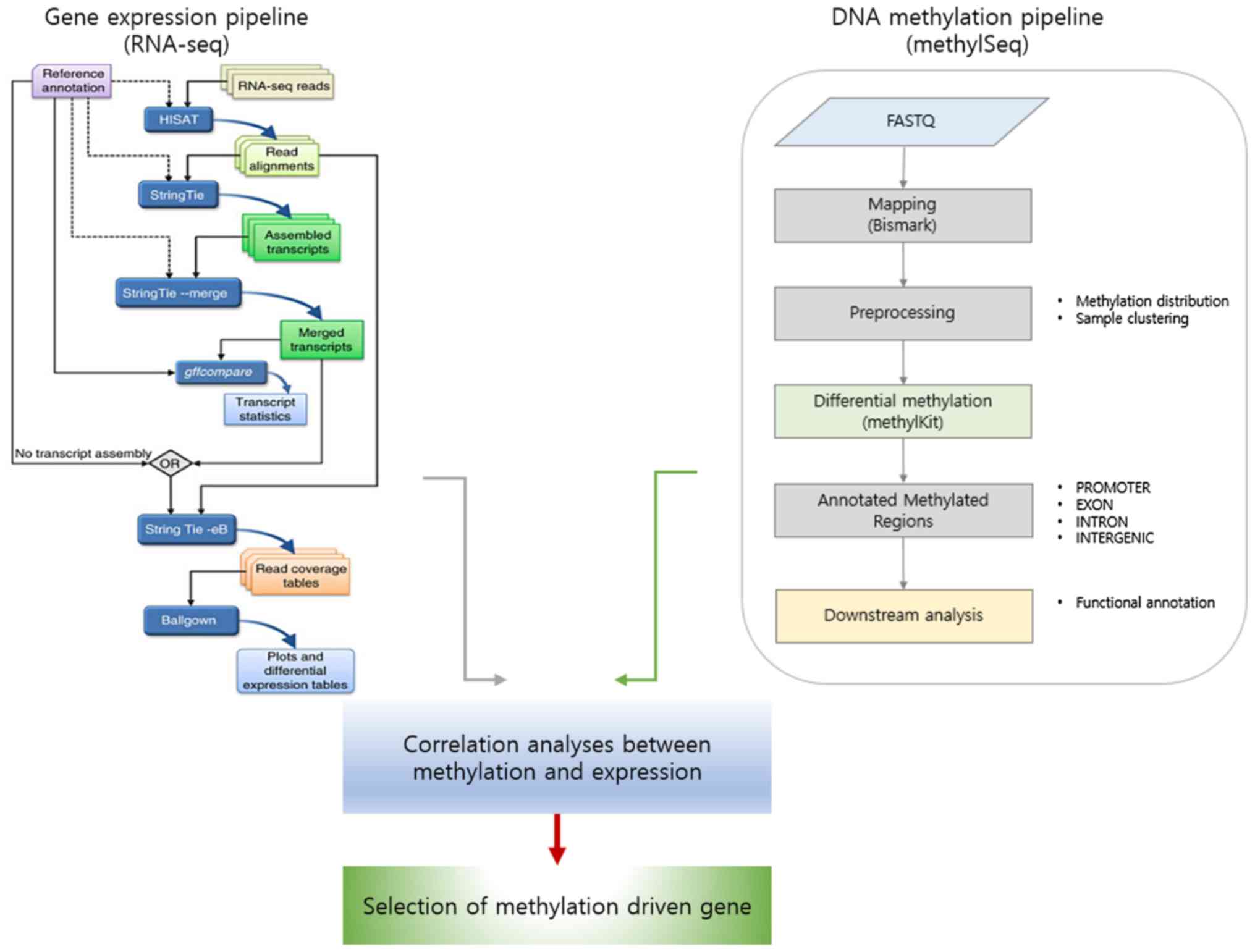
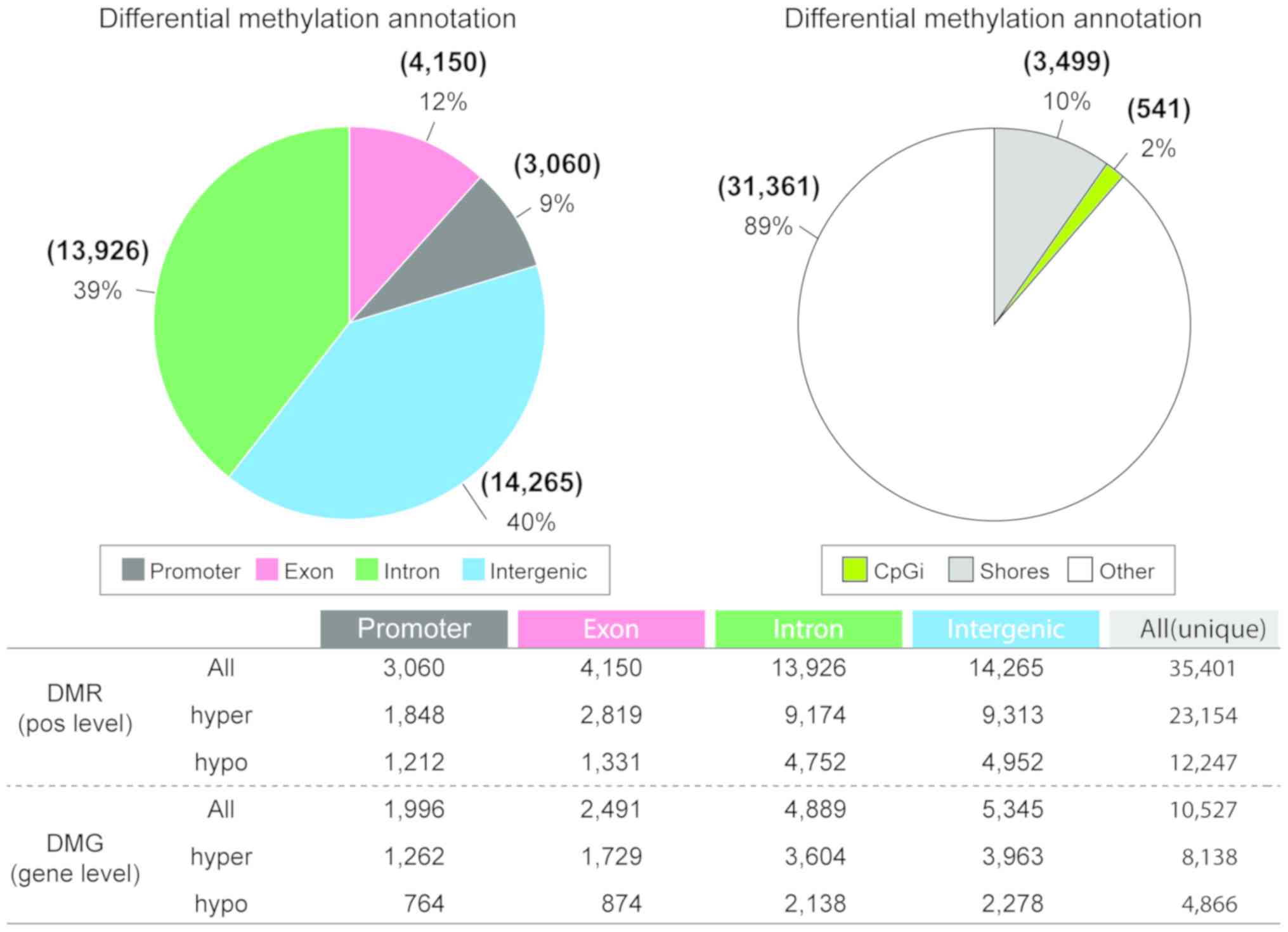
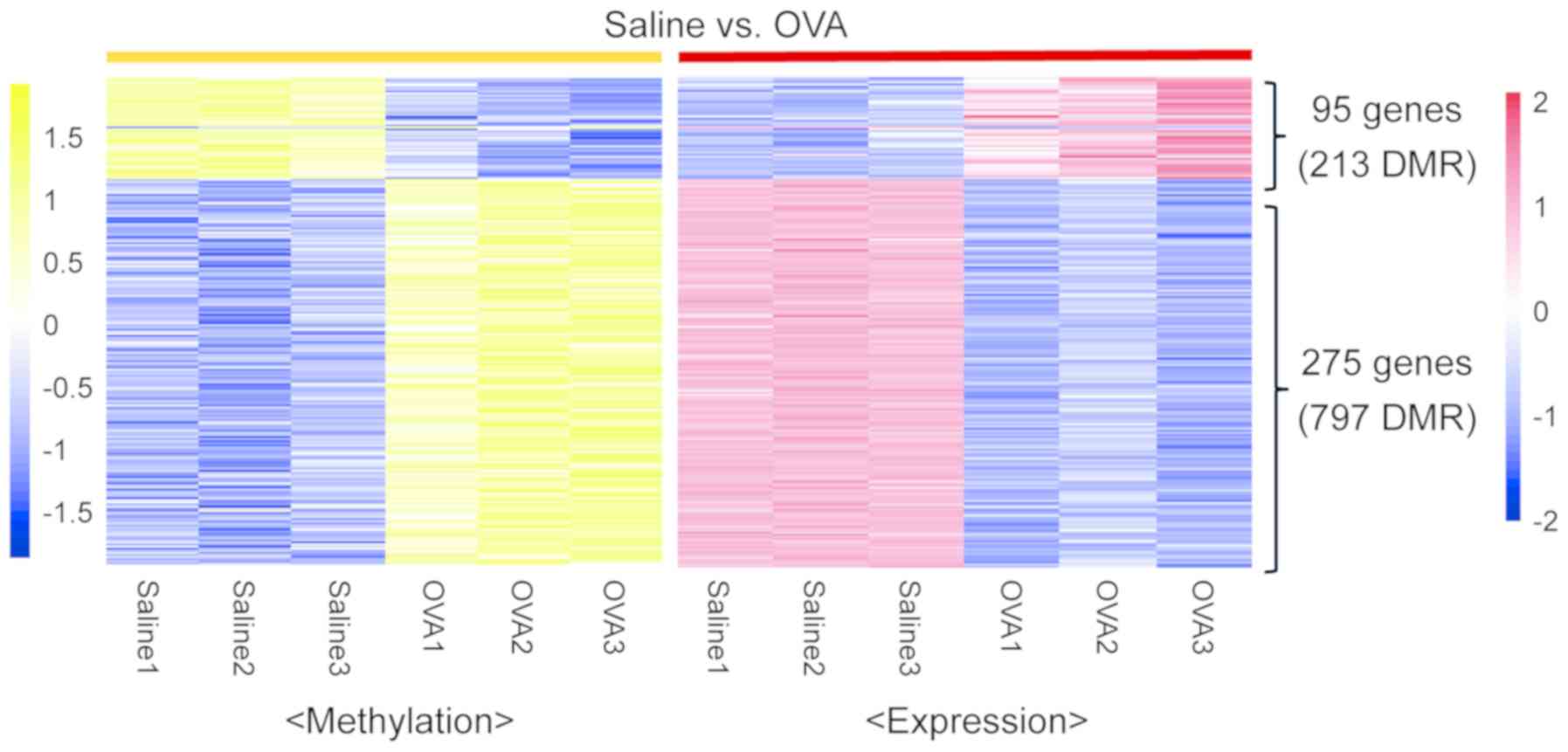
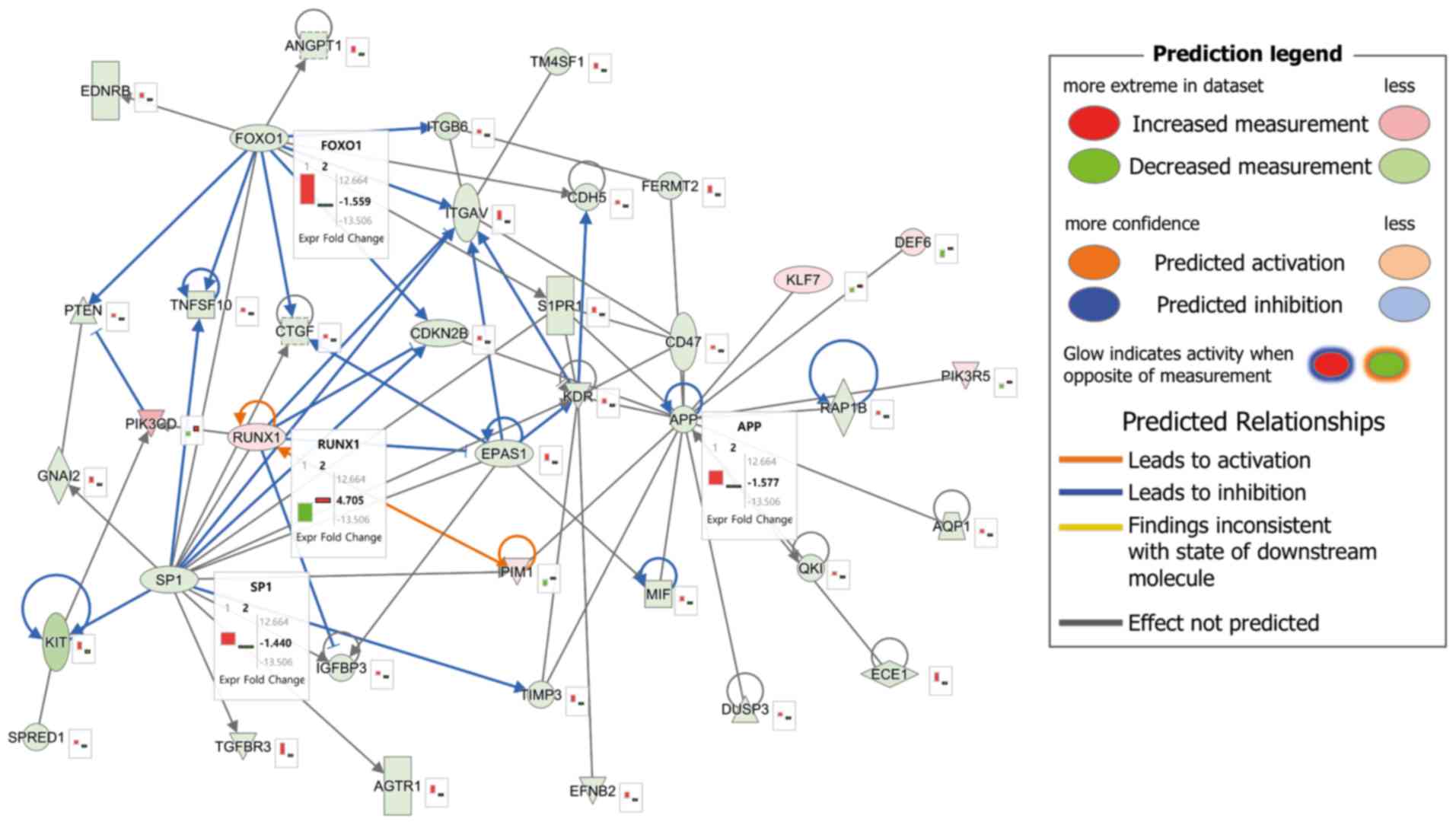
|
1
|
Deliu M, Belgrave D, Sperrin M, Buchan I
and Custovic A: Asthma phenotypes in childhood. Expert Rev Clin
Immunol. 13:705–713. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kanagaratham C and Radzioch D: Allergic
asthma: A summary from genetic basis, mouse study, to diagnosis and
treatment. Curr Pharm Des. 22:6261–6272. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang IV, Lozupone CA and Schwartz DA: The
environment, epigenome, and asthma. J Allergy Clin Immunol.
140:14–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Karmaus W, Ziyab AH, Everson T and
Holloway JW: Epigenetic mechanisms and models in the origins of
asthma. Curr Opin Allergy Clin Immunol. 13:63–69. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kuo CH, Hsieh CC, Lee MS, Chang KT, Kuo HF
and Hung CH: Epigenetic regulation in allergic diseases and related
studies. Asia Pac Allergy. 4:14–18. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Salam MT: Asthma epigenetics. Adv Exp Med
Biol. 795:183–199. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harb H, Alashkar Alhamwe B, Garn H, Renz H
and Potaczek DP: Recent developments in epigenetics of pediatric
asthma. Curr Opin Pediatr. 28:754–763. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chan MA, Ciaccio CE, Gigliotti NM,
Rezaiekhaligh M, Siedlik JA, Kennedy K and Barnes CS: DNA
methylation levels associated with race and childhood asthma
severity. J Asthma. 54:825–832. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lund RJ, Osmala M, Malonzo M, Lukkarinen
M, Leino A, Salmi J, Vuorikoski S, Turunen R, Vuorinen T, Akdis C,
et al: Atopic asthma after rhinovirus-induced wheezing is
associated with DNA methylation change in the SMAD3 gene promoter.
Allergy. 73:1735–1740. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zahiruddin AS, Grant JA and Sur S: Role of
epigenetics and DNA-damage in asthma. Curr Opin Allergy Clin
Immunol. 18:32–37. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Potaczek DP, Harb H, Michel S, Alhamwe BA,
Renz H and Tost J: Epigenetics and allergy: From basic mechanisms
to clinical application. Epigenomics. 9:539–571. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liang L, Willis-Owen SAG, Laprise C, Wong
KCC, Davies GA, Hudson TJ, Binia A, Hopkin JM, Yang IV, Grundberg
E, et al: An epigenome-wide association study of total serum
immunoglobulin E concentration. Nature. 520:670–674. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Haspeslagh E, Debeuf N, Hammad H and
Lambrecht BN: Murine models of allergic asthma. Methods Mol Biol.
1559:121–136. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sagar S, Akbarshahi H and Uller L:
Translation value of animal models of asthma: Challenges and
promises. Eur J Pharmacol. 759:272–277. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kianmeher M, Ghorani V and Boskabady MH:
Animal model of asthma, various methods and measured parameters: A
methodological review. Iran J Allergy Asthma Immunol. 15:445–465.
2016.PubMed/NCBI
|
|
16
|
Wu CJ, Yang CY, Chen YH, Chen CM, Chen LC
and Kuo ML: The DNA methylation inhibitor 5-azacytidine increases
regulatory T cells and alleviates airway inflammation in
ovalbumin-sensitized mice. Int Arch Allergy Immunol. 160:356–364.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu XF, Hu QY, Liang LF, We L, Gu WZ, Tang
LL, Fu LC and Du LZ: Epigenetics of hyper-responsiveness to
allergen challenge following intrauterine growth retardation rat.
Respir Res. 15:1372014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baek SJ, Chun JM, Kang TW, Seo YS, Kim SB,
Seong B, Jang Y, Shin GH and Kim C: Identification of epigenetic
mechanisms involved in the anti-asthmatic effects of Descurainia
sophia seed extract based on a multi-omics approach. Molecules.
23:28792018. View Article : Google Scholar
|
|
19
|
Chun JM, Lee AR, Kim HS, Lee AY, Gu GJ,
Moon BC and Kwon BI: Peucedanum japonicum extract attenuates
allergic airway inflammation by inhibiting Th2 cell activation and
production of pro-inflammatory mediators. J Ethnopharamcol.
211:78–88. 2018. View Article : Google Scholar
|
|
20
|
Martin M: Cutadapt removes adapter
sequences from high-throughput sequencing reads. EMBnet J.
172011.
|
|
21
|
Kim D, Langmead B and Salzberg SL: HISAT:
A fast spliced aligner with low memory requirements. Nat Methods.
12:357–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fu J, Frazee AC, Collado-Torres L, Jaffe
AE and Leek JT: ballgown: Flexible, isoform-level differential
expression analysis. R package version 2.6.0. 2016.
|
|
23
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nature Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
25
|
Huang Da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Akalin A, Kormaksson M, Li S,
Garrett-Bakelman FE, Figueroa ME, Melnick A and Mason CE:
methylKit: A comprehensive R package for the analysis of
genome-wide DNA methylation profiles. Genome Biol. 13:R872012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Akalin A, Franke V, Vlahoviček K, Mason C
and Schübeler D: Genomation: A toolkit to summarize, annotate and
visualize genomic intervals. Bioinformatics. 31:1127–1129. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kent WJ, Sugnet CW, Furey TS, Roskin KM,
Pringle TH, Zahler AM and Haussler D: The human genome browser at
UCSC. Genome Res. 12:996–1006. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
The Gene Ontology Consortium, . The Gene
Ontology Resource: 20 years and still GOing strong. Nucleic Acids
Res. 47:D330–D338. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shin NR, Kwon HJ, Ko JW, Lee IC, Kim JC,
Kim SH and Shin IS: S-allyl cysteine reduces eosinophilic airway
inflammation and mucus overproduction on ovalbumin-induced allergic
asthma model. Int Immunopharmacol. 68:124–130. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aqhasafari P, George U and Pidaparti R: A
review of inflammatory mechanism in airway disease. Inflamm Res.
68:59–74. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lambrecht BN, Hammad H and Fahy JV: The
cytokines of asthma. Immunity. 50:975–991. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Khorasanizadeh M, Eskian M, Gelfand EW and
Rezaei N: Mitogen-activated protein kinases as therapeutic targets
for asthma. Pharmacol Ther. 174:112–126. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shin NR, Ryu HW, Ko JW, Park SH, Yuk HJ,
Kim HJ, Kim JC, Jeong S and Shin IS: Artemisia argyi attenuates
airway inflammation in ovalbumin-induced asthmatic animals. J
Ethnopharmacol. 209:108–115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chung S, Lee TJ, Reader BF, Kim JY, Lee
YG, Park GY, Karpurapu M, Ballinger MN, Qian F, Rusu L, et al:
FoxO1 regulates allergic asthmatic inflammation through regulating
polarization of the macrophage inflammatory phenotype. Oncotarget.
7:17532–17546. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Haley KJ, Lasky-Su J, Manoli SE, Smith LA,
Shahsafaei A, Weiss ST and Tantisira K: RUNX transcription factors:
Association with pediatric asthma and modulated by maternal
smoking. Am J Physiol Lung Cell Mol Physiol. 301:L693–L701. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kumawat K, Menzen MH, Slegtenhorst RM,
Halayko AJ, Schmidt M and Gosens R: TGF-β-induced WNT-5A expression
in airway smooth muscle cells via Sp1 and β-catenin. PLoS One.
9:e948012014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu Y, Brossard M, Sarnowski C, Vaysse A,
Moffatt M, Margaritte-Jeannin P, Llinares-Lopez F, Dizier MH,
Lathrop M, Cookson W, et al: Network-assisted analysis of GWAS data
identifies a functionally-relevant gene module for childhood-onset
asthma. Sci Rep. 7:9382017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ikezoe K, Oga T, Honda T, Hara-Chikuma M,
Ma X, Tsuruyama T, Uno K, Fuchikami J, Tanizawa K, Handa T, et al:
Aquaporin-3 potentiates allergic airway inflammation in
ovalbumin-induced murine asthma. Sci Rep. 6:257812016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kran CM, Deng B, Mutyam V, McDonald CA,
Pazdziorko S, Masson L, Goldman S, Kasaian M, Chaudhary D, Williams
C and Ho MW: Altered regulation of aquaporin gene expression in
allergen and IL-13-induced mouse models of asthma. Cytokines.
46:111–118. 2009. View Article : Google Scholar
|