Introduction
Glioma is a type of cancer that originates from
glial cells in the brain and spinal cord (1). Among gliomas, glioblastoma (GBM) is
the most prevalent and has the highest mortality, with a median
survival time of <15 months (2)
and a 5% 5-year survival rate post-diagnosis worldwide, according
to clinical data collected before 2015 (3). Previous studies have focused on the
metabolic changes of GBM (4–8).
Isocitrate dehydrogenases (IDHs) are a group of enzymes that
catalyze isocitrate oxidative decarboxylation to produce
α-ketoglutarate (α-KG) and CO2, that have been reported
to exhibit altered activities in GBM due to site mutagenesis and
expression level changes (4). The
IDHs involved in isocitrate oxidative decarboxylation are crucial
for providing metabolic substrates and energy, and regulating the
cellular redox status (5–7). Furthermore, α-KG generated from
isocitrate oxidative decarboxylation is essential for the
α-KG-dependent function of dioxygenase, which is important for DNA
and histone demethylation and DNA repair (8).
Previous studies have revealed that the
dysregulation of isocitrate dehydrogenase 1 (IDH1) activity, which
is caused by site mutations and expression changes, contributed to
the occurrence and progression of Ollier disease and Maffucci
syndrome (9,10), spondyloenchondromatosis with
D-2-hydroxyglutaric aciduria (11), glioblastoma (12–15),
acute myeloid leukemia (16) and
early skin tumorigenesis (17),
amongst others. Among IDH1 mutations, arginine to histidine
substitution at the 132th codon (R132H) is the most prevalent
(13). The IDH1 R132H mutant
facilitates the reduction of α-KG to D-2-hydroxyglutarate (D-2HG)
using NADPH, resulting in a decrease in NADPH and α-KG, and an
accumulation of D-2HG (18,19).
These alterations lead to extensive epigenetic modification
changes, metabolic imbalance, dysregulation of reactive oxygen
species and oncogenic substance accumulation (20,21).
IDH1 mutations have primarily been discovered in low-grade glioma
(LGG), secondary GBM and acute myeloid leukemia (AML); however,
they are rare in primary GBM (15). In addition to IDH1 mutations,
aberrant IDH1 expression has been correlated with cancer
progression and metastasis and whether IDH1 is upregulated or
downregulated varies in different types of cancer (13–15).
IDH1 is downregulated in early skin cancer, resulting in a higher
vulnerability of skin tumorigenesis to tumor-inducing substances
(17). In contrast, IDH1
expression was reported to be elevated in numerous types of cancer.
For instance, IDH1 is upregulated in non-small cell lung carcinoma
(NSCLC) (22). The knockdown of
IDH1 using short hairpin (sh)RNAs impedes the growth and
proliferation of NSCLC cells (22). IDH1 is also upregulated in 65% of
primary GBM cases (15). The
inhibition of IDH1 by shRNAs or chemical molecules hinders the
growth of GBM cells and extends the survival of mice with tumor
xenografts (15). Despite the
established roles of IDH1 in cancer, the function of wild-type IDH1
in primary GBM cell migration remains unclear.
Materials and methods
Cell culture
The human GBM cell line U-87 MG (U87; glioblastoma
of unknown origin) was purchased from The Cell Bank of Type Culture
Collection of the Chinese Academy of Sciences and was authenticated
by Short Tandem Repeat profiling as described previously (23). U87 cells were cultured in DMEM
(HyClone; Cytiva) supplemented with 10% FBS (Clark Bioscience), 100
U/ml penicillin and 100 µg/ml streptomycin (Biosharp Life Sciences)
at 37°C in a cell culture incubator (Thermo Forma 4131; Thermo
Fisher Scientific, Inc.) with 5% CO2.
To determine the effect of α-Ketoglutarate (α-KG;
Sigma-Aldrich; Merck KGaA) on cell migration, U87 cells were
treated with 1 or 2.5 mM α-KG supplemented-medium during culture at
37°C for 24 h. To investigate the effect of the IDH1/α-KG axis on
the PI3K/AKT/mTOR signaling pathway, rapamycin (MedChemExpress), an
mTOR-specific inhibitor, was used to treat wild-type or
IDH1-overexpressing U87 cell cultures at 37°C for 24 h to block the
PI3K/AKT/mTOR signaling pathway. Mock control-treated cells
[treated with DMSO (1:200)] were used as controls.
Plasmids and cell transfection
IDH1 overexpression plasmids (pcDNA3-cMyc-IDH1) were
generated by inserting IDH1 coding gene fragments into pcDNA3
vectors (Invitrogen; Thermo Fisher Scientific, Inc.). The IDH1
coding gene fragment was amplified from the human cDNA library,
which was produced by the reverse transcription of total RNA
extracted from 293T cells, using the following primer pairs:
Forward (containing the BamHI enzyme site and the cMyc tag
sequence),
5′-CCGGATCCGCCACCATGGAGCAGAAGCTGATCTCAGAGGAGGACCTGATGTCCAAAAAAATCAGTGGCG-3′
and reverse (containing the EcoRI enzyme site),
5′-CGCGAATTCTTAAAGTTTGGCCTGAGCTAGT-3′. The amplified fragment was
then ligated into the pcDNA3 vector at the BamHI and
EcoRI sites. pcDNA3-cMyc-IDH1 plasmids were sequenced to
confirm its validity using T7 and SP6 general sequencing primers at
General Biosystems, Inc. The pcDNA3 empty vector was used as a
control. IDH1 overexpression (OE; IDH1-OE) and control (Ctrl) cell
lines were constructed by stably transfecting U87 cells with IDH1
overexpression plasmids (pcDNA3-cMyc-IDH1) and empty vectors
(pCDNA3), respectively. IDH1 overexpression was verified by western
blotting with antibodies against the cMyc tag and IDH1.
A total of 2 IDH1 shRNA plasmids (shIDH1-1 and
shIDH1-2) were produced by inserting shRNA sequences designed
against IDH1 into a pLL4.0 vector, which was produced by replacing
the GFP expression cassette of the pLL3.7 vector (cat. no. 11795;
Addgene, Inc.) with a Neomycin expression cassette. The shRNA
sequences were placed downstream of the U6 promoter in the pLL4.0
vector. The shRNAs were designed using the siRNA at WHITEHEAD
website (http://jura.wi.mit.edu/bioc/siRNAext) and were
synthesized at General Biosystems, Inc. The sense and antisense
strands were annealed into double-stranded DNA fragments, which
were subsequently phosphorylated. The phosphorylated shRNA
fragments were ligated into the pLL4.0 vector at the HpaI
and XhoI sites. The shRNA plasmids were sequenced to confirm
their validity. Scramble shRNA (shScramble) was used as a control.
The sequences of the shRNA sense and antisense strands are listed
in Table SI.
The plasmid transfections were performed using 1.5
µg plasmid/well and Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol, upon the cells reaching 70–90% confluence
(8.0×105 cells/well). Following transfection at 37°C for
24 h, the culture medium was changed and selection was performed
beginning at 48 h post-transfection to generate stable cell lines;
400 µg/ml geneticin (Sigma-Aldrich; Merck KGaA) or 1 µg/ml
puromycin (Sangon Biotech Co., Ltd.) was added to the culture
medium of cells transfected with the pCDNA3- or pLL4.0-based
plasmid.
Western blotting
U87 cell lysates were extracted using Cell Lysis
Buffer (Beyotime Institute of Biotechnology) supplemented with
protease inhibitors (cOmplete, Mini, EDTA-free Protease Inhibitor
Cocktail; Roche Diagnostics), according to the manufacturer's
protocol. Protein concentrations were measured using the BCA
protein assay kit (Biosharp Life Sciences), according to the
manufacturer's protocol, and were adjusted to the same
concentration in each set of experiments. Protein samples (4
µg/lane) were subjected to electrophoresis on 10% SDS-PAGE gels and
then transferred onto PVDF membranes. The membranes were blocked
with 5% bovine serum albumin (Sigma-Aldrich; Merck KGaA) at room
temperature for 1 h and incubated with primary antibodies overnight
at 4°C. The membranes were then incubated with horseradish
peroxidase (HRP)-conjugated secondary antibodies at room
temperature for 1.5 h and reacted with chemiluminescent substrates
(Biosharp Life Sciences). The signals were captured by the Tanon
5200 Imaging system (Tanon 5200; Tanon Science and Technology Co.,
Ltd.) and the expression levels were analyzed using ImageJ 1.52a
software (National Institutes of Health). The antibodies and their
dilutions were as follows: cMyc (1:1,000; Thermo Fisher Scientific,
Inc.; cat. no. A21280), IDH1 (1:1,000; Hangzhou HuaAn Biotechnology
Co., Ltd.; cat. no. EM40705), GAPDH (1:2,000; Biosharp Life
Sciences; cat. no. BL006B), phosphorylated (p)-AKT1 (Ser473;
1:1,000; Invitrogen; Thermo Fisher Scientific, Inc.; cat. no.
MA120325), AKT (1:1,000; Invitrogen; Thermo Fisher Scientific,
Inc.; cat. no. 44609G), mTOR (1:1,000; Cell Signaling Technology,
Inc.; cat. no. 2983), p-mTOR (Ser2448; 1:1,000; Cell Signaling
Technology, Inc.; cat. no. 9205), goat anti-mouse HRP-conjugated
immunoglobulin (Ig) G (1:2,000; Biosharp Life Sciences; cat. no.
BL001A) and donkey anti-rabbit HRP-conjugated IgG (1:2,000;
Invitrogen; Thermo Fisher Scientific, Inc.; cat. no. 31458).
Total RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from cells using the Total
RNA Isolation reagent (Biosharp Life Sciences). Reverse
transcription was performed using the FastKing RT kit (Tiangen
Biotech Co., Ltd.), according to the manufacturer's protocol, and
qPCR was performed using the Powerup SYBR Master mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol, on an RT-qPCR machine (Bio-Rad CFX96
Touch; Bio-Rad Laboratories, Inc.). The following thermocycling
conditions were used for the qPCR: Pre-culture at 50°C for 120 sec;
initial denaturation at 95°C for 120 sec; 40 cycles of annellation
at 95°C for 15 sec and elongation at 60°C for 60 sec; and a default
dissociation step of 95°C for 15 sec and then heated from 60–95°C
in incremental steps of 0.2°C for 15 sec. GAPDH served as an
internal control. Gene expression levels were quantified using the
2−ΔΔCq method (24).
The primers used for RT-qPCR (IDH1 and GAPDH) are listed in
Table SI.
Transwell migration assay
Transwell migration assays were performed as
previously reported (25).
Transwell inserts with 8.0-µm pore polycarbonate membranes (Corning
Inc.) and 24-well plates were utilized. U87 cells were initially
seeded at 2.5×104 cells/well in a 100 µl serum-free
DMEM, supplemented with 100 U/ml penicillin and 100 µg/ml
streptomycin, in the upper chamber. DMEM supplemented with 10% FBS,
100 U/ml penicillin and 100 µg/ml streptomycin was plated into the
lower chambers. Following incubation for 20 h at 37°C in a cell
culture incubator with 5% CO2, the migratory cells were
fixed with 4% paraformaldehyde at room temperature for 10 min and
then 100% methanol for 10 min at room temperature. Next, the cells
were stained with 0.05% crystal violet for 30 min at room
temperature. Images were captured using the bright field channel of
an Olympus IX71 fluorescence microscope (magnification, ×100;
Olympus Corporation).
Cell wound healing assay
U87 cells were seeded in 6-well plates at a seeding
density of 6×105 cells/well and cultured overnight at
37°C in a cell culture incubator with 5% CO2. Wounds
were generated by scratching cells with 0–200 µl pipette tips.
Following scratching, cells were briefly washed three times with
PBS and cultured in DMEM for 48 h. The cells were harvested at 0
and 48 h post-scratch. The cells were washed twice with cold PBS
and fixed with chilled methanol for 10 min on ice. Cells were
incubated with a staining solution that contained 0.05% crystal
violet and 25% methanol for 20 min at room temperature and washed
with distilled water. Images were captured with the bright field
channel of an Olympus IX71 fluorescence microscope (magnification,
×40; Olympus Corporation). The width of the wounds was measured by
Canvas X software (version 19; Canvas GFX) and the migratory
distances were calculated using the following equation: Scratch
width at 0 h-scratch width at 48 h. All migratory distances were
normalized to the migratory distances of the control groups at 0
h.
α-KG measurement
α-KG levels in U87 cells were measured using the
a-Ketoglutarate Colorimetric/Fluorometric Assay kit (BioVision,
Inc.) according to the manufacturer's protocol. The values were
determined by measuring absorbance at 570 nm using a TECAN
Microplate Reader (Tecan Group, Ltd.).
TCGA data analysis
TCGA data analysis was performed using the UALCAN
website (ualcan.path.uab.edu/index.html) (26). GBM samples were selected for
analysis; the analysis included 156 primary GBM samples and 5
normal samples. The transcript per million (TPM) of IDH1, AKT,
PTEN, CDK2, Myc, MDM2, SNAIL2, N-cadherin, Vimentin, TWIST1, ZEB1
and RAC1 in the above samples were analyzed using the UALCAN
website and plotted as boxplots.
Statistical analysis
Experiments were performed in triplicate. All data
were normalized to the controls and presented as the mean ±
standard deviation, unless otherwise stated. Student's t-tests were
used for two-group comparisons and one-way ANOVA, followed by
post-hoc Tukey tests, was used for multiple-group comparisons.
P<0.05 was considered to indicate a statistically significant
difference. Significant differences in the statistical analyses are
labeled as ‘*’ in all the figures. All statistical analyses were
performed using GraphPad Prism software (version 8; GraphPad
Software, Inc.).
Results
Downregulation of IDH1 inhibits
primary GBM cell migration
Previously, numerous studies have revealed that IDH1
site mutations, particularly the IDH1 R132H mutation, promoted
tumorigenesis (13,18–21).
However, in primary GBM, IDH1 mutations were not the main
cancer-causing factor (15). The
analysis of TCGA data demonstrated a significant increase in IDH1
expression in samples from patients with primary GBM compared with
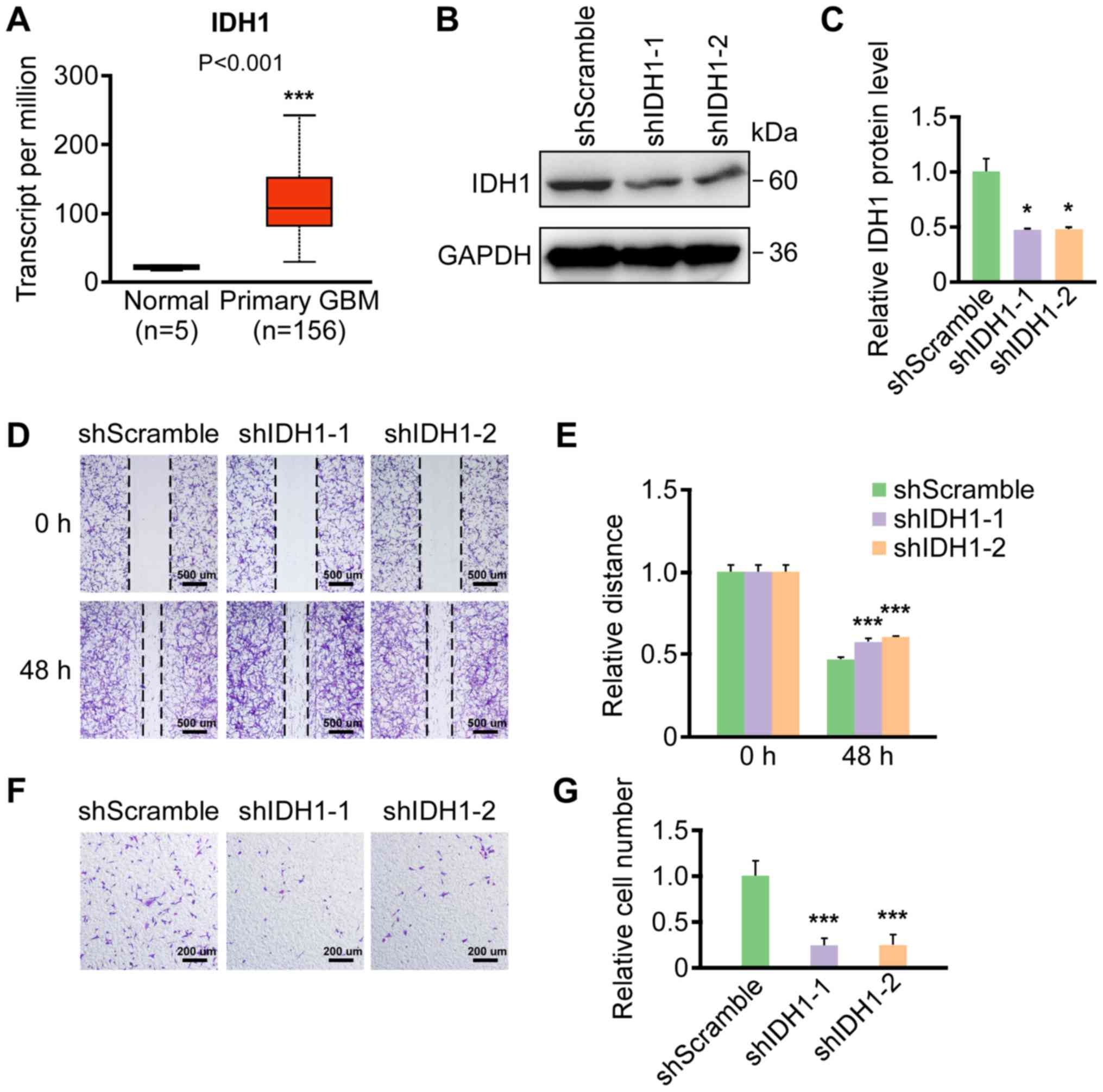
adjacent normal samples (Fig. 1A).
IDH1 knockdown and scramble control cell lines were constructed by
stably transfecting U87 cells with two IDH1 shRNA expression
plasmids (shIDH1-1 or shIDH1-2) or shScramble expression plasmids,
respectively. IDH1 knockdown was verified by western blotting
(Fig. 1B and C) and RT-qPCR
(Fig. S1A). Wound healing assays
were performed on IDH1 knockdown and scramble shRNA control cells.
At 48 h post-scratch, shIDH1-1 and shIDH1-2 groups demonstrated
relatively delayed migration compared with the scramble shRNA group
(Fig. 1D and E). Additionally,
Transwell migration assays were performed with the same cell lines,
the results of which demonstrated that IDH1 knockdown had fewer
cells that successfully migrated through the membrane of the
Transwell inserts (Fig. 1F and G).
In summary, the results indicated that IDH1 downregulation
repressed the migration of primary GBM cells.
Upregulation of IDH1 promotes primary
GBM cell migration
As the downregulation of IDH1 repressed primary GBM
cell migration, the effect of ectopic IDH1 expression on primary
GBM cell migration was investigated. The results revealed that cMyc
was strongly expressed in the IDH1-OE group; however, it was not
detected in the Ctrl group (Fig.
2A). Furthermore, IDH1 expression was significantly increased
in the IDH1-OE group compared with the Ctrl group (Fig. 2B). Similarly, IDH1 mRNA levels were
increased in the IDH1-OE group, as indicated by RT-qPCR (Fig. S1B). IDH1 expression did not
increase markedly, which may be due to IDH1 being endogenously
abundant in U87 cells (15). A
wound healing assay was then performed on IDH1-OE and control cell
lines. Significantly faster migration was observed in the IDH1-OE
group (Fig. 2C and D). The results
of the Transwell migration assays also demonstrated that IDH1
overexpression promoted U87 cell migration (Fig. 2E and F). In conclusion, IDH1
overexpression facilitated the migration of primary GBM cells.
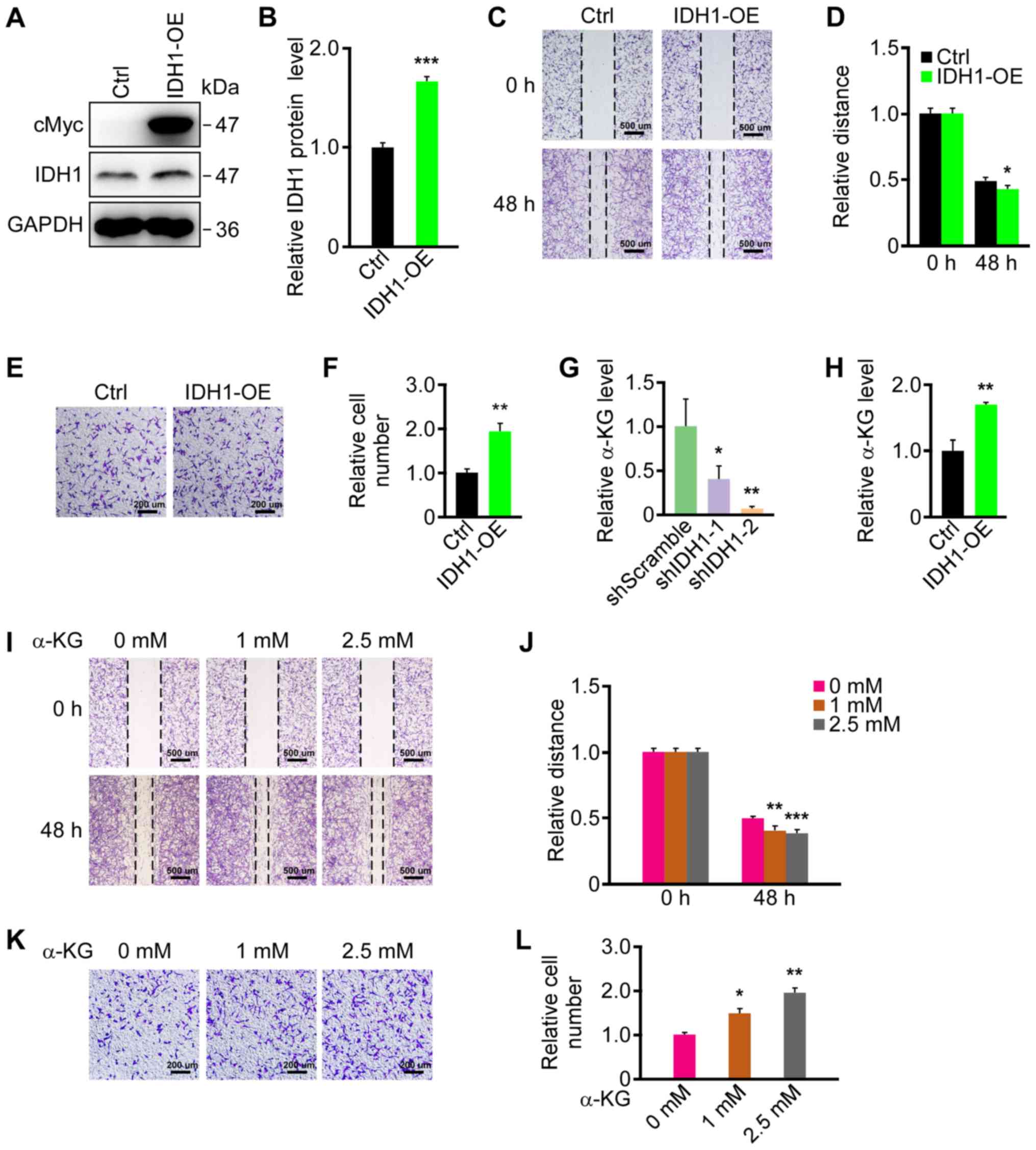 | Figure 2.IDH1 overexpression and α-KG
treatment promoted primary GBM cell migration. (A) IDH1
overexpression was verified by western blotting. (B)
Semi-quantification of expression levels from part (A). (C) IDH1
overexpression promoted the migration of U87 cells as demonstrated
by wound healing assays. (D) Semi-quantification of the relative
migratory distances in part (C). (E) IDH1 overexpression promoted
the migration of U87 cells as revealed by Transwell migration
assays. (F) Semi-quantification of relative cell numbers in part
(E). (G) α-KG was downregulated by IDH1 knockdown in U87 cells. (H)
α-KG was upregulated by IDH1 overexpression in U87 cells. (I) α-KG
promoted the migration of U87 cells in wound-healing assays in a
dose-dependent manner. (J) Semi-quantification of relative
migratory distances in part (I). (K) α-KG promoted the migration of
U87 cells in a dose-dependent manner as demonstrated by Transwell
migration assays. α-KG treatment lasted for 24 h. (L)
Semi-quantification of relative cell numbers in part (K).
*P<0.05, **P<0.01, ***P<0.001 vs. shScramble/Ctrl/0 mM.
IDH1, isocitrate dehydrogenase 1; α-KG, α-ketoglutarate; GBM,
glioblastoma; Ctrl, control; IDH1-OE, IDHI1 overexpression cell
line; shScramble, scramble shRNA; shIDH-1/2, IDH1 shRNA plasmids
1/2; shRNA, short hairpin RNA. |
IDH1 regulates primary GBM cell
migration by affecting αKG levels
Since IDH1 catalyzes the conversion of isocitrate to
α-KG and CO2, whether α-KG levels in primary GBM cells
were associated with IDH1 expression was examined. α-KG levels were
significantly downregulated in the IDH1 knockdown groups (Fig. 2G) and significantly upregulated
when IDH1 was overexpressed (Fig.
2H). Whether α-KG levels affected primary GBM cell migration
was then investigated. U87 cells were treated with 1 and 2.5 mM
α-KG in the medium during culture. The migration of the U87 cells
was promoted by the addition of α-KG in a dose-dependent manner in
the wound healing and Transwell assays (Fig. 2I-L). These results indicated that
α-KG levels may mediate the changes in migration in primary GBM
cells caused by IDH1 level alterations.
PI3K/AKT/mTOR pathway activity is
enhanced in primary GBM
Primary GBM is a type of cancer with the highest
mortality partly due to its relatively high migration rate
(27,28). TCGA database analysis revealed that
PI3K/AKT/mTOR pathway activity was significantly increased in
primary GBM. AKT and phosphatase and tensin homolog, which are key
components of the PI3K/AKT/mTOR pathway, were significantly
upregulated and downregulated in primary GBM, respectively
(Fig. S2A). The expression of the
genes downstream of the PI3K/AKT/mTOR pathway and whose expression
is regulated by the pathway was then examined. Cyclin-dependent
kinase 2, Myc and mouse double minute 2 homolog were significantly
upregulated in primary GBM, which are proteins that promote the
cell cycle and repress apoptosis (Fig. S2B) (29). Snail family transcriptional
repressor 2, N-cadherin and vimentin were also significantly
upregulated, which facilitate the epithelial-mesenchymal transition
(EMT) process (Fig. S2C).
Additionally, twist-related protein 1, zinc finger E-box-binding
homeobox 1 and Ras-related C3 botulin toxin substrate 1 were
upregulated, which are proteins that enhance cell migration and
metastasis (Fig. S2D) (29–31).
The results indicated that the PI3K/AKT/mTOR pathway was
hyperactivated in primary GBM, which is associated with the cell
cycle, apoptosis, EMT, cell migration and metastasis processes
(32–36).
PI3K/AKT/mTOR pathway activity is
altered by changes in IDH1 and α-KG levels in primary GBM
cells
As PI3K/AKT/mTOR pathway activity was increased in
primary GBM, whether IDH1 and α-KG expression affected the
PI3K/AKT/mTOR pathway was investigated. The results of western
blotting revealed that IDH1 knockdown and overexpression resulted
in the downregulation and upregulation of p-AKT (Ser473) and the
p-mTOR (Ser2448), respectively, in primary GBM cells (Fig. 3A-D and G-J). By treating primary
GBM cells with 1 and 2.5 mM α-KG, p-AKT (Ser473) and p-mTOR
(Ser2448) were significantly upregulated in a dose-dependent manner
(Fig. 3E, F and K, L). The results
indicated that IDH1 and α-KG levels regulated PI3K/AKT/mTOR pathway
activity in primary GBM cells.
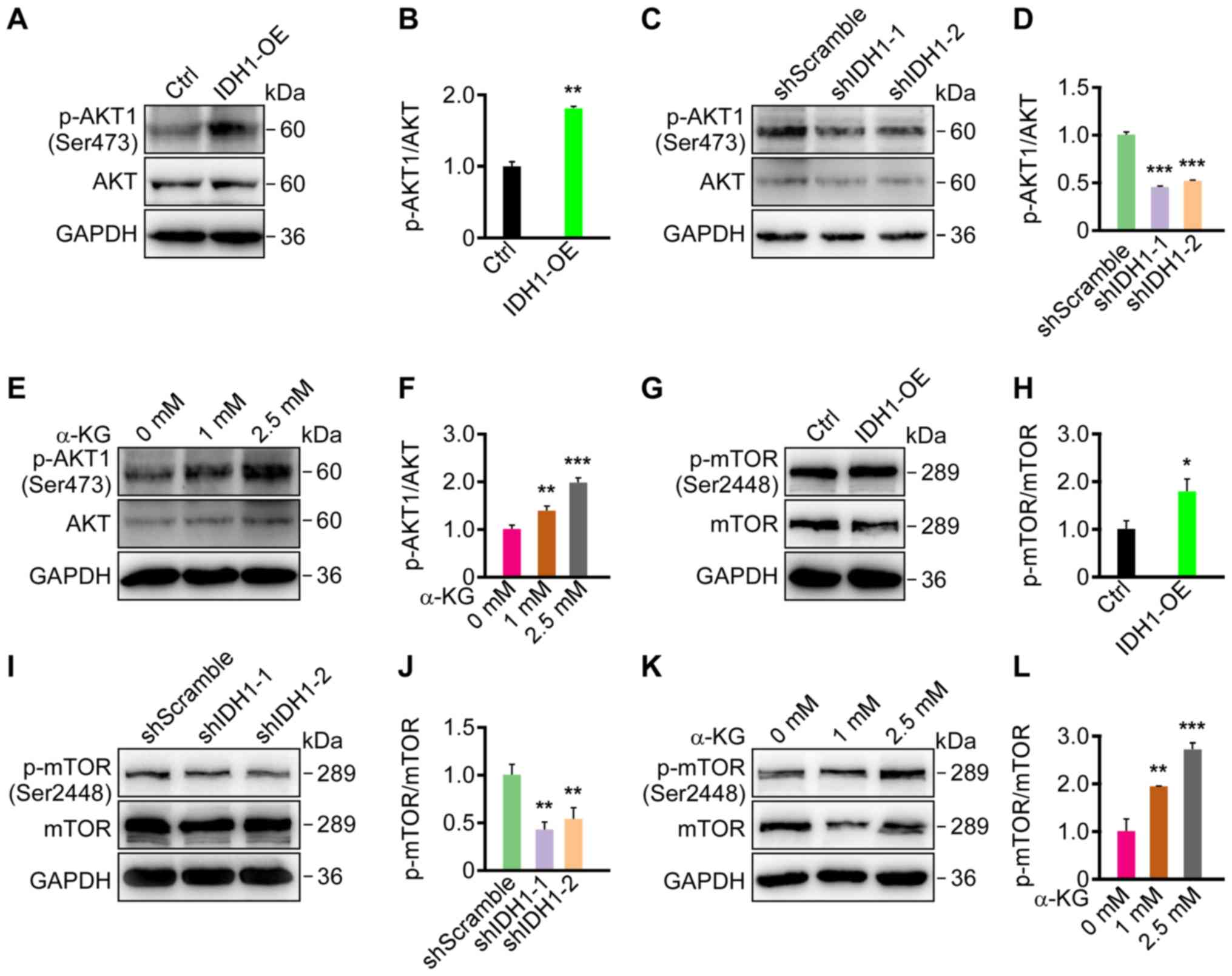 | Figure 3.Phosphoinositide 3-kinase/AKT/mTOR
pathway activity was altered by changes in the levels of IDH1 and
α-KG in primary GBM cells. (A) p-AKT (Ser473) was promoted by IDH1
overexpression in U87 cells. (B) Semi-quantification of the
expression levels from part (A). (C) p-AKT (Ser473) was decreased
by IDH1 knockdown in U87 cells. (D) Semi-quantification of
expression levels from part (C). (E) p-AKT (Ser473) was promoted by
α-KG treatment in a dose-dependent manner in U87 cells. (F)
Semi-quantification of the expression levels from part (E). (G)
p-mTOR (Ser2448) was promoted by IDH1 overexpression in U87 cells.
(H) Semi-quantification of the expression levels from (G). (I)
p-mTOR (Ser2448) was repressed by IDH1 knockdown in U87 cells. (J)
Semi-quantification of the expression levels from part (I). (K)
p-mTOR (Ser2448) was promoted by α-KG treatment in a dose-dependent
manner in U87 cells. α-KG treatment lasted for 24 h. (L)
Semi-quantification of the expression levels from part (K).
*P<0.05, **P<0.01, ***P<0.001 vs. shScramble/Ctrl/0 mM. p,
phosphorylated; AKT, protein kinase B; IDH1, isocitrate
dehydrogenase 1; α-KG, α-ketoglutarate; GBM, glioblastoma; Ser,
serine; Ctrl, control; IDH1-OE, IDHI1 overexpression cell line;
shRNA, short hairpin RNA; shScramble, scramble shRNA; shIDH-1/2,
IDH1 shRNA plasmids 1/2. |
The IDH1/α-KG axis regulates primary
GBM cell migration through the PI3K/AKT/mTOR pathway
As PI3K/AKT/mTOR pathway activity was regulated by
changes in the IDH1 and α-KG levels, whether IDH1/α-KG regulated
primary GBM cell migration by modulating the PI3K/AKT/mTOR pathway
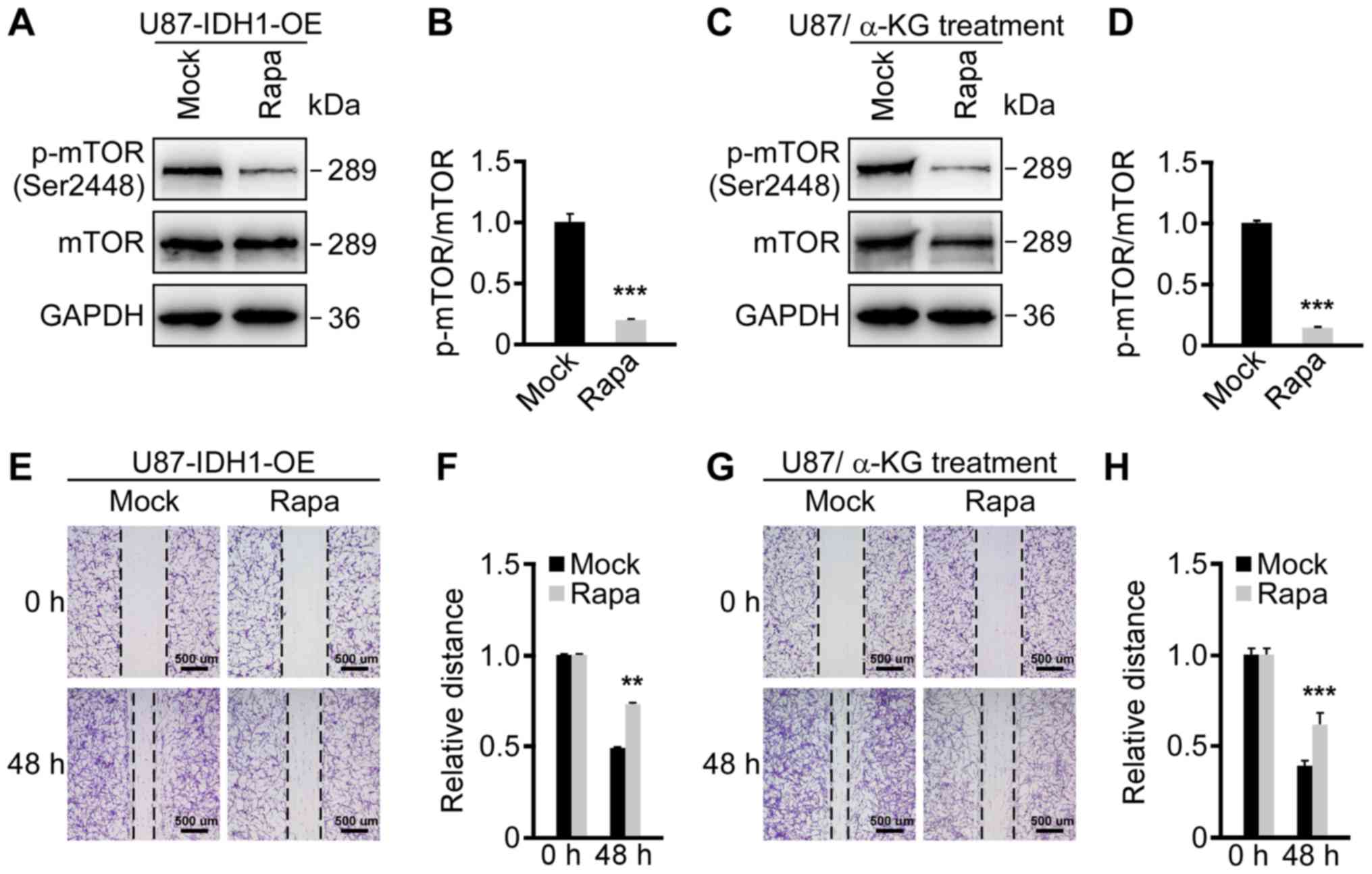
was examined. Wild-type U87 cells were treated with rapamycin,
which is an mTOR-specific inhibitor, to block the PI3K/AKT/mTOR
pathway. Mock control-treated cells were used as controls. Western
blotting results reported that p-mTOR (Ser2448) was significantly
inhibited by rapamycin treatment compared with controls (Fig. S3A and B). Additionally, rapamycin
treated U87 cells exhibited significantly delayed cell migration
compared with controls, indicating that blocking the PI3K/AKT/mTOR
pathway led to the repression of cell migration (Fig. S3C and D). Furthermore,
IDH1-overexpressing U87 cells and α-KG-treated U87 cells were
treated with rapamycin. Western blotting results demonstrated that
p-mTOR (Ser2448) was repressed by rapamycin in both treatment
groups (Fig. 4A-D). The increased
cell migration caused by IDH1 overexpression and α-KG
supplementation were also reversed following rapamycin treatment
(Fig. 4E-H). The results indicated
that the IDH1/α-KG axis regulated primary GBM cell migration by
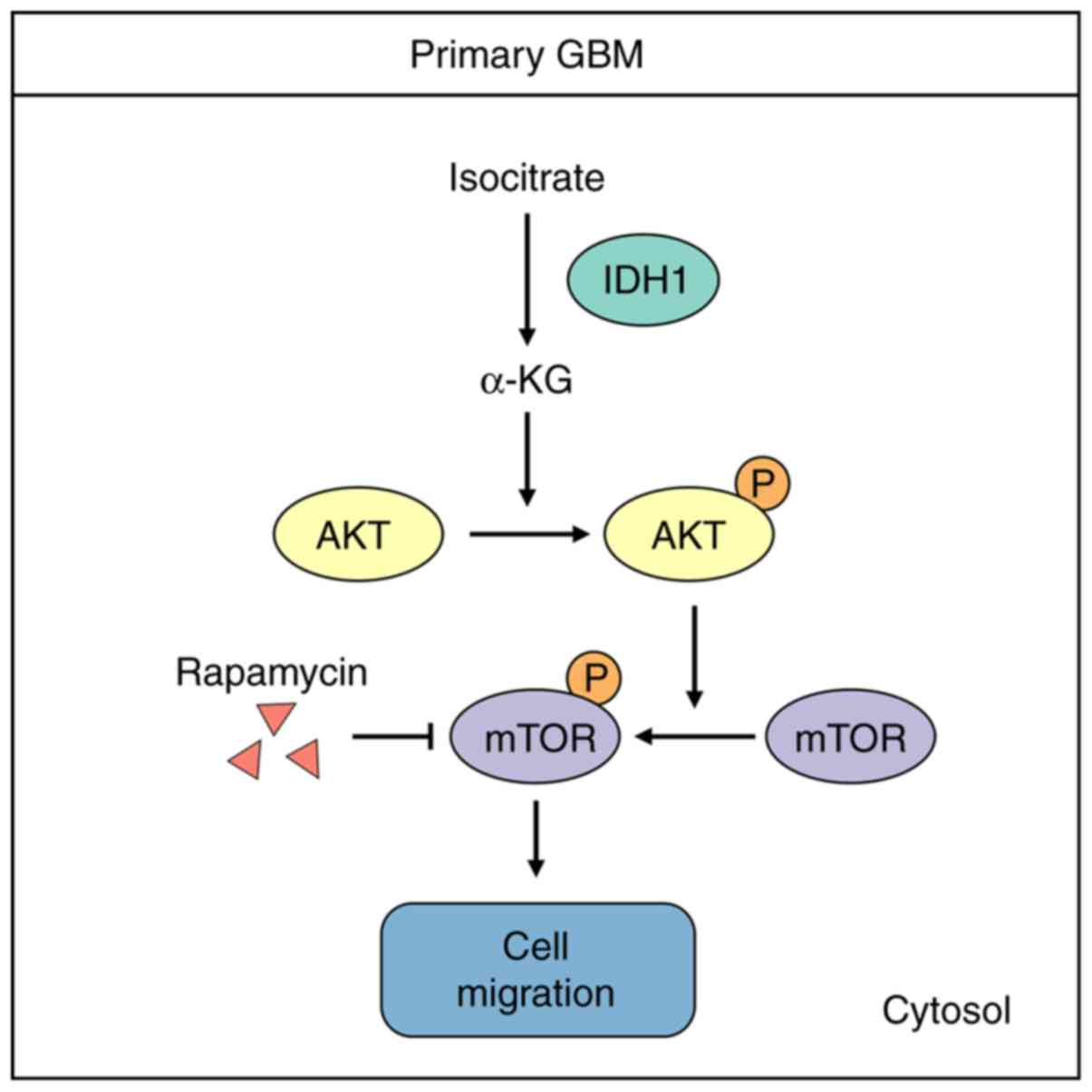
modulating the PI3K/AKT/mTOR pathway (Fig. 5).
Discussion
IDH1 is an important enzyme in cell metabolism that
catalyzes the oxidative decarboxylation of isocitrate to produce
α-KG, NADPH and CO2 (37). The roles of site-mutated IDH1 in
tumorigenesis are well established (13,18–21);
however, the function of wild-type IDH1 in cancer has not been
studied extensively. Clinical data has demonstrated that
site-mutated IDH1 was mainly detected in LGG, secondary GBM and
AML; however, it was observed in only 5% of patients with primary
GBM (15). According to TCGA
database, IDH1 expression was significantly upregulated in primary
GBM, which indicated that alterations in wild-type IDH1 levels may
contribute to primary GBM tumorigenesis. This result was consistent
with that of a previous study, where wild-type IDH1 was upregulated
in primary GBM, instead of being mutated (15). Thus, whether upregulated IDH1
expression was crucial to primary GBM progression was subsequently
investigated. Calvert et al (15) reported that IDH1 suppression via
shRNA or specific inhibitors inhibited primary GBM growth and
facilitated cellular differentiation. However, the role of IDH1 in
primary GBM cell migration remains elusive. Considering that
primary GBM is a type of cancer that exhibits relatively high
migratory abilities (27,28), excess IDH1 was hypothesized to
contribute to primary GBM migration. The current study discovered
that IDH1 knockdown or overexpression led to repressed or improved
cell migration, respectively.
α-KG is primarily produced by IDH1 via oxidative
decarboxylation (38,39). Therefore, whether α-KG mediated the
effect of wild-type IDH1 on primary GBM cell migration was
investigated. Cellular α-KG levels were positively associated with
changes in IDH1 levels. By treating U87 cells with different
concentrations of α-KG, dose-dependent increases in the migration
rates of primary GBM cells were observed. However, to the best of
our knowledge, there is no applicable method to directly reduce
α-KG levels in live cells, which impeded the current study to
examine the effect of decreased α-KG on cell migration. The most
common way to reduce α-KG levels in live cells is to repress
enzymes that catalyze the production of α-KG, such as IDH1.
Therefore, the current study investigated the effect of IDH1
knockdown and the results matched expectations. Thus, the changes
in the migration of primary GBM cells mediated by changes in IDH1
levels may occur by altering α-KG levels.
The PI3K/AKT/mTOR pathway regulates multiple
cellular events, including growth, proliferation, motility and
survival, which are often dysregulated in cancer (40). Although numerous previous studies
(41–43) have reported the effect of the IDH1
R132H mutation on the PI3K/AKT/mTOR pathway, the correlation
remains unclear. While IDH1 R132H and D-2HG were discovered to
inhibit the PI3K/AKT/mTOR pathway in human glioma samples (41), another previous study reported that
IDH1 R132H and 2-HG promoted the PI3K/AKT/mTOR pathway, resulting
in upregulated glioma migration (44,45).
Despite previous reports on mutated IDH1, the function of wild-type
IDH1 and α-KG levels on the PI3K/AKT/mTOR pathway remain unclear.
The current study revealed that PI3K/AKT/mTOR pathway activity was
enhanced by IDH1 overexpression and α-KG treatment, and repressed
by IDH1 knockdown. To further investigate whether the IDH1/α-KG
axis regulated primary GBM cell migration by modulating the
PI3K/AKT/mTOR pathway, rapamycin treatment combined with IDH1
overexpression or α-KG supplementation was employed. The results
demonstrated that the increased cell migration of primary GBM cells
was reversed, indicating that the IDH1/α-KG axis regulated cell
migration in primary GBM cells via the PI3K/AKT/mTOR pathway.
Despite the mechanism revealed in the present study,
there are areas of research that require further study. Firstly, as
the results of the current study demonstrated that IDH1 may be a
potential therapeutic target or diagnostic marker in primary GBM,
further in vivo investigations are required prior to
clinical application. Secondly, although α-KG levels were reported
to regulate the PI3K/AKT/mTOR pathway in primary GBM cells, the
detailed mechanism of how α-KG affected the PI3K/AKT/mTOR pathway
remains, to the best of our knowledge, unknown and should be
investigated in future studies.
In conclusion, the current study discovered that the
wild-type IDH1/α-KG axis regulated primary GBM cell migration
through the PI3K/AKT/mTOR pathway. In contrast to numerous previous
reports that focused on the roles of IDH1 mutations in
tumorigenesis (13,18–21),
the present study was, to the best of our knowledge, the first
study on the function of wild-type IDH1 in primary GBM cell
migration. Moreover, the results demonstrated that α-KG was the
intermediate molecule between IDH1 and the PI3K/AKT/mTOR pathway.
This mechanism reported by the current study expanded the
understanding of the process of primary GBM tumorigenesis and may
be beneficial for therapy against primary GBM.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The current study was funded by the National Natural
Science Foundation of China (grant no. 31701289), Anhui Provincial
Natural Science Foundation (grant no. 1808085QH234), Anhui
Provincial Funding Scheme to Outstanding Innovative Programs by
Returned Scholars (grant no. 2019LCX003), Educational Commission of
Anhui Province of China (grant nos. KJ2017A319 and KJ2019A0498),
Foundation for High-level Talents in Higher Education of Anhui
Province of China [grant no. (2017)51] and the Anhui Normal
University (grant no. 2017XJJ38; start-up funds to XS).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XS, SW, JZ, ML, FX, AW, YL and GZ collected and
analyzed the data. XS and SW conceptualized the current study. AW
and YL prepared materials. XS, SW and GZ wrote the original
manuscript. XS reviewed and edited the manuscript. XS and GZ
supervised the current study. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mamelak AN and Jacoby DB: Targeted
delivery of antitumoral therapy to glioma and other malignancies
with synthetic chlorotoxin (TM-601). Expert Opin Drug Deliv.
4:175–186. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang K, Niu L, Bai Y and Le W:
Glioblastoma: Targeting the autophagy in tumorigenesis. Brain Res
Bull. 153:334–340. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gallego O: Nonsurgical treatment of
recurrent glioblastoma. Curr Oncol. 22:e273–e281. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bergaggio E and Piva R: Wild-type IDH
enzymes as actionable targets for cancer therapy. Cancers (Basel).
11:5632019. View Article : Google Scholar
|
|
5
|
Kirkman HN, Galiano S and Gaetani GF: The
function of catalase-bound NADPH. J Biol Chem. 262:660–666.
1987.PubMed/NCBI
|
|
6
|
Itsumi M, Inoue S, Elia AJ, Murakami K,
Sasaki M, Lind EF, Brenner D, Harris IS, Chio IIC, Afzal S, et al:
Idh1 protects murine hepatocytes from endotoxin-induced oxidative
stress by regulating the intracellular NADP(+)/NADPH ratio. Cell
Death Differ. 22:1837–1845. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee SM, Koh HJ, Park DC, Song BJ, Huh TL
and Park JW: Cytosolic NADP(+)-dependent isocitrate dehydrogenase
status modulates oxidative damage to cells. Free Radic Biol Med.
32:1185–1196. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gagné LM, Boulay K, Topisirovic I, Huot ME
and Mallette FA: Oncogenic activities of IDH1/2 mutations: From
epigenetics to cellular signaling. Trends Cell Biol. 27:738–752.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pansuriya TC, van Eijk R, d'Adamo P, van
Ruler M, Kuijjer ML, Oosting J, Cleton-Jansen AM, van Oosterwijk
JG, Verbeke SLF, Meijer D, et al: Somatic mosaic IDH1 and IDH2
mutations are associated with enchondroma and spindle cell
hemangioma in ollier disease and maffucci syndrome. Nat Genet.
43:1256–1261. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pansuriya TC, Kroon HM and Bovee JV:
Enchondromatosis: Insights on the different subtypes. Int J Clin
Exp Pathol. 3:557–569. 2010.PubMed/NCBI
|
|
11
|
Struys EA, Salomons GS, Achouri Y, Van
Schaftingen E, Grosso S, Craigen WJ, Verhoeven NM and Jakobs C:
Mutations in the D-2-hydroxyglutarate dehydrogenase gene cause
D-2-hydroxyglutaric aciduria. Am J Hum Genet. 76:358–360. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Parsons DW, Jones S, Zhang X, Lin JCH,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yan H, Parsons DW, Jin G, McLendon R,
Rasheed BA, Yuan W, Kos I, Haberle IB, Jones S, Riggins GJ, et al:
IDH1 and IDH2 mutations in gliomas. N Engl J Med. 360:765–773.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Balss J, Meyer J, Mueller W, Korshunov A,
Hartmann C and von Deimling A: Analysis of the IDH1 codon 132
mutation in brain tumors. Acta Neuropathol. 116:597–602. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Calvert AE, Chalastanis A, Wu Y, Hurley
LA, Kouri FM, Bi Y, Kachman M, May JL, Bartom E, Hua Y, et al:
Cancer-associated IDH1 promotes growth and resistance to targeted
therapies in the absence of mutation. Cell Rep. 19:1858–1873. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ma QL, Wang JH, Wang YG, Hu C, Mu QT, Yu
MX, Wang L, Wang DM, Yang M, Yin XF, et al: High IDH1 expression is
associated with a poor prognosis in cytogenetically normal acute
myeloid leukemia. Int J Cancer. 137:1058–1065. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Robbins D, Wittwer JA, Codarin S, Circu
ML, Aw TY, Huang TT, Van Remmen H, Richardson A, Wang DB, Witt SN,
et al: Isocitrate dehydrogenase 1 is downregulated during early
skin tumorigenesis which can be inhibited by overexpression of
manganese superoxide dismutase. Cancer Sci. 103:1429–1433. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
DiNardo CD, Jabbour E, Ravandi F,
Takahashi K, Daver N, Routbort M, Patel KP, Brandt M, Pierce S,
Kantarjian H and Manero GG: IDH1 and IDH2 mutations in
myelodysplastic syndromes and role in disease progression.
Leukemia. 30:980–984. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dang L, White DW, Gross S, Bennett BD,
Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et
al: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate.
Nature. 462:739–744. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan
S, Wahab OA, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al:
IDH mutation impairs histone demethylation and results in a block
to cell differentiation. Nature. 483:474–478. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim
SH, Ito S, Yang C, Wang P, Xiao NT, et al: Oncometabolite
2-hydroxyglutarate is a competitive inhibitor of
alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 19:17–30.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tan F, Jiang Y, Sun N, Chen Z, Lv Y, Shao
K, Li N, Qiu B, Gao Y, Li B, et al: Identification of isocitrate
dehydrogenase 1 as a potential diagnostic and prognostic biomarker
for non-small cell lung cancer by proteomic analysis. Mol Cell
Proteomics. 11:M111 008821. 2012.
|
|
23
|
Reid Y, Storts D, Riss T and Minor L:
Authentication of human cell lines by STR DNA profiling analysis.
Assay Guidance Manual (Internet) Bethesda (MD): Sittampalam GS,
Grossman A, Brimacombe K, Arkin M, Auld D, Austin CP, Baell J,
Bejcek B, Caaveiro JMM, Chung TDY, et al: Eli Lilly & Company
and the National Center for Advancing Translational Sciences; 2013,
May
1–2013
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
van de Merbel AF, van der Horst G, Buijs
JT and van der Pluijm G: Protocols for migration and invasion
studies in prostate cancer. Methods Mol Biol. 1786:67–79. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Rodriguez IP, Chakravarthi BV and Varambally S:
UALCAN: A portal for facilitating tumor subgroup gene expression
and survival analyses. Neoplasia. 19:649–658. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Holland EC: Gliomagenesis: Genetic
alterations and mouse models. Nat Rev Genet. 2:120–129. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nakada M, Nakada S, Demuth T, Tran NL,
Hoelzinger DB and Berens ME: Molecular targets of glioma invasion.
Cell Mol Life Sci. 64:458–478. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ersahin T, Tuncbag N and Cetin-Atalay R:
The PI3K/AKT/mTOR interactive pathway. Mol Biosyst. 11:1946–1954.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Popolo A, Pinto A, Daglia M, Nabavi SF,
Farooqi AA and Rastrelli L: Two likely targets for the anti-cancer
effect of indole derivatives from cruciferous vegetables:
PI3K/Akt/mTOR signalling pathway and the aryl hydrocarbon receptor.
Semin Cancer Biol. 46:132–137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee HJ, Venkatarame Gowda Saralamma V, Kim
SM, Ha SE, Raha S, Lee WS, Kim EH, Lee SJ, Heo JD and Kim GS:
Pectolinarigenin induced cell cycle arrest, autophagy, and
apoptosis in gastric cancer cell via PI3K/AKT/mTOR signaling
pathway. Nutrients. 10:10432018. View Article : Google Scholar
|
|
34
|
Zhang H, Xu HL, Wang YC, Lu ZY, Yu XF and
Sui DY: 20(S)-protopanaxadiol-induced apoptosis in MCF-7 breast
cancer cell line through the inhibition of PI3K/AKT/mTOR signaling
pathway. Int J Mol Sci. 19:10532018. View Article : Google Scholar
|
|
35
|
Larue L and Bellacosa A:
Epithelial-mesenchymal transition in development and cancer: Role
of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene.
24:7443–7454. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vanhaesebroeck B, Guillermet-Guibert J,
Graupera M and Bilanges B: The emerging mechanisms of
isoform-specific PI3K signalling. Nat Rev Mol Cell Biol.
11:329–341. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Geisbrecht BV and Gould SJ: The human PICD
gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent
isocitrate dehydrogenase. J Biol Chem. 274:30527–30533. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dimitrov L, Hong CS, Yang C, Zhuang Z and
Heiss JD: New developments in the pathogenesis and therapeutic
targeting of the IDH1 mutation in glioma. Int J Med Sci.
12:201–213. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xu X, Zhao J, Xu Z, Peng B, Huang Q,
Arnold E and Ding J: Structures of human cytosolic NADP-dependent
isocitrate dehydrogenase reveal a novel self-regulatory mechanism
of activity. J Biol Chem. 279:33946–33957. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Janku F, Yap TA and Meric-Bernstam F:
Targeting the PI3K pathway in cancer: Are we making headway? Nat
Rev Clin Oncol. 15:273–291. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Birner P, Pusch S, Christov C, Mihaylova
S, Uzeir KT, Natchev S, Schoppmann SF, Tchorbanov A, Streubel B,
Tuettenberg J and Guentchev M: Mutant IDH1 inhibits PI3K/Akt
signaling in human glioma. Cancer. 120:2440–2447. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Noushmehr H, Weisenberger DJ, Diefes K,
Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP,
Bhat KP, et al: Identification of a CpG island methylator phenotype
that defines a distinct subgroup of glioma. Cancer Cell.
17:510–522. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bralten LB, Kloosterhof NK, Balvers R,
Sacchetti A, Lapre L, Lamfers M, Leenstra S, Jonge Hd, Kros JM,
Jansen EEW, et al: IDH1 R132H decreases proliferation of glioma
cell lines in vitro and in vivo. Ann Neurol. 69:455–463. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu H, Zhang Y, Chen J, Qiu J, Huang K, Wu
M and Xia C: IDH1 R132H mutation enhances cell migration by
activating AKT-mTOR signaling pathway, but sensitizes cells to 5-FU
treatment as nadph and gsh are reduced. PLoS One. 12:e01690382017.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Carbonneau M, Gagné LM, Lalonde ME,
Germain MA, Motorina A, Guiot MC, Secco B, Vincent EE, Tumber A,
Hulea L, et al: The oncometabolite 2-hydroxyglutarate activates the
mTOR signalling pathway. Nat Commun. 7:127002016. View Article : Google Scholar : PubMed/NCBI
|