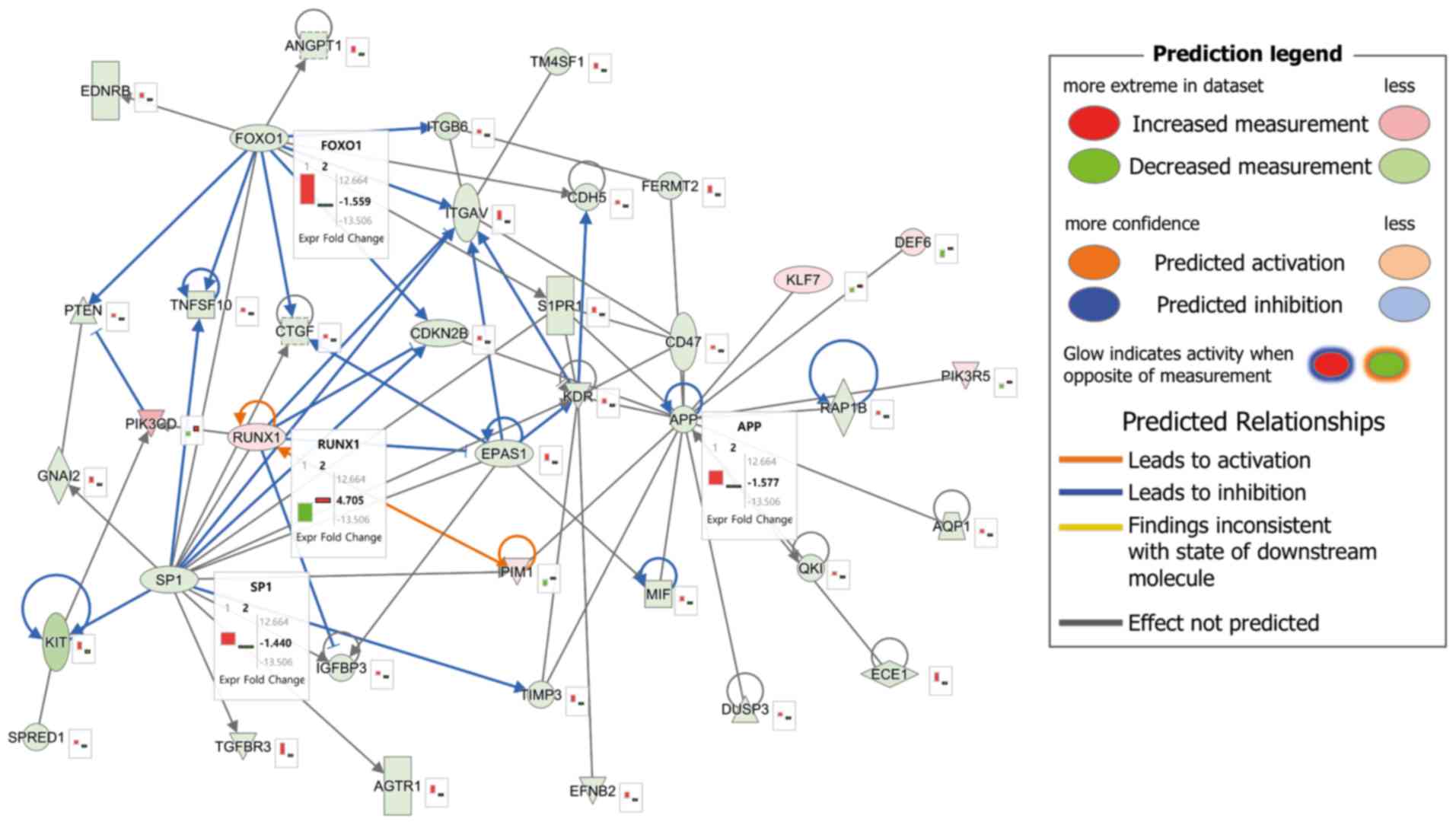
An integrated network analysis of DNA methylation
and gene expression was conducted to understand the interactions
between the genes related to asthma development. The predicted
pathway is presented in Fig. 4.
There were four hub genes, consisting of three upregulated genes
[forkhead box O1 (FOXO1), SP1 transcription factor
(SP1) and amyloid β precursor protein (APP)] and one
downregulated gene [RUNX family transcription factor 1
(RUNX1)], which demonstrated the association between DNA
methylation and gene expression. These genes were closely
interconnected nodes in the IPA module, and were functionally
significant. Therefore, FOXO1, SP1, APP and RUNX1
were identified as key integrated network regulators in the
development of asthma.
Discussion
The purpose of the present study was to conduct an
in-depth analysis of DNA methylation and gene expression in the
lung tissues of an OVA-induced asthma mouse model. The important
features of DNA methylation in asthma development and how they
differ from those of gene expression were investigated. The present
results identified important genes underlying asthma development,
and provided an incorporated network of DNA methylation and gene
expression in mice with OVA-induced asthma.
Histopathology was conducted, and the eosinophil
count and IL-4 level were determined, as indicators for
establishment of asthma in animals. In the development of asthma,
IL-4 leads to eosinophil-rich airway inflammation, elevated
immunoglobulin E production and mucus hypersecretion by goblet
cells (15). In addition,
eosinophilia is considered as an important feature in allergic
asthma. Activated eosinophils, as a result of various allergens,
can lead to changes in levels of reactive oxygen species, cationic
granule proteins, growth factors and cytokines (32). Therefore, resulting in the
aggravation of allergic responses, such as airway inflammation and
mucus hypersecretion (13). In the
present study, the experimental model demonstrated that the
eosinophil count and IL-4 were increased with inflammatory cell
infiltration into lung tissue in comparison with the normal
controls. This suggested that allergic asthma was successfully
established in the animal model.
The present study primarily profiled the genome-wide
gene and methylation expression patterns of lung tissue from
OVA-induced asthmatic animals described by Baek et al
(18) in order to examine the
effect of methylation on asthma development. GO analysis
demonstrated that the candidate genes were associated with
cytokines/chemokines, MAPKs and oxidative stress signaling
pathways. Asthma is characterized by airway inflammation,
hyper-responsiveness and mucus overproduction (32); airway inflammation is induced by
various factors, including cytokines, chemokines, reactive oxygen
species and MAPK signaling pathways, all of which accelerate the
development of asthma (33).
Cytokines and chemokines induce asthmatic pathophysiological
alterations, such as eosinophilia, smooth muscle contraction and
mucus hyperproduction (34). MAPK
signaling also plays a crucial role in the development of asthma,
and MAPK phosphorylation induces airway inflammation and mucus
production by activating epidermal growth factor signaling
(35).
GO analysis identified a strong enrichment of terms
associated with acute myeloid leukemia, chemokines, focal adhesion
and vascular smooth muscle contraction signaling pathways.
Furthermore, FOXO1, RUNX1, SP1 and APP were
identified as closely connected hub genes in the integrated
networks of DNA methylation and gene expression affecting the
development of asthma. FOXO1 promotes inflammation by
increasing the expression of several pro-inflammatory genes,
including IL-1B, IL-6 and IL-13, and exacerbates type
2 immune allergic airway inflammation in response to allergen
challenge (36). In contrast,
RUNX1 mediates the normal maturation of immune system
functions in various organisms; for example, RUNX1 promotes
T helper type 1 cell development by activating IL-4
silencers, reducing IL-4 expression, and elevating
interferon-γ expression (37);
therefore, RUNX1 dysfunction activates T helper type 2 cell
immune responses and induces asthmatic phenotypes (37). SP1 is the transcription
factor of WNT-5A expression in airway smooth muscle cells,
and its activity and expression are regulated by the
phosphorylation of MAPKs, such as p38 and JNK (38).
Recently, it was identified that APP was
associated with childhood-onset asthma in a network-assisted
analysis of genome-wide association studies of asthma (39). Of the four genes, previous studies
have demonstrated that FOXO1 and RUNX1 are correlated
with the development of asthma (37,38).
However, to the best of the authors' knowledge, SP1 and
APP have not been studied extensively and therefore thee are
insufficient data to correlate these genes with the development of
asthma. In particular, in the case of APP, only clinical
values were provided and to the best of the authors' knowledge, no
studies have been performed on the association with the development
of asthma. The results of the present study may differ from those
of previous studies. However, these differences are considered to
be due to various experimental conditions, such as transgenic,
knockout mice and stimulants. For example, in the case of
aquaporin-3, the elevation of its expression induced the
development of asthma in a previous study (40). However, in another previous study,
aquaporin-3 expression was not altered compared with normal
controls in the OVA-induced asthma model and was only upregulated
in an IL-13-induced asthma model (41). As such, small differences may be
identified depending on the respective experimental conditions.
Therefore, further studies are needed to elucidate the association
between these two genes and the development of asthma.
In summary, the present study focused on RNA-DNA
methylation, which is one of the factors that can affect RNA
expression in the development of asthma. In the present study, the
genes that were highly associated with both RNA expression and DNA
methylation were analyzed in the OVA-induced asthma model.
FOXO1, RUNX1, SP1 and APP were identified as closely
connected hub genes in an integrated network of DNA methylation and
gene expression affecting the development of asthma. The results of
the present study suggested that the modulation of these four genes
could contribute to the development of asthma.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by The Application
Development of Standardized Herbal Resources (grant no. KSN1911420)
from The Korea Institute of Oriental Medicine.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JSK and ISS drafted the manuscript. ISS, JSK and CK
conceived and designed the study. NRS, JYN, JSK and ISS analyzed
the data. JSK and CK reviewed and edited the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by The
Institutional Animal Care and Use Committee of Korea Institute of
Oriental Medicine (approval no. 17-073) and the animals were cared
for in accordance with The National Institutes of Health Guide for
the Care and Use of Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Deliu M, Belgrave D, Sperrin M, Buchan I
and Custovic A: Asthma phenotypes in childhood. Expert Rev Clin
Immunol. 13:705–713. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kanagaratham C and Radzioch D: Allergic
asthma: A summary from genetic basis, mouse study, to diagnosis and
treatment. Curr Pharm Des. 22:6261–6272. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang IV, Lozupone CA and Schwartz DA: The
environment, epigenome, and asthma. J Allergy Clin Immunol.
140:14–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Karmaus W, Ziyab AH, Everson T and
Holloway JW: Epigenetic mechanisms and models in the origins of
asthma. Curr Opin Allergy Clin Immunol. 13:63–69. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kuo CH, Hsieh CC, Lee MS, Chang KT, Kuo HF
and Hung CH: Epigenetic regulation in allergic diseases and related
studies. Asia Pac Allergy. 4:14–18. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Salam MT: Asthma epigenetics. Adv Exp Med
Biol. 795:183–199. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harb H, Alashkar Alhamwe B, Garn H, Renz H
and Potaczek DP: Recent developments in epigenetics of pediatric
asthma. Curr Opin Pediatr. 28:754–763. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chan MA, Ciaccio CE, Gigliotti NM,
Rezaiekhaligh M, Siedlik JA, Kennedy K and Barnes CS: DNA
methylation levels associated with race and childhood asthma
severity. J Asthma. 54:825–832. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lund RJ, Osmala M, Malonzo M, Lukkarinen
M, Leino A, Salmi J, Vuorikoski S, Turunen R, Vuorinen T, Akdis C,
et al: Atopic asthma after rhinovirus-induced wheezing is
associated with DNA methylation change in the SMAD3 gene promoter.
Allergy. 73:1735–1740. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zahiruddin AS, Grant JA and Sur S: Role of
epigenetics and DNA-damage in asthma. Curr Opin Allergy Clin
Immunol. 18:32–37. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Potaczek DP, Harb H, Michel S, Alhamwe BA,
Renz H and Tost J: Epigenetics and allergy: From basic mechanisms
to clinical application. Epigenomics. 9:539–571. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liang L, Willis-Owen SAG, Laprise C, Wong
KCC, Davies GA, Hudson TJ, Binia A, Hopkin JM, Yang IV, Grundberg
E, et al: An epigenome-wide association study of total serum
immunoglobulin E concentration. Nature. 520:670–674. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Haspeslagh E, Debeuf N, Hammad H and
Lambrecht BN: Murine models of allergic asthma. Methods Mol Biol.
1559:121–136. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sagar S, Akbarshahi H and Uller L:
Translation value of animal models of asthma: Challenges and
promises. Eur J Pharmacol. 759:272–277. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kianmeher M, Ghorani V and Boskabady MH:
Animal model of asthma, various methods and measured parameters: A
methodological review. Iran J Allergy Asthma Immunol. 15:445–465.
2016.PubMed/NCBI
|
|
16
|
Wu CJ, Yang CY, Chen YH, Chen CM, Chen LC
and Kuo ML: The DNA methylation inhibitor 5-azacytidine increases
regulatory T cells and alleviates airway inflammation in
ovalbumin-sensitized mice. Int Arch Allergy Immunol. 160:356–364.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu XF, Hu QY, Liang LF, We L, Gu WZ, Tang
LL, Fu LC and Du LZ: Epigenetics of hyper-responsiveness to
allergen challenge following intrauterine growth retardation rat.
Respir Res. 15:1372014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baek SJ, Chun JM, Kang TW, Seo YS, Kim SB,
Seong B, Jang Y, Shin GH and Kim C: Identification of epigenetic
mechanisms involved in the anti-asthmatic effects of Descurainia
sophia seed extract based on a multi-omics approach. Molecules.
23:28792018. View Article : Google Scholar
|
|
19
|
Chun JM, Lee AR, Kim HS, Lee AY, Gu GJ,
Moon BC and Kwon BI: Peucedanum japonicum extract attenuates
allergic airway inflammation by inhibiting Th2 cell activation and
production of pro-inflammatory mediators. J Ethnopharamcol.
211:78–88. 2018. View Article : Google Scholar
|
|
20
|
Martin M: Cutadapt removes adapter
sequences from high-throughput sequencing reads. EMBnet J.
172011.
|
|
21
|
Kim D, Langmead B and Salzberg SL: HISAT:
A fast spliced aligner with low memory requirements. Nat Methods.
12:357–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fu J, Frazee AC, Collado-Torres L, Jaffe
AE and Leek JT: ballgown: Flexible, isoform-level differential
expression analysis. R package version 2.6.0. 2016.
|
|
23
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nature Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
25
|
Huang Da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Akalin A, Kormaksson M, Li S,
Garrett-Bakelman FE, Figueroa ME, Melnick A and Mason CE:
methylKit: A comprehensive R package for the analysis of
genome-wide DNA methylation profiles. Genome Biol. 13:R872012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Akalin A, Franke V, Vlahoviček K, Mason C
and Schübeler D: Genomation: A toolkit to summarize, annotate and
visualize genomic intervals. Bioinformatics. 31:1127–1129. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kent WJ, Sugnet CW, Furey TS, Roskin KM,
Pringle TH, Zahler AM and Haussler D: The human genome browser at
UCSC. Genome Res. 12:996–1006. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
The Gene Ontology Consortium, . The Gene
Ontology Resource: 20 years and still GOing strong. Nucleic Acids
Res. 47:D330–D338. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shin NR, Kwon HJ, Ko JW, Lee IC, Kim JC,
Kim SH and Shin IS: S-allyl cysteine reduces eosinophilic airway
inflammation and mucus overproduction on ovalbumin-induced allergic
asthma model. Int Immunopharmacol. 68:124–130. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aqhasafari P, George U and Pidaparti R: A
review of inflammatory mechanism in airway disease. Inflamm Res.
68:59–74. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lambrecht BN, Hammad H and Fahy JV: The
cytokines of asthma. Immunity. 50:975–991. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Khorasanizadeh M, Eskian M, Gelfand EW and
Rezaei N: Mitogen-activated protein kinases as therapeutic targets
for asthma. Pharmacol Ther. 174:112–126. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shin NR, Ryu HW, Ko JW, Park SH, Yuk HJ,
Kim HJ, Kim JC, Jeong S and Shin IS: Artemisia argyi attenuates
airway inflammation in ovalbumin-induced asthmatic animals. J
Ethnopharmacol. 209:108–115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chung S, Lee TJ, Reader BF, Kim JY, Lee
YG, Park GY, Karpurapu M, Ballinger MN, Qian F, Rusu L, et al:
FoxO1 regulates allergic asthmatic inflammation through regulating
polarization of the macrophage inflammatory phenotype. Oncotarget.
7:17532–17546. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Haley KJ, Lasky-Su J, Manoli SE, Smith LA,
Shahsafaei A, Weiss ST and Tantisira K: RUNX transcription factors:
Association with pediatric asthma and modulated by maternal
smoking. Am J Physiol Lung Cell Mol Physiol. 301:L693–L701. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kumawat K, Menzen MH, Slegtenhorst RM,
Halayko AJ, Schmidt M and Gosens R: TGF-β-induced WNT-5A expression
in airway smooth muscle cells via Sp1 and β-catenin. PLoS One.
9:e948012014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu Y, Brossard M, Sarnowski C, Vaysse A,
Moffatt M, Margaritte-Jeannin P, Llinares-Lopez F, Dizier MH,
Lathrop M, Cookson W, et al: Network-assisted analysis of GWAS data
identifies a functionally-relevant gene module for childhood-onset
asthma. Sci Rep. 7:9382017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ikezoe K, Oga T, Honda T, Hara-Chikuma M,
Ma X, Tsuruyama T, Uno K, Fuchikami J, Tanizawa K, Handa T, et al:
Aquaporin-3 potentiates allergic airway inflammation in
ovalbumin-induced murine asthma. Sci Rep. 6:257812016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kran CM, Deng B, Mutyam V, McDonald CA,
Pazdziorko S, Masson L, Goldman S, Kasaian M, Chaudhary D, Williams
C and Ho MW: Altered regulation of aquaporin gene expression in
allergen and IL-13-induced mouse models of asthma. Cytokines.
46:111–118. 2009. View Article : Google Scholar
|