Introduction
Pulmonary fibrosis is a devastating pathological
condition associated with various chronic lung diseases,
characterized by aberrant extracellular matrix (ECM) synthesis and
accumulation, which disrupts normal alveolar architecture,
compromises oxygen diffusion, and eventually leads to respiratory
failure and death (1,2). The key pathogenic factor of fibrotic
remodeling in chronic lung injury is the formation of fibroblastic
foci, whereby lung fibroblasts transdifferentiate into
myofibroblasts in response to fibrotic stimulation (3). Fibroblast-to-myofibroblast
transdifferentiation is associated with increased expression of
α-smooth muscle actin (α-SMA) and periostin, and contributes to the
excessive production of ECM (4,5).
Therefore, an improved understanding of the molecular basis of lung
fibroblast transdifferentiation and the identification of novel
therapeutic targets are critical for treating pulmonary
fibrosis.
Transforming growth factor-β (TGF-β) has been
identified as the key driver of myofibroblast activation and plays
a crucial role in regulating the initiation and progression of
pulmonary fibrosis (6,7). In response to TGF-β stimulation, Smad
proteins are phosphorylated and translocate from the cytoplasm into
the nucleus, where they promote the transcription of the fibrotic
gene program (8,9). In addition to the canonical
Smad-dependent pathway, TGF-β can also induce pulmonary
fibrogenesis via Smad-independent pathways, including
mitogen-activated protein kinase (MAPK) pathways (10). The P38 MAPKs (P38) branch of
kinases is one of the downstream axes of MAPK pathways and has been
proven to be essential for organ fibrosis, particularly pulmonary
fibrosis (11–13). Previous studies have observed that
P38 was activated in human fibrotic lung tissue and fibroblast
cells (14). Furthermore, P38
inhibition significantly ameliorated pulmonary fibrosis and
prevented the irreversible decline in pulmonary function (15–17).
Mitogen-activated protein kinase kinase 6 (MAP2K6) is a type of
MAPKs kinase and functions as an upstream activator of P38 in the
pathogenesis of fibrotic remodeling (18). Thus, targeting these signaling
pathways may help to develop efficacious strategies to prevent
fibrotic remodeling in the lung.
MicroRNAs (miRNAs/miRs) are an abundant class of
evolutionarily conserved, single-stranded, non-coding RNAs that are
approximately 19–22 nucleotides in length and act to control gene
expression by targeting mRNAs for degradation or translational
repression (19,20). Increasing evidence has defined
miRNAs as fundamental regulators of gene expression at the
post-transcriptional level that have diverse functional roles in
fibrotic diseases, particularly pulmonary fibrosis (21,22).
Xiao et al (23),
previously found that miR-29 was downregulated in fibrotic
lungs and therapeutic delivery of miR-29 mimics alleviated
bleomycin-induced pulmonary fibrosis in mice (24). Liang et al (25), revealed that miR-26a could
directly inhibit connective tissue growth factor (Ctgf) expression,
and then diminish the proliferation and activation of lung
fibroblasts. Moreover, deletion of Dicer-1 (an integral miRNA
processing component) in lung fibroblasts decreased the biogenesis
of mature miRNAs, thereby promoting myofibroblast
transdifferentiation and collagen synthesis (26). These data revealed that miRNAs were
critical in the regulation of pulmonary fibrosis. miR-375
was initially identified as a tumor-suppressive factor, and
miR-375 silencing promoted the proliferation, invasion and
metastasis of cancer cells (27).
Numerous studies have demonstrated that miR-375 may be
downregulated in lung cancer cells, and that its expression could
be negatively associated with advanced disease stage and lymphatic
metastasis; however, its function in pulmonary fibrosis remains
unclear (28,29). Consequently, the present study
aimed to investigate the role and potential mechanism of
miR-375 in TGF-β-dependent transdifferentiation of lung
fibroblasts.
Materials and methods
Reagents and antibodies
Recombinant human TGF-β protein (active; cat. no.
ab50036) and SB203580 (P38 inhibitor; cat. no. ab120162) were
obtained from Abcam. The mimic (cat. no. miR10000728-1-5) and
inhibitor (cat. no. miR20000728-1-5) of miR-375 and their
negative controls [mimic control (MControl, cat. no.
miR1N0000001-1-5) and inhibitor control (IControl, cat. no.
miR2N0000001-1-5)] were synthesized by Guangzhou RiboBio Co., Ltd.
The small interfering RNA (siRNA) against Map2k6
(siMapk2k6; cat. no. sc-35913) was purchased from Santa Cruz
Biotechnology, Inc. and scramble control RNA (siRNA, cat. no.
sc-37007) was used as the control. Primary antibodies against the
following proteins were obtained from Abcam: α-SMA (cat. no.
ab32575, 1:1,000 dilution), periostin (cat. no. ab14041, 1:1,000
dilution), β-actin (cat. no. ab8226, 1:1,000 dilution),
phosphorylated (p)-Smad3 (cat. no. ab52903, 1:1,000 dilution, total
(t)-Smad3 (cat. no. ab40854, 1:1,000 dilution) and MAP2K6 (cat. no.
ab33866, 1:1,000 dilution). Anti-p-AKT (cat. no. 4060, 1:1,000
dilution), anti-t-AKT (cat. no. 4691, 1:1,000 dilution), anti-p-P38
(cat. no. 4511, 1:1,000 dilution) and anti-t-P38 (cat. no. 9212,
1:1,000 dilution) were purchased from Cell Signaling Technology,
Inc.
Cell culture and treatment
CCD-19Lu normal human lung fibroblasts were
purchased from the American Type Culture Collection (ATCC) and were
cultured in complete Eagle's minimum essential medium (EMEM;
ATCC® 30–2003™) containing 10% heat-inactivated fetal
bovine serum (FBS, ATCC® 30–2021™) in a cellular
incubator (5% CO2, 37°C), as previously described
(30). Cells were synchronized in
serum-free medium for 12 h after achieving 50–60% confluence, and
were then stimulated with 1, 3, 5, 10 and 15 ng/ml TGF-β for 24 h
or 10 ng/ml TGF-β for 6, 12, 24, 48 and 72 h, in order to assess
the effects of TGF-β stimulation on miR-375 expression. To
investigate the role of miR-375 in vitro, cells
(3×105/ml) were pre-treated with miR-375 mimic
(25 nM), inhibitor (50 nM) or their negative controls (Mcontrol, 25
nM; Icontrol, 50 nM) at 37°C for 4 h using
Lipofectamine® RNAiMAX reagent (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's
instructions. Subsequently, the cells were cultured in fresh EMEM
supplemented with 10% FBS for an additional 24 h before incubation
with TGF-β (10 ng/ml) for another 48 h (31–33).
For P38 inhibition, CCD-19Lu cells were incubated with the P38
inhibitor, SB203580 (10 µM; 37°C) at 1 h prior to TGF-β
stimulation. MAP2K6 knockdown was performed using siMap2k6
at 48 h before TGF-β stimulation, and the efficiency was verified
by reverse transcription-quantitative PCR (RT-qPCR). Briefly, cells
(3×105/ml) were transfected with siMap2k6 (50 nM)
or siRNA (50 nM) at 37°C for 4 h using Lipofectamine®
RNAiMAX reagent as previously described (34).
RT-qPCR
Total RNA was extracted from the cultured cells
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions,
and was then reverse transcribed to cDNA using the High-Capacity
cDNA Reverse Transcription kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.), as previously described (35,36).
The expression levels of fibrotic markers, collagen type I α1
(Col1α1), Col3α1, Ctgf and fibronectin (Fn)
were quantified with SYBR® Premix EX Taq™ (Takara
Biotechnology Co., Ltd.) and normalized to GAPDH.
Quantification of miRNAs was performed using a TaqMan MicroRNA
Assay kit (Applied Biosystems; Thermo Fisher Scientific, Inc.) and
U6 small nucleolar RNA was used for normalization, as previously
described (37). The thermocycling
conditions were as follows: Initial denaturation at 95°C for 10
min, followed by 40 cycles at 95°C for 2 sec, 60°C for 20 sec and
70°C for 10 sec. The primer sequences were as follows:
Col1α1 forward, 5′-GAGGGCCAAGACGAAGACATC-3′ and reverse,
5′-CAGATCACGTCATCGCACAAC-3′; Col3α1 forward,
5′-GGAGCTGGCTACTTCTCGC-3′ and reverse, 5′-GGGAACATCCTCCTTCAACAG-3′;
Ctgf forward, 5′-CAGCATGGACGTTCGTCTG-3′ and reverse,
5′-AACCACGGTTTGGTCCTTGG-3′; Fn forward,
5′-CGGTGGCTGTCAGTCAAAG-3′ and reverse, 5′-AAACCTCGGCTTCCTCCATAA-3′;
miR-375 forward, 5′-AGTGTCGTCAGAAAGAACGAACGGC-3′ and
reverse, 5′-CTCAACTGGTGTCGTGGAGTC-3′; and U6 forward,
5′-CTCGCTTCGGCAGCACA-3′ and reverse,
5′-AACGCTTCACGAATTTGCGT-3′.
Western blotting
CCD-19Lu lung fibroblasts were lysed in RIPA lysis
buffer (50 mM Tris-HCl, 0.5% NP-40, 250 mM NaCl, 5 mM EDTA and 50
mM NaF) and protein isolation was performed as previously described
(38,39). After quantification using the Rapid
Gold BCA Protein Assay kit (Pierce; Thermo Fisher Scientific,
Inc.), a total of 50 µg proteins were then loaded onto 10% SDS-PAGE
gels for separation. Subsequently, the proteins were transferred
onto PVDF membranes, which were blocked with 5% skimmed milk at
room temperature for 1 h and incubated with the indicated primary
antibodies overnight at 4°C. Finally, the proteins were labelled
with horseradish peroxidase-conjugated secondary antibodies
(1:10,000; cat. no. GB23303; Servicebio, Inc.) at room temperature
for 1 h and scanned using a ChemiDoc Touch Imaging system (Bio-Rad
Laboratories, Inc.) in the presence of a ECL reagent (cat. no.
G2020-25ML; Servicebio, Inc.). Data were analyzed using the Image
Lab software (v6.0.0 Build 25; Bio-Rad Laboratories, Inc.)
Bioinformatic prediction
The online database TargetScanHuman (Release v7.2;
http://www.targetscan.org/vert_72/)
was used for target prediction and analysis of miR-375.
Luciferase reporter assay
The luciferase reporter assay was performed as
described previously (33,40). Briefly, the wild type (WT)
3′-untranslated region (UTR) of Map2k6 containing the
putative miR-375 binding site or a mutant (MUT; the seed
region of the binding site was mutated) 3′-UTR sequence was cloned
into the pGL3 Basic vector (Promega Corporation). Subsequently, the
WT or MUT reporter plasmid (200 ng) was co-transfected into cells
(3×105/ml) with miR-375 mimic (25 nM) or the
negative control using Lipofectamine® RNAiMAX reagent at
37°C. Cells were then assayed for luciferase activity 36 h
post-transfection with the Dual-Light Chemiluminescent Reporter
Gene Assay system (Titertek-Berthold).
Statistical analysis
Quantitative data are presented as the mean ±
standard deviation (SD) and were analyzed by SPSS 23.0 software
(IBM Corp.). Comparisons between two groups were determined by a
unpaired Student's t-test, whereas one-way analysis of variance
(ANOVA) followed by Tukey's post-hoc test was used to compare the
differences among multiple groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
miR-375 inhibition aggravates
TGF-β-dependent transdifferentiation of lung fibroblasts
To investigate the role of miR-375 in the
development of myofibroblast transdifferentiation and pulmonary
fibrosis, the present study initially detected miR-375
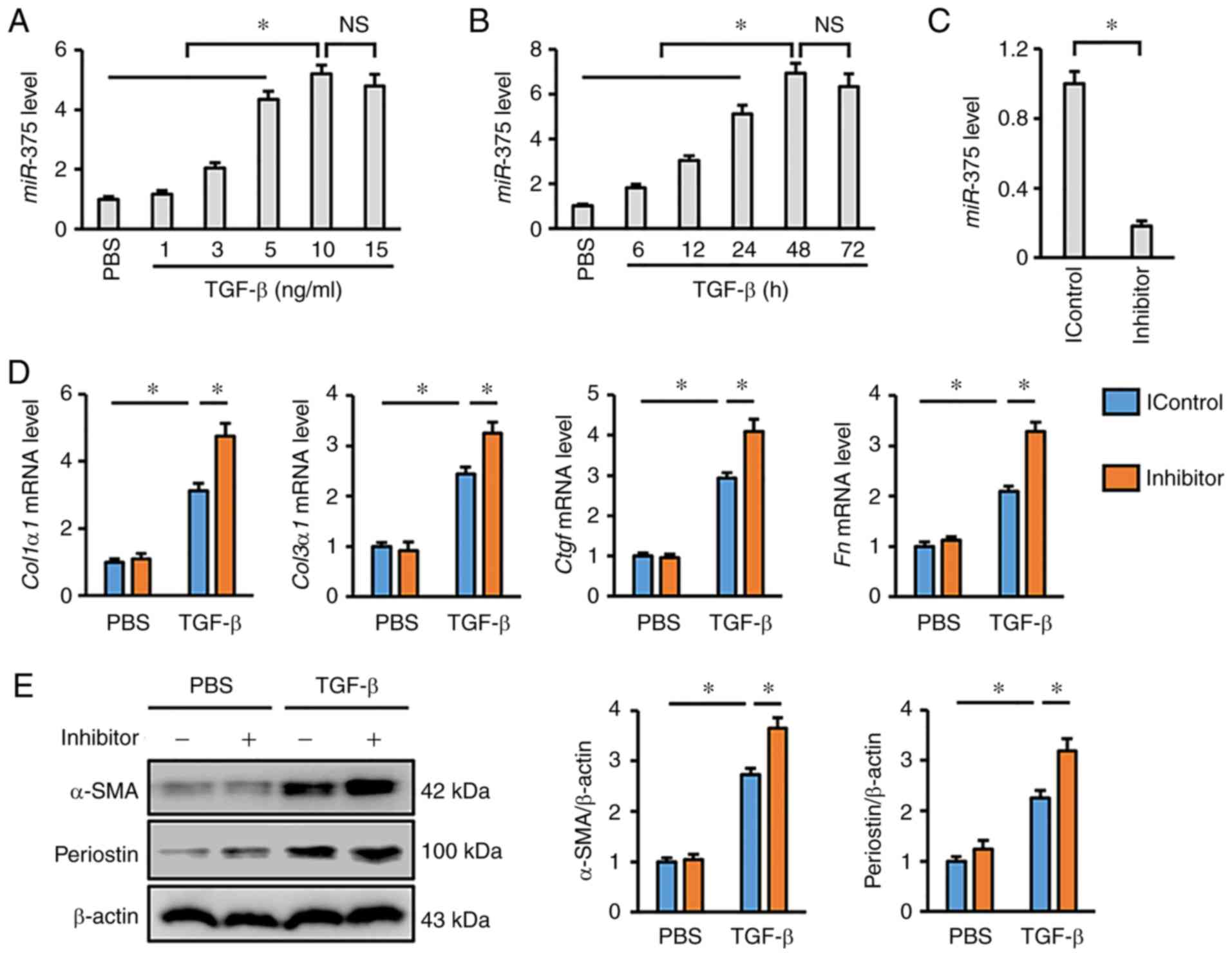
expression in lung fibroblasts after TGF-β stimulation. As depicted
in Fig. 1A, TGF-β treatment
significantly increased the expression of miR-375 in human
lung fibroblasts in a dose-dependent manner. In addition,
miR-375 expression was also progressively elevated from 6 to
48 h after TGF-β incubation compared with the PBS control group
(Fig. 1B). Moreover, it was found
that treatment with 10 ng/ml TGF-β for 48 h was sufficient to
trigger optimal miR-375 upregulation in vitro;
therefore, this time course and concentration were selected for
further experiments (Fig. 1A and
B). A chemically modified inhibitor was used to suppress
miR-375 expression in cultured cells, and the data revealed
that miR-375 was downregulated by 82% in vitro
post-transfection with this inhibitor (Fig. 1C). As shown in Fig. 1D, the mRNA expression levels of the
fibrotic markers, Col1α1, Col3α1, Ctgf and Fn, were
notably upregulated in response to miR-375 inhibition.
Increased expression levels of α-SMA and periostin in response to
TGF-β are known to be hallmarks of myofibroblast
transdifferentiation (41).
Consistently, miR-375 inhibition was observed to exacerbate
TGF-β-induced upregulation of α-SMA and periostin (Fig. 1E). Overall, it was concluded that
miR-375 was upregulated in response to fibrotic stimulation,
and that miR-375 inhibition aggravated TGF-β-dependent
transdifferentiation of lung fibroblasts.
miR-375 activation prevents lung
fibroblast transdifferentiation in response to TGF-β
stimulation
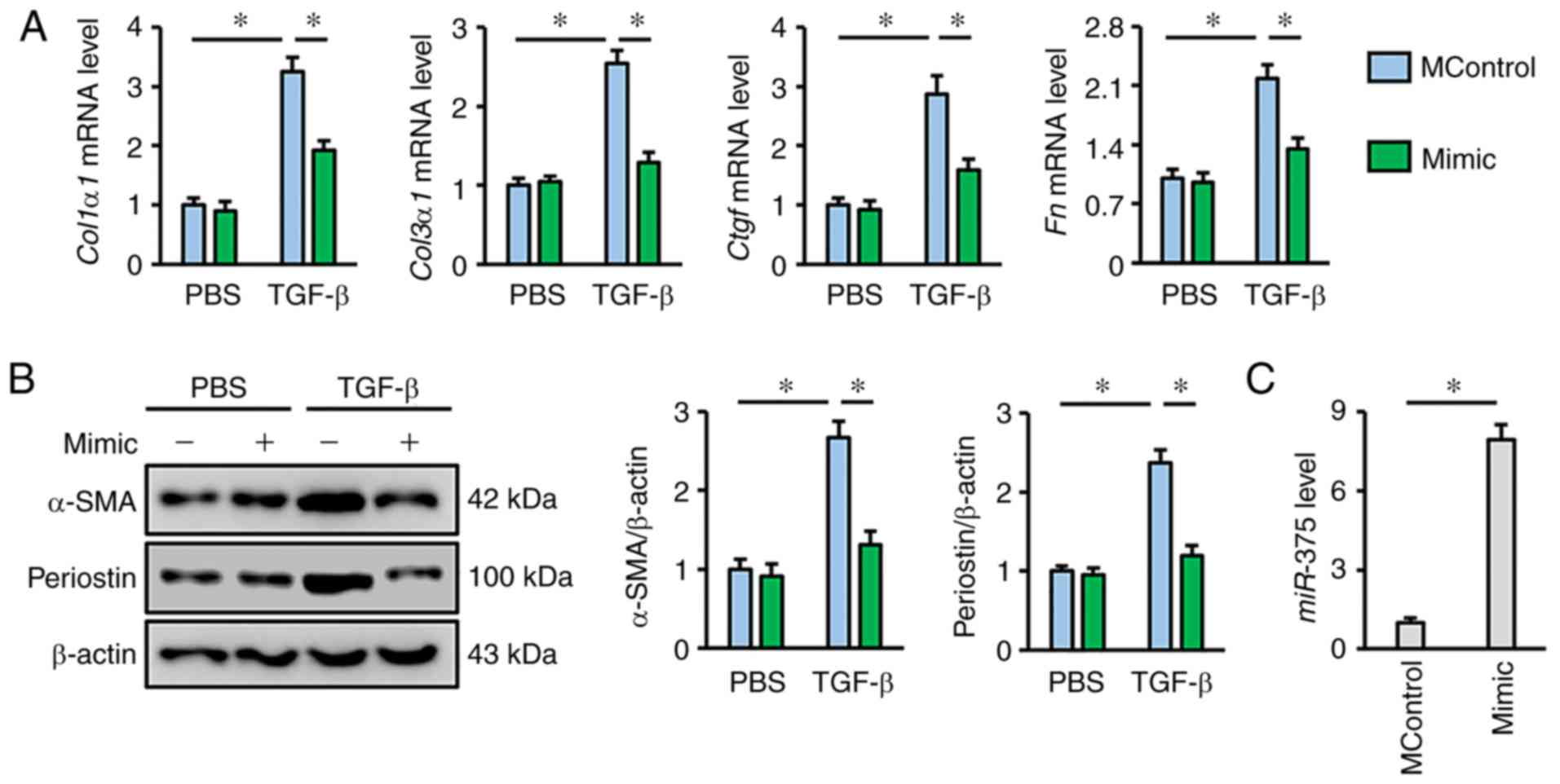
The present study also determined whether
miR-375 activation could prevent lung fibroblast
transdifferentiation in the presence of TGF-β, which is of more
interest in clinical situations as it is a crucial pathogenic
factor during fibrotic remodeling. As shown in Fig. 2A, miR-375 mimic transfection
markedly suppressed TGF-β-triggered collagen synthesis in lung
fibroblasts, as evidenced by the decreased mRNA expression levels
of Col1α1, Col3α1, Ctgf and Fn. The expression levels
of α-SMA and periostin were also suppressed following treatment
with TGF-β in miR-375 mimic-transfected lung fibroblasts,
but not in cells transfected with MControl (Fig. 2B). The results also revealed that
transfection with the miR-375 mimic caused a 7.94-fold
increase in miR-375 expression in vitro compared with
the MControl group (Fig. 2C).
Taken together, these data indicated that miR-375 helped to
negatively regulate lung fibroblast transdifferentiation, which
occurred in response to stimulation with TGF-β.
miR-375 blocks TGF-β-dependent
transdifferentiation of lung fibroblasts via suppressing P38
activation
The present study then attempted to explore the
possible mechanism through which miR-375 exerted its
beneficial effect on lung fibroblast transdifferentiation. It has
previously been reported that the Smad-dependent pathway is the
most common signaling axis in the progression of myofibroblast
transdifferentiation and pulmonary fibrosis (42). The present study detected the
phosphorylation of Smad3, which is known as the crucial node for
the Smad-dependent pathway. Notably, it was observed that
transfection with the miR-375 mimic did not alter Smad3
phosphorylation in the presence or absence of TGF-β (Fig. 3A and B). Previous studies have
suggested that miR-375 may be involved in regulating the AKT
pathway, which is a well-established, pro-fibrotic kinase cascade
(43,44). As shown in Fig. 3A and B, TGF-β stimulation resulted
in elevated levels of AKT phosphorylation; however, the
miR-375 mimic did not affect AKT activation. MAPK pathways,
particularly the P38 kinase branch have been reported to be
essential for pulmonary fibrosis (13); therefore, the present study sought
to evaluate the phosphorylation status of P38. As exhibited in
Fig. 3C-E, the miR-375
mimic decreased, whereas the miR-375 inhibitor increased
TGF-β-elicited P38 phosphorylation. To further corroborate the role
of P38, lung fibroblasts were pre-treated with SB203580 to inhibit
P38 activity as aforementioned. The data indicated that P38
inhibition abolished the adverse effect of the miR-375
inhibitor on lung fibroblast transdifferentiation, as determined by
the decreased mRNA expression levels of fibrotic markers (Fig. 3F). In Fig. 3F, cells in the second group were
treated with TGF-β and IControl to ensure that the inhibitor had no
off-target effects. These data demonstrated that the P38 pathway
may be responsible for the miR-375-mediated anti-fibrotic
effect.
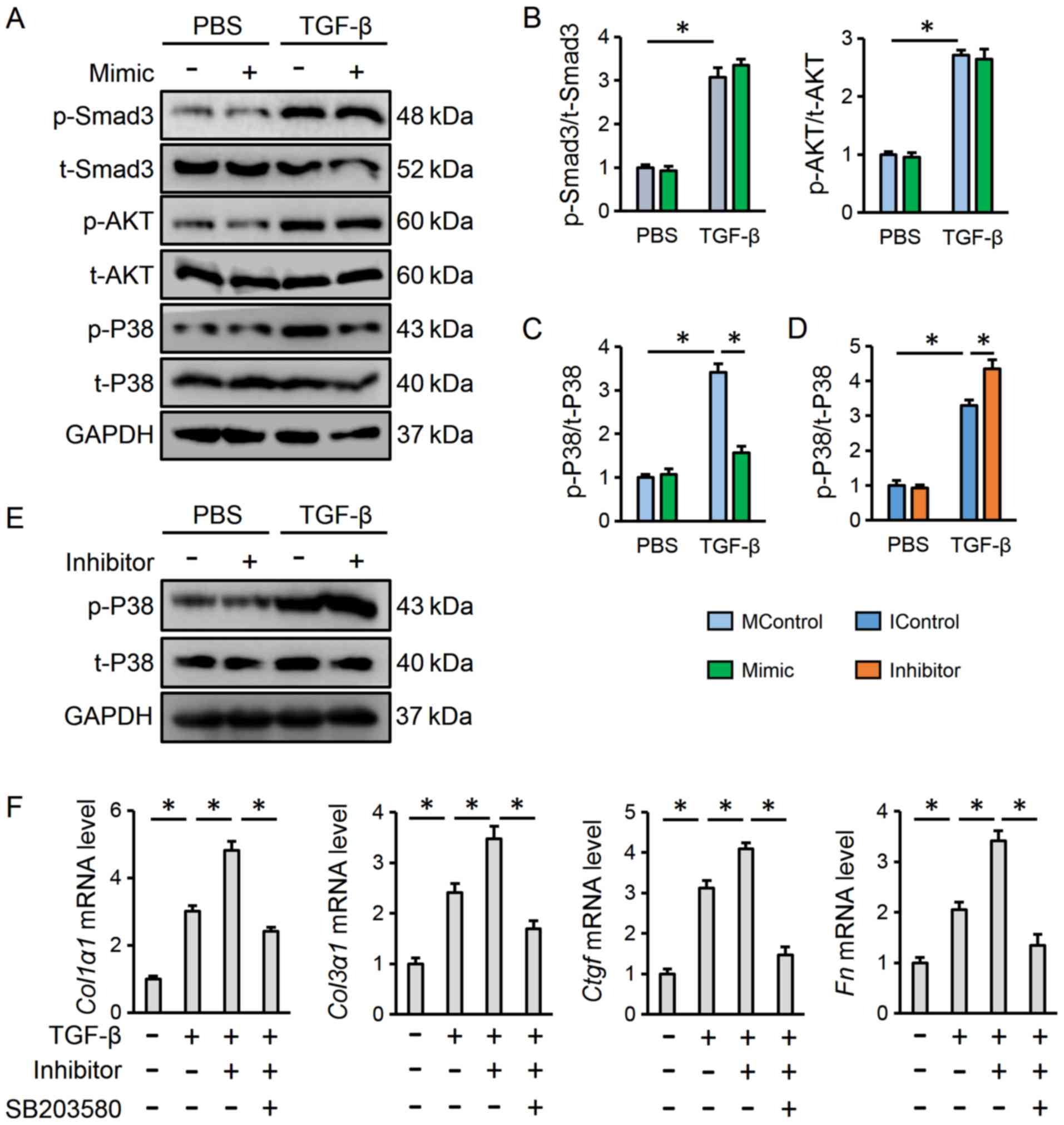 | Figure 3.miR-375 blocks TGF-β-dependent
transdifferentiation of lung fibroblasts via suppressing P38
activation. (A-C) Representative western blot and statistical
analysis post-transfection with the miR-375 mimic (n=6). (D
and E) Representative western blot and statistical analysis
post-transfection with the miR-375 inhibitor (n=6). (F)
Relative mRNA expression levels of fibrotic markers in human lung
fibroblasts treated with miR-375 or SB203580 (n=6). Results
are expressed as the mean ± SD. *P<0.05. miR, microRNA;
TGF-β, transforming growth factor-β; p-, phosphorylated; t-, total;
P38, P38 mitogen-activated protein kinases; MControl, mimic
control; IControl, inhibitor control; Ctgf, connective
tissue growth factor; Col1α1, collagen type I α1;
Col3α1, collagen type III α1; Fn, fibronectin. |
miR-375 suppresses P38 activation via
directly inhibiting MAP2K6
The present study finally attempted to clarify how
miR-375 regulates the P38 pathway in TGF-β-treated lung
fibroblasts. It is well-accepted that miRNAs negatively regulate
gene expression via binding to the 3′-UTR of target mRNAs (20). The possible targets for
miR-375 were predicted using the TargetScan database, and
the study became focused on MAP2K6, which is the key upstream
kinase for P38 activation. As shown in Fig. 4A, a putative binding site of
miR-375 was found in the 3′-UTR of Map2k6. To further
investigate whether MAP2K6 was a direct target of miR-375, a
luciferase reporter assay was performed. The results revealed that
miR-375 mimic incubation markedly decreased luciferase
activity in fibroblasts co-transfected with the WT 3′-UTR of
Map2k6; however, no alteration in luciferase activity was
detected when the putative binding sequence was mutated (Fig. 4B). Similarly, it was observed that
the protein expression levels of MAP2K6 were downregulated by the
miR-375 mimic, and upregulated by the miR-375
inhibitor in lung fibroblasts following TGF-β treatment (Fig. 4C and D). To further confirm the
role of MAP2K6 in P38 activation by miR-375, MAP2K6 expression was
knocked down with siMap2k6 (Fig. 4E). As expected, the upregulation of
fibrotic markers induced by miR-375 inhibitor transfection
was completely abrogated in Map2k6-deficient lung
fibroblasts (Fig. 4F). In Fig. 4F, cells in the second group were
treated with TGF-β and IControl, whereas cells in the third group
were treated with TGF-β, Inhibitor and siRNA to ensure that the
inhibitor had no off-target effects. These findings showed that
miR-375 may suppress P38 activation via directly inhibiting
MAP2K6.
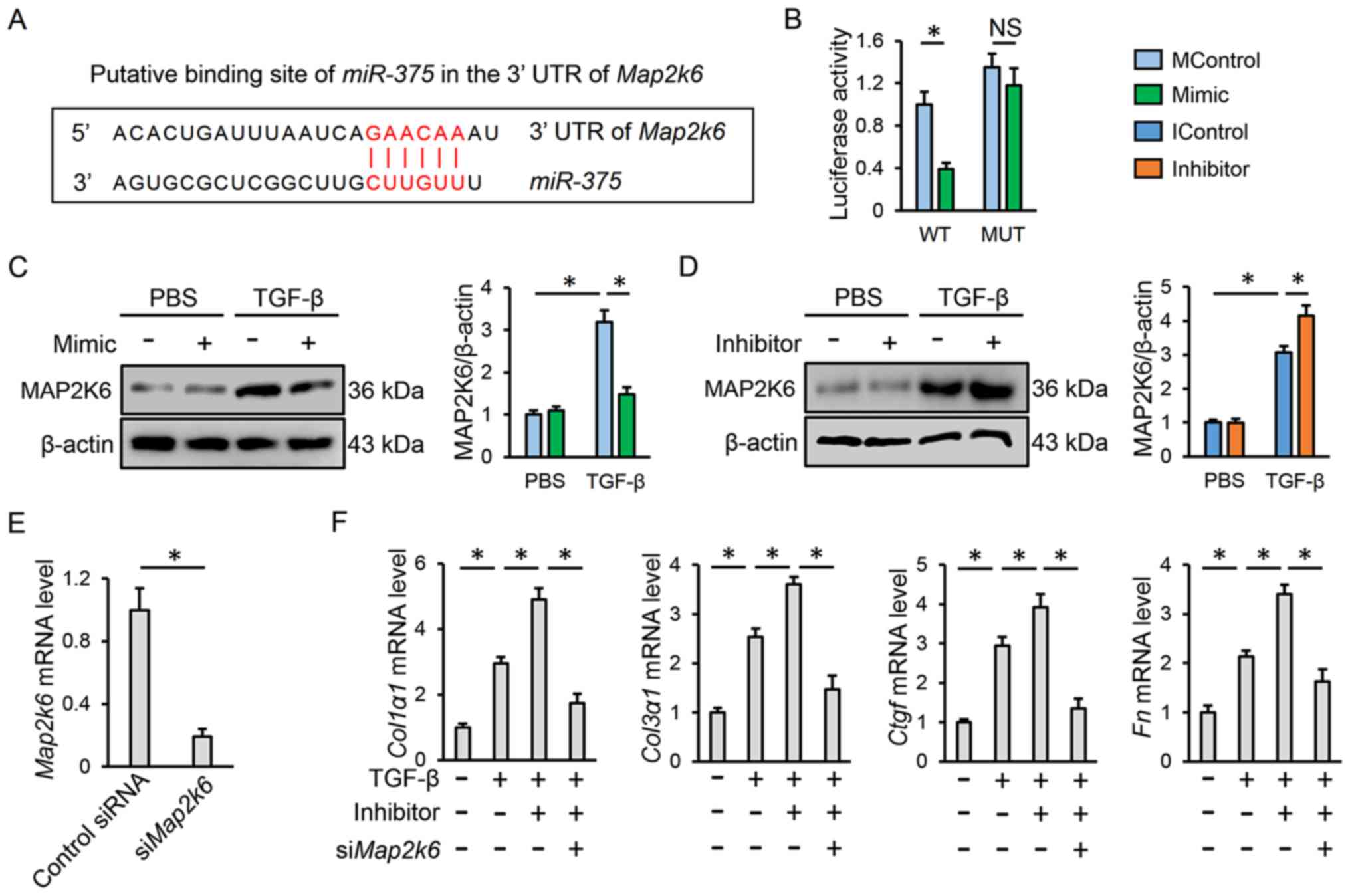 | Figure 4.miR-375 suppresses P38
activation by directly inhibiting MAP2K6. (A) Putative binding site
of miR-375 in the 3′-UTR of Map2k6. (B) Statistical
analysis of the luciferase reporter assay with WT or MUT 3′-UTR of
Map2k6 (n=8). (C and D) Representative western blot and
statistical analysis (n=6). (E) Transfection efficiency of
siMap2k6 in human lung fibroblasts (n=6). (F) Relative mRNA
expression levels of fibrotic markers in human lung fibroblasts
transfected with miR-375 inhibitor or siMap2k6 (n=6).
Results are expressed as the mean ± SD. *P<0.05. NS no
significance; miR, microRNA; MControl, mimic control;
IControl, inhibitor control; UTR, untranslated region; WT, wild
type; MUT, mutant; si, small interfering RNA; MAP2K6,
mitogen-activated protein kinase 6; TGF-β, transforming growth
factor-β; Ctgf, connective tissue growth factor;
Col1α1, collagen type I α1; Col3α1, collagen type III
α1; Fn, fibronectin. |
Discussion
Myofibroblasts are the primary cell type responsible
for the regulation of ECM homeostasis and pulmonary fibrosis. It
has been reported that myofibroblasts can be derived from multiple
cell sources, such as endothelial cells, epithelial cells and
pericytes (3,45). However, resident fibroblasts have
been identified as the major contributor of myofibroblasts in the
lung (46). In response to
fibrotic stimulation, lung fibroblasts are recruited to
fibroblastic foci and then transdifferentiate into myofibroblasts,
which promote ECM synthesis and deposition (2,3). In
the present study, it was found that miR-375 was
significantly upregulated in TGF-β-treated lung fibroblasts, which
in turn inhibited the MAP2K6/P38 signaling axis and subsequently
prevented myofibroblast transdifferentiation in vitro.
Accordingly, inhibition of P38 or genetic manipulation of
Map2k6 abrogated the miR-375 inhibitor-mediated
aggravating effect on myofibroblast transdifferentiation. To the
best of our knowledge, this study is the first to define
miR-375 as a negative regulator of lung fibroblast
transdifferentiation.
miRNAs have previously been identified as key
endogenous modulators of various signaling pathways or gene
networks, which have been implicated in the development of multiple
fibrotic diseases, including pulmonary fibrosis (23,24).
miR-375 was initially proposed as a tumor-suppressing miRNA,
and knockdown of miR-375 expression promoted the invasion
and metastasis of cancer cells (27). Similarly, Shao et al
(29) and others (28) demonstrated that miR-375 was
decreased in lung cancer samples, which was associated with a poor
prognosis in patients. In addition, Zhang et al (47) found that miR-375 could
reduce the secretion of surfactants in the lungs via reorganizing
the cytoskeleton, without affecting surfactant synthesis or the
formation of lamellar bodies. These data indicated that
miR-375 may be essential for the pathophysiological outcome
of lung diseases. Herein, it was found that miR-375 was
upregulated in lung fibroblasts after TGF-β treatment, and
miR-375 overexpression significantly prevented myofibroblast
transdifferentiation. In line with the present study, the results
from Sheng et al (48),
indicated that miR-375 activation markedly compromised the
pro-fibrotic function of mesenchymal stem cells and delayed the
wound-healing process in mice. These results provide robust
evidence for preventing myofibroblast transdifferentiation and
pulmonary fibrosis via targeting miR-375 in lung
fibroblasts.
Scientific understanding of the cellular pathways
involved in pulmonary fibrosis is not yet complete; however, it is
known that activation of the Smad-dependent pathway plays a
critical role in accelerating fibrotic remodeling in the lung
(25). Upon TGF-β stimulation, the
two TGF-β receptor isoforms form a heterodimer and then promote
Smad3 phosphorylation and nuclear translocation, which ultimately
transduces fibrotic stimuli to the nucleus (8). However, it was found in the present
study that miR-375 did not alter Smad3 phosphorylation. In
addition to the Smad-dependent pathway, numerous non-canonical
Smad-independent pathways are essential for the initiation and
progression of pulmonary fibrosis, including the AKT and MAPK
pathways (11,12). Herein, it was observed that AKT
phosphorylation was not affected by miR-375 mimic
transfection of TGF-β-stimulated lung fibroblasts, but
miR-375 overexpression distinctly attenuated P38 activation.
Furthermore, P38 inhibition abolished the protective effect of
miR-375 on TGF-β-induced myofibroblast transdifferentiation
and collagen synthesis. P38 kinase was previously recognized as a
nodal signaling effector in fibrogenesis, and various pro-fibrotic
signals converge on P38 kinase to trigger programmed fibroblast to
myofibroblast transdifferentiation and the fibrotic response within
the lung tissue (15). Conversely,
P38 suppression resulted in a significant protective effect against
myofibroblast transdifferentiation and pulmonary fibrosis, as this
reduces the expression of fibrotic factors (12). Moreover, in this study, a putative
binding site of miR-375 was found in the 3′-UTR of
Map2k6, which is a well-known upstream activator of P38
kinase, whereas Map2k6 deficiency reduced the anti-fibrotic
effect of miR-375 in vitro.
In conclusion, the present study demonstrated that
miR-375 prevented TGF-β-dependent transdifferentiation of
lung fibroblasts via the MAP2K6/P38 pathway, and suggested that
targeting miR-375 may help to develop therapeutic approaches
for treating pulmonary fibrosis. More empirical studies with
multiple cell lines and animal models should be conducted to
confirm the conclusions of this study.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XHZ and GJK contributed to the conception and design
of the experiments. XHZ, QC, HYS, WLJ and SPX carried out the
experiments. XHZ, QC, JH and GJK analyzed the experimental results
and interpreted the data. XHZ, JH and GJK wrote and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ng B, Dong J, D'Agostino G, Viswanathan S,
Widjaja AA, Lim WW, Ko N, Tan J, Chothani SP, Huang B, et al:
Interleukin-11 is a therapeutic target in idiopathic pulmonary
fibrosis. Sci Transl Med. 11:eaaw12372019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wolters PJ, Collard HR and Jones KD:
Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol.
9:157–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hung C, Linn G, Chow YH, Kobayashi A,
Mittelsteadt K, Altemeier WA, Gharib SA, Schnapp LM and Duffield
JS: Role of lung pericytes and resident fibroblasts in the
pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med.
188:820–830. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin X, Sime PJ, Xu H, Williams MA, LaRussa
L, Georas SN and Guo J: Yin yang 1 is a novel regulator of
pulmonary fibrosis. Am J Respir Crit Care Med. 183:1689–1697. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen Y, Zhao X, Sun J, Su W, Zhang L, Li
Y, Liu Y, Zhang L, Lu Y, Shan H and Liang H: YAP1/Twist promotes
fibroblast activation and lung fibrosis that conferred by miR-15a
loss in IPF. Cell Death Differ. 26:1832–1844. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Celada LJ, Kropski JA, Herazo-Maya JD, Luo
W, Creecy A, Abad AT, Chioma OS, Lee G, Hassell NE, Shaginurova GI,
et al: PD-1 up-regulation on CD4+ T cells promotes
pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1
production. Sci Transl Med. 10:eaar83562018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wei Y, Kim TJ, Peng DH, Duan D, Gibbons
DL, Yamauchi M, Jackson JR, Le Saux CJ, Calhoun C, Peters J, et al:
Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung
and tumor fibrosis. J Clin Invest. 127:3675–3688. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schmierer B and Hill CS: TGFbeta-SMAD
signal transduction: Molecular specificity and functional
flexibility. Nat Rev Mol Cell Biol. 8:970–982. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang X, Ma ZG, Yuan YP, Xu SC, Wei WY,
Song P, Kong CY, Deng W and Tang QZ: Rosmarinic acid attenuates
cardiac fibrosis following long-term pressure overload via
AMPKα/Smad3 signaling. Cell Death Dis. 9:1022018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Budas GR, Boehm M, Kojonazarov B,
Viswanathan G, Tian X, Veeroju S, Novoyatleva T, Grimminger F,
Hinojosa-Kirschenbaum F, Ghofrani HA, et al: ASK1 inhibition halts
disease progression in preclinical models of pulmonary arterial
hypertension. Am J Respir Crit Care Med. 197:373–385. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Avivi-Green C, Singal M and Vogel WF:
Discoidin domain receptor 1-deficient mice are resistant to
bleomycin-induced lung fibrosis. Am J Respir Crit Care Med.
174:420–427. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Molkentin JD, Bugg D, Ghearing N, Dorn LE,
Kim P, Sargent MA, Gunaje J, Otsu K and Davis J:
Fibroblast-specific genetic manipulation of p38 mitogen-activated
protein kinase in vivo reveals its central regulatory role in
fibrosis. Circulation. 136:549–561. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoshida K, Kuwano K, Hagimoto N, Watanabe
K, Matsuba T, Fujita M, Inoshima I and Hara N: MAP kinase
activation and apoptosis in lung tissues from patients with
idiopathic pulmonary fibrosis. J Pathol. 198:388–396. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matsuoka H, Arai T, Mori M, Goya S, Kida
H, Morishita H, Fujiwara H, Tachibana I, Osaki T and Hayashi S: A
p38 MAPK inhibitor, FR-167653, ameliorates murine bleomycin-induced
pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol.
283:L103–L112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cao Y, Liu Y, Ping F, Yi L, Zeng Z and Li
Y: miR-200b/c attenuates lipopolysaccharide-induced early pulmonary
fibrosis by targeting ZEB1/2 via p38 MAPK and TGF-β/smad3 signaling
pathways. Lab Invest. 98:339–359. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Underwood DC, Osborn RR, Bochnowicz S,
Webb EF, Rieman DJ, Lee JC, Romanic AM, Adams JL, Hay DW and
Griswold DE: SB 239063, a p38 MAPK inhibitor, reduces neutrophilia,
inflammatory cytokines, MMP-9, and fibrosis in lung. Am J Physiol
Lung Cell Mol Physiol. 279:L895–L902. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yuan J, Liu H, Gao W, Zhang L, Ye Y, Yuan
L, Ding Z, Wu J, Kang L, Zhang X, et al: MicroRNA-378 suppresses
myocardial fibrosis through a paracrine mechanism at the early
stage of cardiac hypertrophy following mechanical stress.
Theranostics. 8:2565–2582. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qiao J, Zhao J, Chang S, Sun Q, Liu N,
Dong J, Chen Y, Yang D, Ye D, Liu X, et al: MicroRNA-153 improves
the neurogenesis of neural stem cells and enhances the cognitive
ability of aged mice through the notch signaling pathway. Cell
Death Differ. 27:808–825. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Treiber T, Treiber N and Meister G:
Regulation of microRNA biogenesis and its crosstalk with other
cellular pathways. Nat Rev Mol Cell Biol. 20:5–20. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Seo HH, Lee S, Lee CY, Lee J, Shin S, Song
BW, Kim IK, Choi JW, Lim S, Kim SW and Hwang KC: Multipoint
targeting of TGF-β/Wnt transactivation circuit with microRNA 384-5p
for cardiac fibrosis. Cell Death Differ. 26:1107–1123. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sonneville F, Ruffin M, Coraux C,
Rousselet N, Le Rouzic P, Blouquit-Laye S, Corvol H and Tabary O:
MicroRNA-9 downregulates the ANO1 chloride channel and contributes
to cystic fibrosis lung pathology. Nat Commun. 8:7102017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xiao J, Meng XM, Huang XR, Chung AC, Feng
YL, Hui DS, Yu CM, Sung JJ and Lan HY: miR-29 inhibits
bleomycin-induced pulmonary fibrosis in mice. Mol Ther.
20:1251–1260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Montgomery RL, Yu G, Latimer PA, Stack C,
Robinson K, Dalby CM, Kaminski N and van Rooij E: MicroRNA mimicry
blocks pulmonary fibrosis. EMBO Mol Med. 6:1347–1356. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liang H, Xu C, Pan Z, Zhang Y, Xu Z, Chen
Y, Li T, Li X, Liu Y, Huangfu L, et al: The antifibrotic effects
and mechanisms of microRNA-26a action in idiopathic pulmonary
fibrosis. Mol Ther. 22:1122–1133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Herrera J, Beisang DJ, Peterson M, Forster
C, Gilbertsen A, Benyumov A, Smith K, Korenczuk CE, Barocas VH,
Guenther K, et al: Dicer1 deficiency in the idiopathic pulmonary
fibrosis fibroblastic focus promotes fibrosis by suppressing
microRNA biogenesis. Am J Respir Crit Care Med. 198:486–496. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kong KL, Kwong DL, Chan TH, Law SY, Chen
L, Li Y, Qin YR and Guan XY: MicroRNA-375 inhibits tumour growth
and metastasis in oesophageal squamous cell carcinoma through
repressing insulin-like growth factor 1 receptor. Gut. 61:33–42.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen LJ, Li XY, Zhao YQ, Liu WJ, Wu HJ,
Liu J, Mu XQ and Wu HB: Down-regulated microRNA-375 expression as a
predictive biomarker in non-small cell lung cancer brain metastasis
and its prognostic significance. Pathol Res Pract. 213:882–888.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shao Y, Geng Y, Gu W, Huang J, Ning Z and
Pei H: Prognostic significance of microRNA-375 downregulation in
solid tumors: A meta-analysis. Dis Markers. 2014:6261852014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saidi A, Kasabova M, Vanderlynden L,
Wartenberg M, Kara-Ali GH, Marc D, Lecaille F and Lalmanach G:
Curcumin inhibits the TGF-β1-dependent differentiation of lung
fibroblasts via PPARγ-driven upregulation of cathepsins B and L.
Sci Rep. 9:4912019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhu L, Wang J, Kong W, Huang J, Dong B,
Huang Y, Xue W and Zhang J: LSD1 inhibition suppresses the growth
of clear cell renal cell carcinoma via upregulating P21 signaling.
Acta Pharm Sin B. 9:324–334. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang YM, Zheng YF, Yang SY, Yang ZM, Zhang
LN, He YQ, Gong XH, Liu D, Finnell RH, Qiu ZL, et al: MicroRNA-197
controls ADAM10 expression to mediate MeCP2′s role in the
differentiation of neuronal progenitors. Cell Death Differ.
26:1863–1879. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee J, Heo J and Kang H: miR-92b-3p-TSC1
axis is critical for mTOR signaling-mediated vascular smooth muscle
cell proliferation induced by hypoxia. Cell Death Differ.
26:1782–1795. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fujiki K, Inamura H, Sugaya T and Matsuoka
M: Blockade of ALK4/5 signaling suppresses cadmium- and
erastin-induced cell death in renal proximal tubular epithelial
cells via distinct signaling mechanisms. Cell Death Differ.
26:2371–2385. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guo Q, Li C, Zhou W, Chen X, Zhang Y, Lu
Y, Zhang Y, Chen Q, Liang D, Sun T and Jiang C: GLUT1-mediated
effective anti-miRNA21 pompon for cancer therapy. Acta Pharm Sin B.
9:832–842. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang X, Hu C, Kong CY, Song P, Wu HM, Xu
SC, Yuan YP, Deng W, Ma ZG and Tang QZ: FNDC5 alleviates oxidative
stress and cardiomyocyte apoptosis in doxorubicin-induced
cardiotoxicity via activating AKT. Cell Death Differ. 27:540–555.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li X, Tian Y, Tu MJ, Ho PY, Batra N and Yu
AM: Bioengineered miR-27b-3p and miR-328-3p modulate drug
metabolism and disposition via the regulation of target ADME gene
expression. Acta Pharm Sin B. 9:639–647. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang X, Zhu JX, Ma ZG, Wu HM, Xu SC, Song
P, Kong CY, Yuan YP, Deng W and Tang QZ: Rosmarinic acid alleviates
cardiomyocyte apoptosis via cardiac fibroblast in
doxorubicin-induced cardiotoxicity. Int J Biol Sci. 15:556–567.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hu C, Zhang X, Wei W, Zhang N, Wu H, Ma Z,
Li L, Deng W and Tang Q: Matrine attenuates oxidative stress and
cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via
maintaining AMPKα/UCP2 pathway. Acta Pharm Sin B. 9:690–701. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen L, Chen L, Qin Z, Lei J, Ye S, Zeng
K, Wang H, Ying M, Gao J, Zeng S and Yu L: Upregulation of
miR-489-3p and miR-630 inhibits oxaliplatin uptake in renal cell
carcinoma by targeting OCT2. Acta Pharm Sin B. 9:1008–1020. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xiao D, Zhang Y, Wang R, Fu Y, Zhou T,
Diao H, Wang Z, Lin Y, Li Z, Wen L, et al: Emodin alleviates
cardiac fibrosis by suppressing activation of cardiac fibroblasts
via upregulating metastasis associated protein 3. Acta Pharm Sin B.
9:724–733. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hecker L, Vittal R, Jones T, Jagirdar R,
Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ and Thannickal
VJ: NADPH oxidase-4 mediates myofibroblast activation and
fibrogenic responses to lung injury. Nat Med. 15:1077–1081. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Garikipati VNS, Verma SK, Jolardarashi D,
Cheng Z, Ibetti J, Cimini M, Tang Y, Khan M, Yue Y, Benedict C, et
al: Therapeutic inhibition of miR-375 attenuates post-myocardial
infarction inflammatory response and left ventricular dysfunction
via PDK-1-AKT signalling axis. Cardiovasc Res. 113:938–949. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang PX, Cheng J, Zou S, D'Souza AD, Koff
JL, Lu J, Lee PJ, Krause DS, Egan ME and Bruscia EM:
Pharmacological modulation of the AKT/microRNA-199a-5p/CAV1 pathway
ameliorates cystic fibrosis lung hyper-inflammation. Nat Commun.
6:62212015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yao L, Conforti F, Hill C, Bell J,
Drawater L, Li J, Liu D, Xiong H, Alzetani A, Chee SJ, et al:
Paracrine signalling during ZEB1-mediated epithelial-mesenchymal
transition augments local myofibroblast differentiation in lung
fibrosis. Cell Death Differ. 26:943–957. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hoyles RK, Derrett-Smith EC, Khan K,
Shiwen X, Howat SL, Wells AU, Abraham DJ and Denton CP: An
essential role for resident fibroblasts in experimental lung
fibrosis is defined by lineage-specific deletion of high-affinity
type II transforming growth factor β receptor. Am J Respir Crit
Care Med. 183:249–261. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang H, Mishra A, Chintagari NR, Gou D
and Liu L: Micro-RNA-375 inhibits lung surfactant secretion by
altering cytoskeleton reorganization. IUBMB Life. 62:78–83.
2010.PubMed/NCBI
|
|
48
|
Sheng W, Feng Z, Song Q, Niu H and Miao G:
Modulation of mesenchymal stem cells with miR-375 to improve their
therapeutic outcome during scar formation. Am J Transl Res.
8:2079–2087. 2016.PubMed/NCBI
|