Introduction
The p32 protein, also known as hyaluronan-binding
protein 1, is usually localized to the mitochondria (1). However, p32 also acts as a cell
surface receptor for globular head-domain complement 1q (2). p32 was originally recognized in the
nucleus as a pre-mRNA splicing factor SF2-binding protein (3) and reported to be targeted to the
Golgi apparatus (4). Exogenously
epitope-tagged p32 at the N-terminus can be redirected to the
endoplasmic reticulum (ER) and cell surface (5). Functional studies have revealed that
p32 is required for the induction of mitochondria-dependent cell
death (6,7) and p32 knockdown causes a metabolic
shift from oxidative phosphorylation to aerobic glycolysis, which
is poorly tumorigenic (8). In
addition, p32 protein contributes to the morphology of the
mitochondria and ER, as well as cellular metabolism and stress
responses (9). Several potential
p32 ligands have been identified. The p32 protein interacts with
α1B- and α1D-adrenoreceptors, which control expression and cellular
localization (10). p32 has also
been revealed to interact with protein kinase C (PKC) μ, a
regulator of kinase activity and intracellular compartmentalization
(11). Moreover, the p32 protein
can interact with nuclear components such as the lamin B receptor,
as a linker between the nuclear membrane and intranuclear
substructures (12). p32
additionally binds to components of the extracellular matrix,
hyaluronic acid (13) and
vitronectin (14). It is also
involved in cell adhesion and motility, viral proteins, HIV Tat
(15), and EBV EBNA-1 (16), and exerts augmented transcriptional
activation of viral proteins, bacterial surface protein, and InlB,
which are necessary for bacterial invasion into mammalian cells
(17).
Although endothelial nitric oxide synthase (eNOS)
activity is regulated by extracellular signals, subcellular
localization, and protein-protein interactions, Ca2+
mainly contributes to modulation of eNOS activity in these
regulations. Activated Ca2+-bound calmodulin
(Ca2+/CaM) from increased cytosolic Ca2+
([Ca2+]c) levels binds to the canonical CaM-binding
domain in eNOS, which promotes the alignment of the oxygenase and
reductase domains of eNOS and prevents eNOS Thr495 phosphorylation.
Ca2+/CaM also activates
Ca2+/calmodulin-dependent protein kinase II (CaMKII),
which participates in the phosphorylation of eNOS Ser1177 to
increase NO release (18,19). Intracellular Ca2+ can be
transported and stored in the ER, and mitochondria. Ca2+
uptake by these organelles controls cellular Ca2+
homeostasis, regulates the oxidative phosphorylation rate and ATP
synthesis, and attenuates transient [Ca2+]c (20).
Our previous study reported that arginase II
participates in the Ca2+/CaMKII/eNOS signaling cascade
by p32-dependent regulation of [Ca2+]c (21). Downregulation of arginase II
protein using small interfering (si)RNA and genetic deletion was
associated with reduction of p32 protein levels, which resulted in
increased [Ca2+]c and eNOS activation (21). Therefore, the present study
investigated the target organelles of overexpressed p32 and
determined whether the p32 involved in the regulation of
[Ca2+] was associated with Ca2+-dependent
eNOS activation.
Materials and methods
Materials
NG-nitro-L-arginine methyl ester (L-NAME) and
manganese (III) tetrakis (4-benzoic acid) porphyrin chloride
(MnTBAP) were purchased from Calbiochem; Merck KGaA. Anti-sera
against eNOS, phospho-eNOS (Ser1177 and Thr495), phospho-CaMKII,
phospho-AKT (Ser473) and pan-actin were obtained from BD
Biosciences, and antiserum to p32 was obatined from Abcam. The
siRNA against arginase II (siArgII; cat. no. sc-29729) and
scrambled RNA (scm; cat. no. sc-37007) were purchased from Santa
Cruz Biotechnology, Inc. All other chemicals were obtained from
Sigma-Aldrich, Merck KGaA, unless otherwise stated.
Cell culture and animals
Human umbilical vein endothelial cells (HUVECs) were
purchased from Thermo Fisher Scientific, Inc. and maintained (37°C;
5% CO2) in Medium 200 containing low serum growth (5%)
supplement according to the supplier's instructions (Thermo Fisher
Scientific, Inc.). Human epithelial cervical carcinoma (HeLa) and
293A cells were purchased from the American Type Culture Collection
and maintained in DMEM (Thermo Fisher Scientific, Inc.) with 10%
FBS (Thermo Fisher Scientific, Inc.). Male C57BL/6J wild-type (WT;
21 mice; weight, 24±1.2 g; Daehan Biolink, Co., Ltd.) were obtained
at 10 weeks of age and fed a normal diet. Mice were housed on 12 h
dark/light cycle and had free access to food and water. Temperature
and humidity were 24.2±1.5°C and 51.2±3.9%, respectively. This
study adhered to the Guide for the Care and Use of Laboratory
Animals and was approved by the Institutional Review Board of
Kangwon National University.
siRNA treatment and knockdown of
mitochondrial p32
For siRNA transfection, HUVECs were incubated in
starvation medium (DMEM plus 5% FBS and penicillin and streptomycin
50 U/ml) containing siRNA against p32 (sip32; 100 nmol/l;
5′-TGTCTCCGTCGGTGTGCAGC-Cy5- 3′), scramble siRNA (100 nmol/l;
5′-GCTGCACACCGACGGAGACA-Cy5-3′) or no oligonucleotide for 24 h
without a reagent. siRNA against ArgII mRNA (siArgII; 100 nmol/l)
were transfected using the same method.
Electron microscopy (EM)
The p32 gene was cloned into the AfII
and BamHI restriction sites of the pcDNA3
connexin43-GFP-APEX2 plasmid (Addgene) for EM analysis. HeLa cells
were grown on a gridded glass-bottom dish (MatTek Corporation). The
day after seeding the cells (50% confluence), the p32-APEX2 plasmid
(1 µg DNA/60 mm dish) construct was transfected into HeLa cells
with Lipofectamine® 3000 (Thermo Fisher Scientific,
Inc.). After 16–24 h, the cells were fixed on ice using cold 1.5 ml
of fixation solution (1% glutaraldehyde and 1% paraformaldehyde in
0.15 M sodium cacodylate solution, pH 7.0) for 30 min. All
subsequent studies were performed using pre-chilled buffers and
reagents. Fixed cells were washed with 0.15 mM cold sodium
cacodylate buffer three times and then treated for 5 min with
buffer containing 50 mM glycine to quench the unreacted
glutaraldehyde on ice. Staining using 3,3-diaminobenzidine (DAB)
was initiated by adding freshly diluted 1 mg/ml DAB (Sigma-Aldrich;
Merck KGaA) from a stock (10 mg/ml) dissolved in 0.1 M HCl and 10
mM H2O2 in phosphate-buffered saline. After
30 min on ice, the reaction was stopped by removing the DAB
solution, and the cells were washed with 0.15 M cold sodium
cacodylate buffer three times on ice. Post-fixation was performed
using 2% (w/v) osmium tetroxide (OsO4) and 1.5% (w/v)
potassium ferrocyanide in 0.1 M sodium cacodylate buffer for 1 h on
ice. The cells were then rinsed three times for 10 min each in
chilled distilled water, and dehydrated in a graded ethanol series
(50, 60, 70, 80, 90 and 100%) for 15 min each time, then they were
infiltrated into EMbed-812 (Electron Microscopy Sciences) using 1:3
(v/v), 1:1 (v/v), and 3:1 (v/v) resin and anhydrous ethanol for 1
h. The samples were embedded with the 100% resin and incubated
overnight at room temperature. The next day, the samples were
filled again with 100% resin for 3 h at room temperature before
transferring the sample to fresh resin, followed by polymerization
at 60°C for 24 h. The embedded cells were cut with a diamond knife
into 50-nm sections using a ultramicrotome (Leica Microsystems,
Inc.), and images were acquired by transmission electron microscopy
(TEM; Tecnai G2; Thermo Fisher Scientific, Inc.) operating at 120
kV (magnification, ×20,000). The TEM data were acquired using the
Brain Research Core Facilities at the Korea Brain Research
Institute.
Mitochondrial fractionation
Cells and aortic segments were homogenized twice in
subcellular fractionation buffer (250 mM sucrose, 20 mM HEPES, pH
7.4, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, and
protease inhibitors; Roche Diagnostics) for 3 min and centrifuged
at 1,000 × g for 10 min at 4°C to remove cell debris and intact
cells. The supernatants were then centrifuged at 21,000 × g for 45
min at 4°C. The cytosolic (supernatant) and mitochondrial (pellet)
fractions containing 20 µg protein were used for subsequent western
blot analysis. The purity of the fractions was measured using
western blotting for HSP60 and actin, respectively.
Western blot analysis
Proteins of cell lysates were dissolved in SDS-PAGE
sample buffer (Tris-HCl 62.5 mM, pH 6.8, SDS 2%, glycerol 10%, DTT
50 mM, bromophenolblue 0.01%), then resolved by SDS-PAGE on 10%
gels (15 µg/lane; determined by Bradford assay), transferred to
PVDF and the membranes were blocked with 5% skim milk (room
temperature; 1 h). Then, membranes were immunoblotted with a
primary antibody (4°C; 12 h; 1:1,000) and the secondary antibody
(room temperature; 2 h; 1:2,000). Antisera against eNOS (cat. no.
610297) and phospho-eNOS (Ser1177; cat. no. 612392; Thr495; cat.
no. 612707) were acquired from BD Biosciences. p32 (cat. no.
ab24733) antisera were obtained from Abcam. Akt (cat. no. 9272),
phospho-Akt (Ser473; cat. no. 9271), CaMKII (cat. no. 3362),
phospho-CaMKII (Thr286; cat. no. 12716) and pan-actin (cat. no.
4968) antisera were obtained from Cell Signaling Technology, Inc.
Arginase II (cat. no. sc-271443) and HSP60 (cat. no. sc-13115) were
purchased from Santa Cruz Biotechnology, Inc. Pan-actin was used as
a reference protein. Horseradish peroxidase-conjugated antibody
(Goat anti-mouse IgG, cat. no. G-21040; Goat anti-rabbit IgG, cat.
no. G-21234; Thermo Fisher Scientific, Inc.) was detected using an
enhanced ECL-based chemiluminescence system (Thermo Fisher
Scientific, Inc.). Band intensities were analyzed using ImageJ
software (Fiji; National Institutes of Health).
Immunofluorescence staining and
imaging
Cells were washed with PBS, fixed at room
temperature with 3.7% formaldehyde (Fisher Chemical Ltd.) for 15
min and permeabilized with 0.1% Triton X-100 (Sigma-Aldrich; Merck
KGaA). Samples were then washed three times with PBS, blocked at
room temperature with 1% BSA (Sigma-Aldrich) for ≥1 h and incubated
overnight with the indicated p32 primary antibody at 4°C. Samples
were rinsed with PBS three times before conjugation at room
temperature for 1 h with the CY-5-conjugated secondary antibody
(Thermo Fisher Scientific, Inc.; cat. no. A10524). Samples were
imaged using epifluorescence microscopy (magnification, ×10
objective lens) equipped with the Metamorph program 7.6 (Molecular
Devices LLC).
Preparation of p32-expressing
adenovirus
The p32 plasmid, pCMV6-XL5, was purchased from
OriGene Technologies, Inc. and subcloned into the BglII and
KpnI restriction sites of the pCMV-Tag1 plasmid. For virus
generation, full-length p32 was cloned into the BamHI and
XhoI sites of the pENTR-CMV vector that has attL sites for
site-specific recombination using the Gateway Destination Vector
system (Invitrogen; Thermo Fisher Scienfitic, Inc.). The
site-specific recombination between the pENTR-CMV/p32 and the
adenovirus vector, pAd/PL-DEST, was conducted using LR clonase II.
WT Adp32 is an adenovirus encoding full-length human p32. The
adenovirus was amplified in 293A cells and purified using an
Adeno-X™ purification kit (Takara Bio, Inc.) and the multiplicity
of infection was determined using an Adeno-X™ titer kit (Takara
Bio, Inc.). Adp32 was used to treat HUVECs at a concentration of
1×106 pfu/ml. For in vivo mice experiments, the
purified recombinant adenovirus containing 5×109
particles was injected in the tail vein of mice. Adenovirus only as
an empty vector (Ad) was used as an adenoviral control.
Mitochondrial Ca2+
([Ca2+]m), ER Ca2+ ([Ca2+]ER), and
cytosolic Ca2+ ([Ca2+]c) measurements using
confocal microscopy and flow cytometry
Direct assessment of [Ca2+]m content was
peformed using an established loading procedure (21) with Rhod-2 acetoxymethyl (AM, Thermo
Fisher Scientific, Inc.). Briefly, the cells were loaded with 2.5
µM Rhod-2 AM at 37°C for 1 h in starvation medium (M199 and 1% FBS
plus penicillin and streptomycin 50 U/ml). Subsequently, the cells
were washed and incubated (37°C for 30 min) in Tyrode's modified
solution (150 mM NaCl, 4 mM KCl, 2 mM CaCl2, 2 mM
MgCl2, 10 mM HEPES, and 10 mM glucose). For detection of
Rhod-2 AM fluorescence, a 552-nm excitation (Ex) and a 581-nm
emission (Em) filters were used. MitoTracker Green FM (Thermo
Fisher Scientific, Inc.) was added to cells and incubated at 100 nM
for 1 h at 37°C and imaged at 490 nm exitation and 516 nm emission.
To examine the [Ca2+]ER, ER-tracker Red (5 µM; 30 min;
37°C; Thermo Fisher Scientific, Inc.) and Fluo-5N AM (5 µM; 1 h;
37°C; Thermo Fisher Scientific, Inc.) were used to obtain images at
wavelengths of 588/620 nm (Ex/Em) and 488/530 nm (Ex/Em),
respectively. The [Ca2+]c was monitored using Fluo-4 AM
(100 nM; 1 h; Thermo Fisher Scientific) at 494 nm Ex, and 506 nm
Em. The intensity values were normalized according to the samples
fluorescence values after subtraction of background using the
Metamorph software 7.6 (Molecular Probes; Thermo Fisher Scientific,
Inc.).
[Ca2+]m, [Ca2+]ER, and
[Ca2+]c were also determined using flow cytometry
(FACSCalibur; BD Biosciences). The fluorescence intensity for each
sample was determined using CellQuest software 5.1 (BD
Biosciences). The Ca2+ level was determined by comparing
the fold changes in the fluorescence intensities of treated cells
relative to unstained control cells.
Measurement of nitric oxide (NO) and
reactive oxygen species (ROS)
Aortic rings from 10-week-old male C57BL/6 WT mice
were prepared for fluorescent probe labeling of superoxide [1 µM
(final concentration); dihydroethidine (DHE); Abcam; cat. no.
ab236206;] and NO [5 µM (final concentration);
4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-DA);
cat. no. D23844; Thermo Fisher Scientific, Inc.). The fluorescent
intensity was measured for 5 min with 30-sec intervals at 37°C. The
NOS inhibitor, L-NAME (10 µM; 37°C; 30 min) blocked NO production
and MnTBAP (1 µM; 37°C; 30 min), as a ROS scavenger, reduced DHE
fluorescence intensity. Images were acquired using a BX51
epifluorescence microscope (magnification, ×400; Olympus
Corporation). Fluorescence intensity was measured as previously
described (22) using the
Metamorph software 7.6 (Molecular Probes; Thermo Fisher Scientific,
Inc.).
Aortic vascular tension assay
Heparin was administered 1 h before mice were
sacrificed. Mice were anesthetized using inhalant isoflurane (1%),
and the thoracic aorta from the aortic root to the bifurcation of
the iliac arteries was rapidly isolated and cut into 1.5 mm rings.
The aortic rings were placed in ice-cold oxygenated Krebs-Ringer
bicarbonate buffer (118.3 mM NaCl, 4.7 mM KCl, 1.2 mM
MgSO4, 1.6 mM CaCl2, 25 mM NaHCO3,
11.1 mM glucose; pH 7.4) and suspended between two wire stirrups
(150 mm) in a myograph (Multi Myograph System 620; Danish Myo
Technology A/S) containing 10 ml Krebs-Ringer (95% O2,
5% CO2, pH 7.4, 37°C). One stirrup was connected to a
three-dimensional micromanipulator, and the other to a force
transducer. The aortic rings were passively stretched at 10 min
intervals in increments of 100 mg to reach the optimal tone (600
mg). After the aortic rings were stretched to their optimal resting
tone, the contractile response to 60 mM KCl was determined. The
response to a maximal dose of KCl was used to normalize the
responses to agonist across vessel rings. Dose responses to the
vasoconstrictor phenylephrine (PE, 10−9−10−5
M) were assessed, and responses to the vasodilators acetylcholine
(Ach, 10−9−10−5 M) and sodium nitroprusside
(SNP, 10−10−10−6 M) were assessed after
pre-constriction with PE (10−5 M). To further confirm
the NO-dependent vasorelaxation activity, aortic rings were treated
with 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ,
10−5 M), a soluble guanylyl cyclase inhibitor.
Statistical analysis
Each graph represents cumulative data from three
independent experiments performed at least in triplicate. Data were
analyzed using one-way ANOVA with a Bonferroni post hoc test, or
unpaired Student's t-test, or two-way ANOVA with Bonferroni
correction test with Prism 5 software (GraphPad Software, Inc.).
Data are presented either as the mean ± SEM, or mean ± SD where
two-way ANOVA was used. P<0.05 was considered to indicate a
statistically significant difference.
Results
p32 is targeted to the ER and
mitochondria
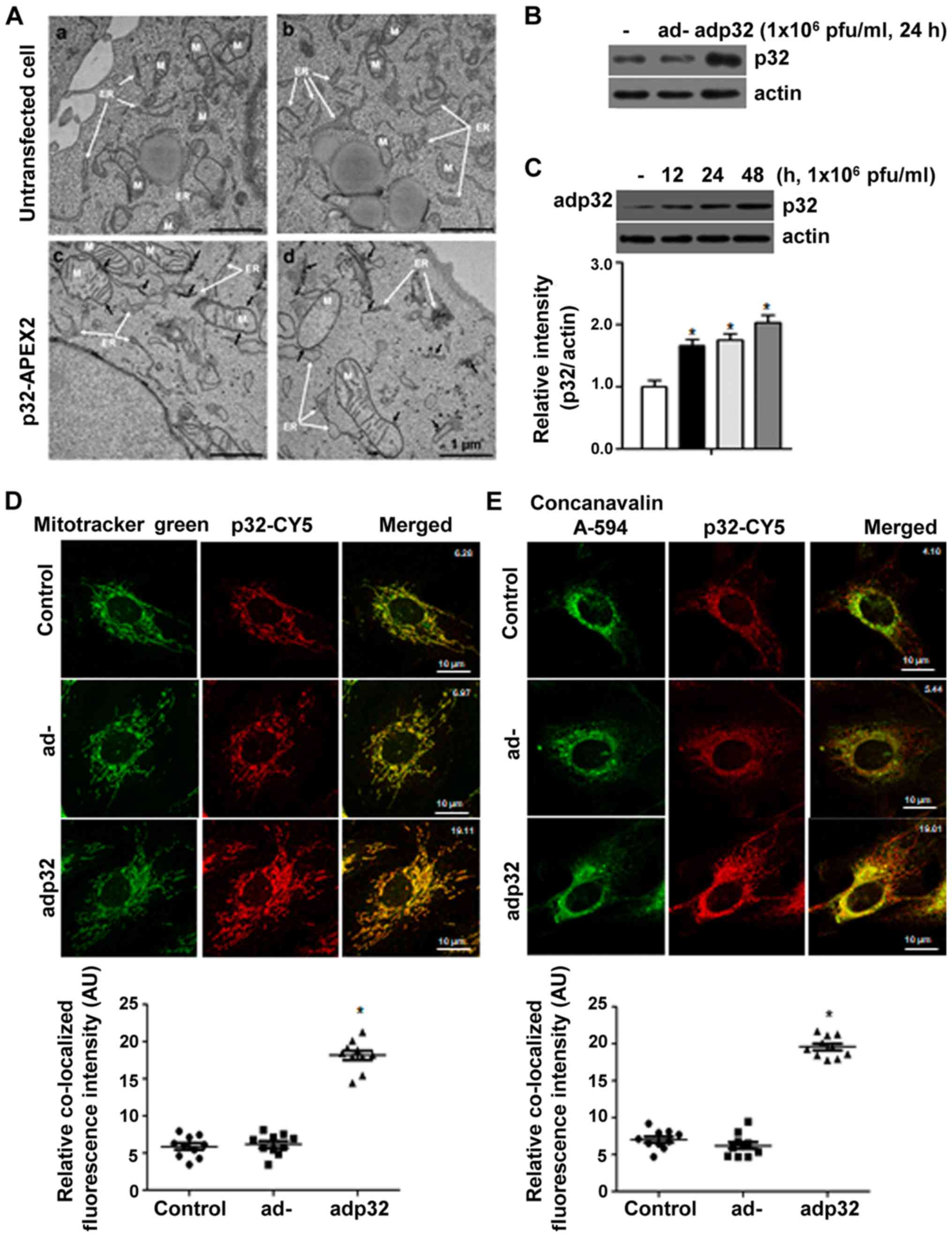
To localize p32, EM analysis was carried out
following overexpression of p32 in HeLa cells. p32 was localized to
the ER, mitochondria and vesicles near to ER. In the untransfected
cells (Fig. 1A-a and -b), the
electron density of the mitochondria and the ER membrane was
constant, but in the p32-APEX2 transfected cells (Fig. 1A-c and -d), strong electron density
was found where DAB was precipitated, according to oxidation by the
APEX2 enzyme. To further confirm these results, p32-expressing
adenovirus (adp32) was generated and used to infect HUVECs. The
protein amount of p32 was increased after adp32 infection (Fig. 1B) in a time-dependent manner
(Fig. 1C). Using
immunocytochemical analysis, p32 after adp32 treatment was targeted
to the mitochondria (Fig. 1D) and
ER (Fig. 1E). Mitotracker green
and concanavalin A-594 were used to indicate mitochondria and ER,
respectively, showing that overexpressed p32 was localized to the
Ca2+ storage organelles (ER and mitochondria).
Additionally, under normal physiological conditions, sip32 did not
affect the change of [Ca2+]ER, although
[Ca2+]m was decreased (Fig. S1). This suggested that p32 was
targeted to mitochondria, and not to ER, under normal conditions
without the presence of a membrane signal sequence.
p32 enhances Ca2+ flux from
the cytosol to the ER and mitochondria, resulting in decreased
[Ca2+]c
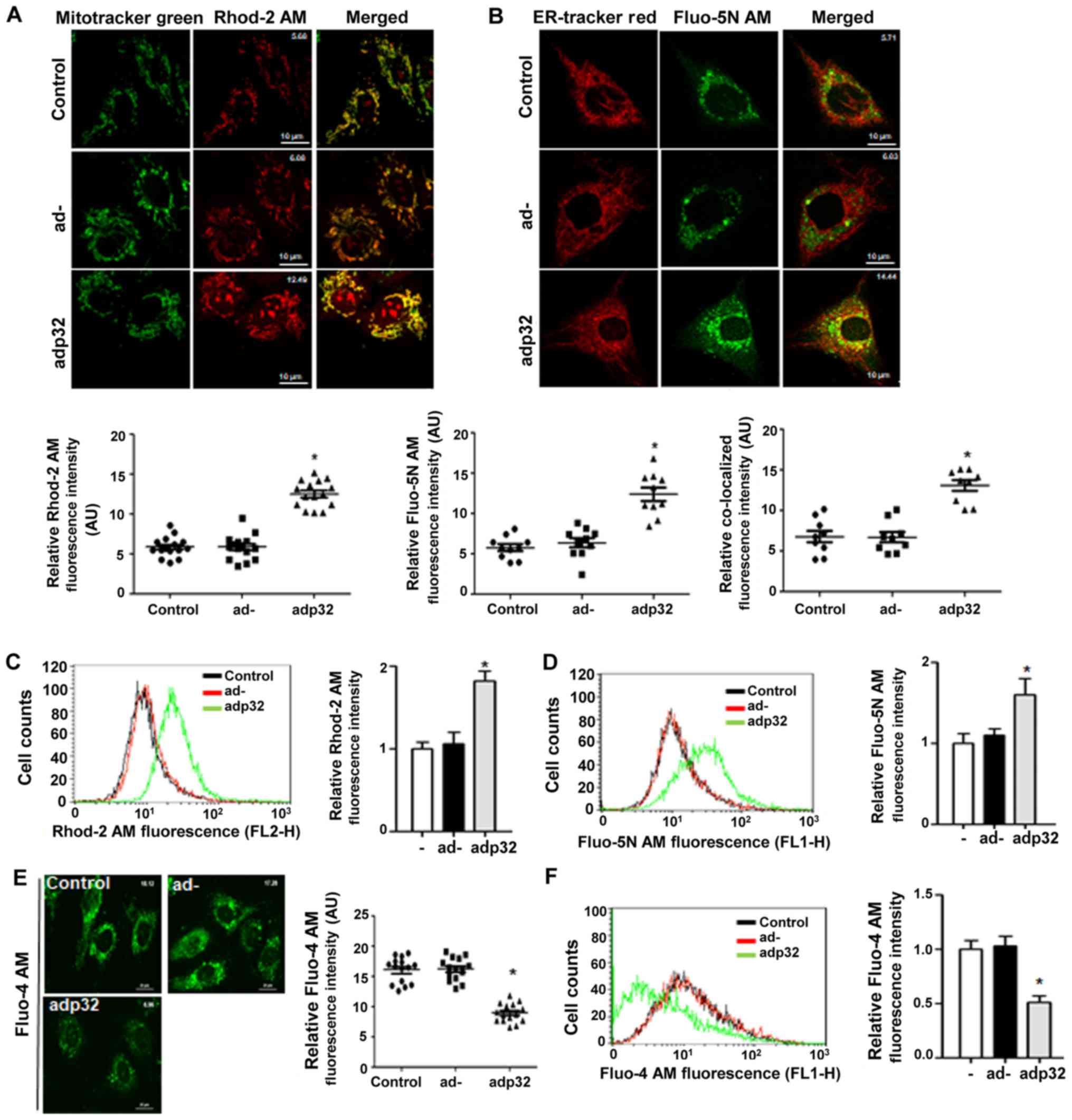
The effect of increased p32 levels on the
Ca2+ concentration in target organelles was then
evaluated by using fluorescent dye Rhod-2 AM as a mitochondrial
Ca2+ indicator, and Fluo-5N AM as an ER Ca2+
indicator. Both [Ca2+]m and [Ca2+]ER were
significantly increased after adp32 treatment (Fig. 2A and B). The fluorescence
intensities of Fluo-5N alone were analyzed and co-localization of
Fluo-5N and ER-tracker red were observed from the microscopic
images of adp32-infected HUVECs (Fig.
2B).
Using flow cytometry, Rhod-2 and Fluo-5N intensities
were significantly increased following adp32 transfection (Fig. 2C and D), indicating that increased
p32 protein in both organelles caused an uptake of Ca2+.
Whether adp32 changed [Ca2+]c was then tested using
Fluo-4 AM. [Ca2+]c was decreased in adp32-treated HUVECs
as observed using both microscopy (Fig. 2E) and flow cytometry (Fig. 2F). Altogether, these findings
indicated that adp32-dependent p32 overexpression decreased
[Ca2+]c by mediating Ca2+ uptake into the ER
and mitochondria.
p32 overexpression attenuates
Ca2+-dependent eNOS phosphorylation and impaired
endothelial function
Because [Ca2+]c plays an important role
in the regulation of eNOS activity through CaMKII-dependent
signaling cascade in endothelial cells (ECs) (21), the Ca2+-dependent
signaling cascade associated with eNOS phosphorylation was next
examined. p32 overexpression attenuated the signaling cascade of
CaMKII/AKT/eNOS Ser1177 phosphorylations and increased eNOS Thr495
phosphorylation without any effect on the proteins levels (Fig. 3A). To determine the effect of adp32
on endothelial function, the adp32 vector was injected into WT
mice. Aortic vessels were isolated from the sacrificed mice to
confirm that the amount of p32 protein was predominantly
overexpressed in ECs, but not smooth muscle cells (SMCs) (Fig. 3B). In aortic endothelia infected
with adp32, NO production was significantly attenuated (Fig. 3C) and ROS generation was
accelerated (Fig. 3D). Using the
vascular tension assay, Ach-induced vasorelaxation responses were
decreased (Fig. 3E), while
PE-dependent dose responses were increased (Fig. 3F). Responses to KCl (Fig. 3G) and the NO donor SNP (Fig. 3H) remained similar across all
groups.
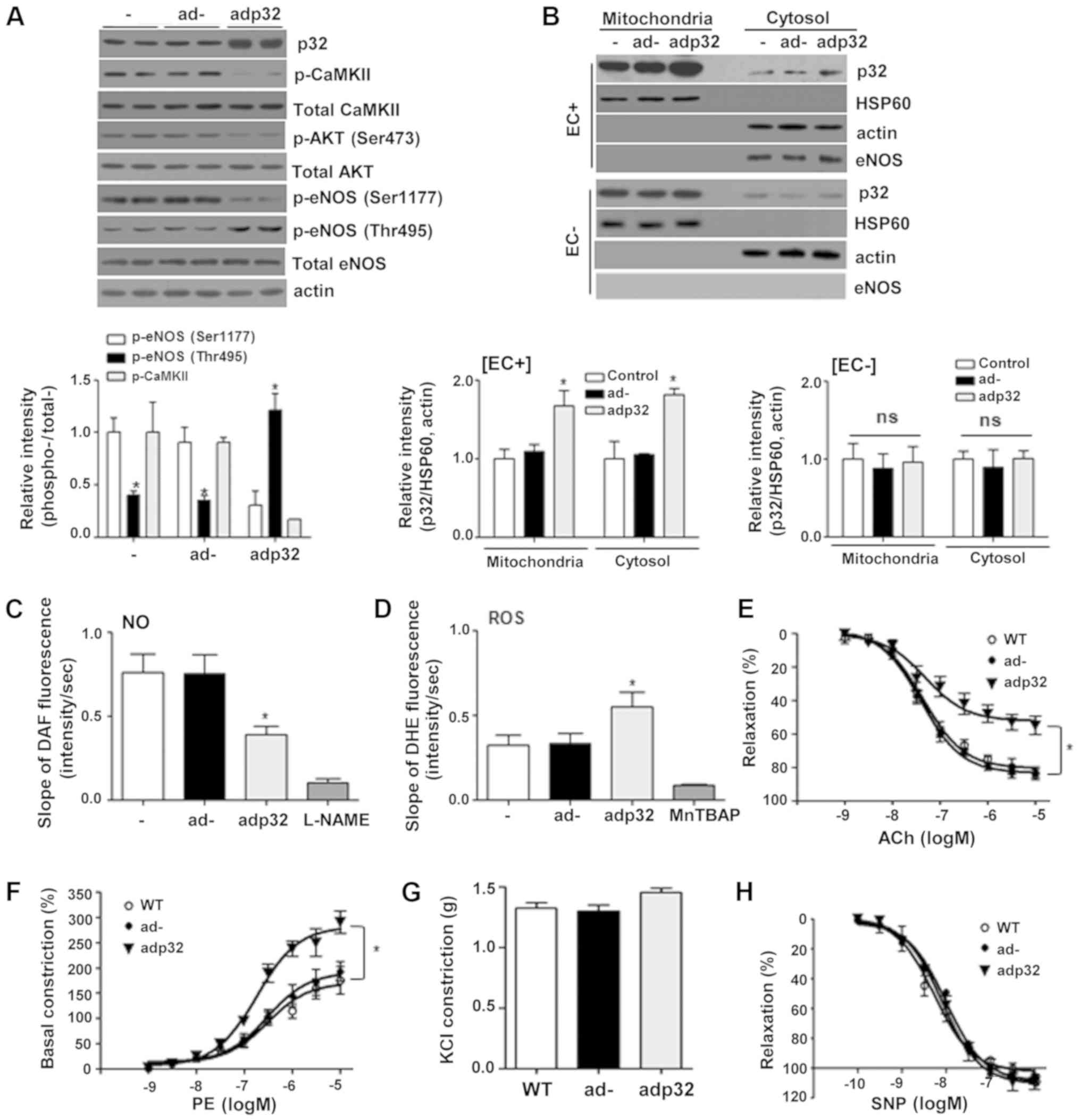 | Figure 3.Adp32 infection impairs endothelial
function attributing to reduced eNOS phosphorylation at Ser1177.
(A) Mice (n=3) injected intravenously with 5×109 pfu
adp32 were sacrificed after 24 h, and p32 expression was increased
in aortas. Increased p32 expression decreased CaMKII/AKT/eNOS
Ser1177 phosphorylations and increased eNOS phosphorylation at
Thr495. Ad- had no effect on this signaling cascade. (B) The
protein level of p32 was increased in the mitochondrial fraction of
adp32-infected aortas, but the p32 protein level was not altered in
the mitochondrial fraction of de-endothelialized vessels. (C) Adp32
infection attenuated NO production and (D) accelerated ROS
generation. L-NAME and MnTBAP were used as a negative control. n=6
aortas from three mice. Using the vascular tension assay,
adp32-infected aortas had decreased responses to (E) Ach and (F)
PE-dependent vasoconstrictive responses were accelerated. The (G
KCl response and (H) SNP dose responses were the same in all
groups. n=3. *P<0.05, vs. ad. eNOS, endothelial nitric oxide
synthase; CaMKII, calcium/calmodulin dependent protein kinase II;
NO, nitric oxide; ROS, reactive oxygen species; ad, adenovirus;
Ach, acetylcholine, PE, phenylephrine; SNP, sodium
nitroprusside. |
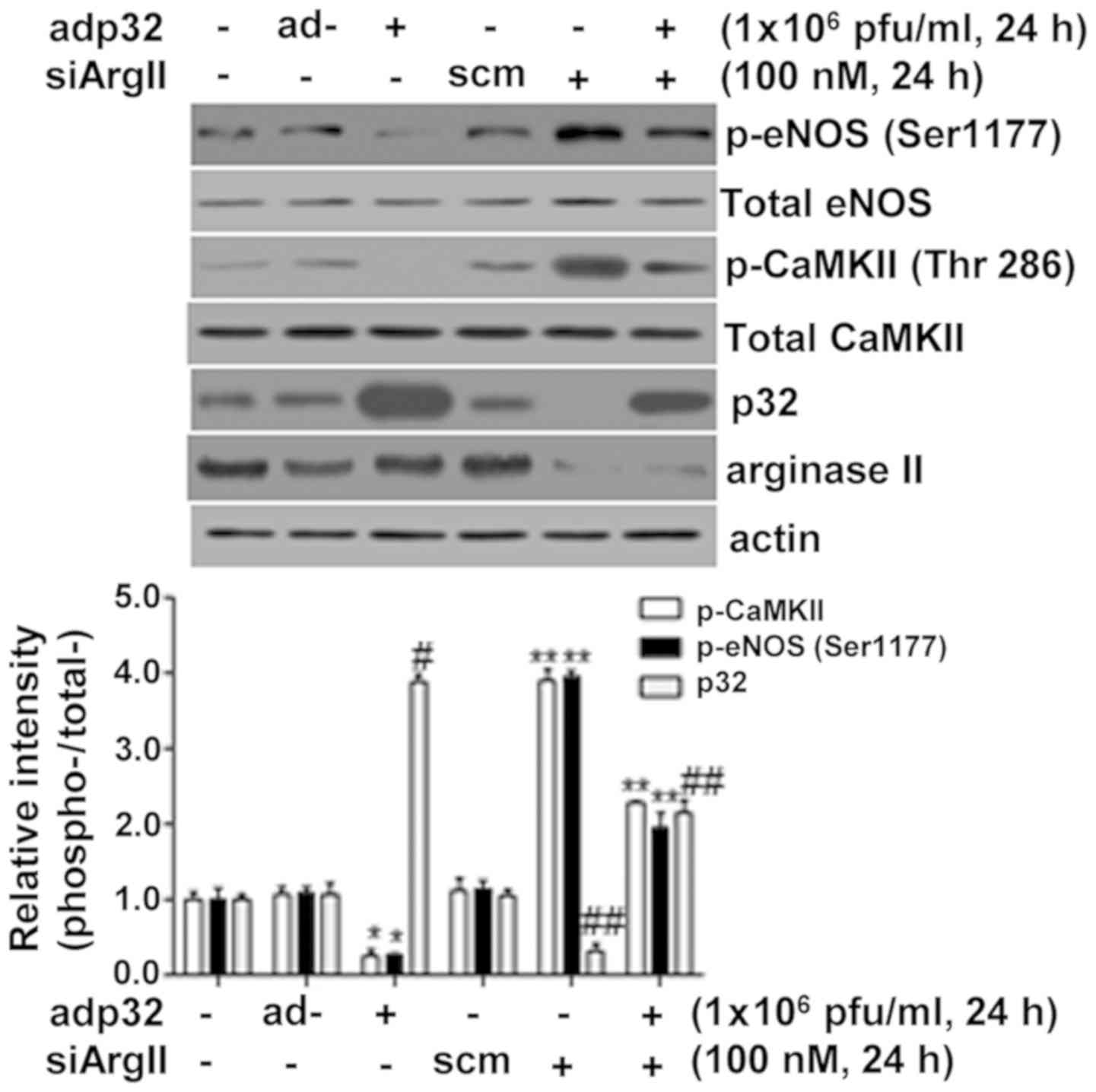
Decreased ArgII protein levels restore
adp32-dependent inactivation of eNOS
Because ArgII protein knockdown using siRNA
decreases the levels of p32 protein in normal physiological
conditions (21), the effect of
siArgII on p32 overexpression following adp32 transfection was then
evaluated. Notably, reduced ArgII protein levels after siArgII
treatment resulted in decreased levels of p32 and increased CaMKII
and eNOS phosphorylation in adp32-treated HUVECs (Fig. 4). These results indicated that
arginase II protein was involved in the regulation of p32
expression.
Discussion
In the present study, overexpressed p32 was
preferentially localized to the ER and mitochondria, and played an
important role in Ca2+ translocation that resulted in
[Ca2+]c regulation. In addition, p32 overexpression
decreased [Ca2+]c, which, in turn attenuated the
Ca2+-dependent signaling cascade involving
CaMKII/AKT/eNOS phosphorylation. Using a vascular functional assay,
it was demonstrated that increased p32 reduced NO production and
accelerated ROS generation in aortic endothelia. This was
accompanied by reduced Ach-induced vasorelaxation and augmented
PE-dependent vasoconstriction. Notably, ArgII knockdown also led to
p32 downregulation.
p32 is exclusively located in mitochondria,
consistent with the mitochondrial targeting sequence (MTS)
contained in its 73 N-terminal amino acid sequence (1,8).
However, in the present study, Flag-p32 overexpression using ad and
plasmid was localized to the ER and mitochondria as revealed in EM
and confocal microscopy. These results are consistent with a
previous study that reported that the addition of an epitope tag to
the N-terminus of p32 blocked the MTS, resulting in redirection due
to the net negative charge of the Flag tag or due to a spacing
alteration within the N-terminus (5). Notably, as indicated by EM imaging,
p32 overexpression was localized to the ER membrane, mitochondrial
membrane, and vesicles. p32 targeting to the ER membrane can be
explained by: i) A GFP-APEX2-dependent disturbance of p32 MTS; ii)
alterations of protein interactions in the ER; iii) redirection
attributed to ER stress; iv) possible membrane signals in p32; and
v) signal sequences directed to the secretory pathway (23).
p32 is a multifunctionally and multicompartmentally
(targted to various organelles) conserved eukaryotic protein. In
our previous study (21), the
binding ability of p32 to Ca2+ was assessed,
demonstrating that spermine produced from arginase activity
directly bound to p32 to facilitate Ca2+ release.
Knockdown of p32 resulted in reduced intracellular ATP levels,
restored endothelial function in ECs and decreased ROS generation
in aortic vessels (21). p32
protein levels were upregulated in ECs (21) and SMCs (24) of ApoE−/− mice fed an
high cholesterol diet (HCD), which was associated with the
impairment of the intracellular Ca2+ signaling cascade.
p32 in mitochondria also plays a key functional role in
Ca2+ movement from the cytosol to mitochondria in ECs
and SMCs (21,24). p32 overexpression in mitochondria
and the ER consistently increases in Ca2+ concentrations
in both organelles, while and [Ca2+]c is reciprocally
reduced. These results suggested that p32 itself functioned as a
channel-like protein, and not as a component of the Ca2+
channel complex, based on the EM images, which localized it on the
membrane, as well as measurements of Ca2+ concentrations
in the p32-targeted ER. Because [Ca2+]c is an important
regulator of eNOS in ECs (25),
p32 overexpression induced decreases in [Ca2+]c that
resulted in attenuated eNOS Ser1177 phosphorylation through the
CaMKII/AKT signaling cascade, and enhanced eNOS Thr495
phosphorylation. These results correlated with endothelial NO
production and reduced Ach-dependent vasorelaxation, and augmented
PE-induced vasoconstriction. Therefore, p32 could represent a new
therapeutic target for vascular disorders such as atherosclerosis
in which NO production is interrupted and detrimental ROS
generation is promoted by endothelial dysfunction.
ArgII is the principal extrahepatic isoform in human
and mouse aortic ECs, and provides L-ornithine for the synthesis of
polyamines associated with cell proliferation and differentiation
(26,27). ArgII is also induced by hypoxia,
lipopolysaccharide, and tumor necrosis factor α (28–30),
and increased ArgII activity in ECs has recently been linked to
other disorders in animal models, including aging (31), ischemic reperfusion injury
(32,33), hypertension (34,35),
ballon injury (36) and
atherosclerosis (22). Because p32
plays an essential role in the regulation of [Ca2+]c,
the stability of p32 is important for the maintenance of
intracellular Ca2+ homeostasis (21). A novel mechanism can therefore be
proposed, in which ArgII is associated with p32 stability.
Consistent with this possibility, siRNA knockdown of ArgII resulted
in decreased p32 protein.
In conclusion, p32 adenoviral overexpression
predominantly localized to the mitochondria and ER membrane and
accelerated Ca2+ movement into these organelles, which
was associated with a decrease in [Ca2+]c. p32
overexpression also decreased the Ca2+-dependent
signaling cascade involved in eNOS activation and CaMKII/AKT/eNOS
Ser1177 phosphorylation, and decreased NO production and increased
the generation of ROS. Increased levels of p32 induced endothelial
dysfunction and impaired Ach-stimulated vasorelaxation responses.
Furthermore, the present study also suggests that ArgII protein
plays an important role in the maintenance of p32 stability.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank to Miss H.A. Kim
(the Central Laboratory of Kangwon National University) for
technical assistance with the instruments.
Funding
The present study was supported by The Basic Science
Research Program of The National Research Foundation of Korea
funded by The Ministry of Education, Science and Technology (grant
nos. 2016M3A9B6903185 and 2018R1D1A1B07047959) and by a 2019
research grant (PoINT) from Kangwon National University program. Ji
Young Mun and Minkyo Jung were supported by The Korea Brain
Research Institute funded by The Ministry of Science and
Information and Communications Technology (grant. no.
19-BR-01-08).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KC, BHK, HYY, BJY and MJ performed the experiments.
BHJ, MHW and YMK analyzed the data and wrote the manuscript. JYM,
HKL and SR designed the study and wrote the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All experiments were performed with the approval of
the Ethics Committee of Kangwon National University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ghebrehiwet B, Lim BL, Peerschke EI,
Willis AC and Reid KB: Isolation, cDNA cloning, and overexpression
of a 33-kD cell surface glycoprotein that binds to the globular
‘heads’ of C1q. J Exp Med. 179:1809–1821. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Krainer AR, Mayeda A, Kozak D and Binns G:
Functional expression of cloned human splicing factor SF2: Homology
to RNA-binding proteins, U1 70K, and Drosophila splicing
regulators. Cell. 66:383–394. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Muta T, Kang D, Kitajima S, Fujiwara T and
Hamasaki N: p32 protein, a splicing factor 2-associated protein, is
localized in mitochondrial matrix and is functionally important in
maintaining oxidative phosphorylation. J Biol Chem.
272:24363–24370. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sengupta A, Banerjee B, Tyagi RK and Datta
K: Golgi localization and dynamics of hyaluronan binding protein 1
(HABP1/p32/C1QBP) during the cell cycle. Cell Res. 15:183–186.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van Leeuwen HC and O'Hare P: Retargeting
of the mitochondrial protein p32/gC1Qr to a cytoplasmic compartment
and the cell surface. J Cell Sci. 114:2115–2123. 2001.PubMed/NCBI
|
|
6
|
Itahana K and Zhang Y: Mitochondrial p32
is a critical mediator of ARF-induced apoptosis. Cancer Cell.
13:542–553. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sunayama J, Ando Y, Itoh N, Tomiyama A,
Sakurada K, Sugiyama A, Kang D, Tashiro F, Gotoh Y, Kuchino Y, et
al: Physical and functional interaction between BH3-only protein
Hrk and mitochondrial pore-forming protein p32. Cell Death Differ.
11:771–781. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fogal V, Richardson AD, Karmali PP,
Scheffler IE, Smith JW and Ruoslahti E: Mitochondrial p32 protein
is a critical regulator of tumor metabolism via maintenance of
oxidative phosphorylation. Mol Cell Biol. 30:1303–1318. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hu M, Crawford SA, Henstridge DC, Ng IH,
Boey EJ, Xu Y, Febbraio MA, Jans DA and Bogoyevitch MA: p32 protein
levels are integral to mitochondrial and endoplasmic reticulum
morphology, cell metabolism and survival. Biochem J. 453:381–391.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pupo AS and Minneman KP: Specific
interactions between gC1qR and alpha1-adrenoceptor subtypes. J
Recept Signal Transduct Res. 23:185–195. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Storz P, Hausser A, Link G, Dedio J,
Ghebrehiwet B, Pfizenmaier K and Johannes FJ: Protein kinase C
[micro] is regulated by the multifunctional chaperon protein p32. J
Biol Chem. 275:24601–24607. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Simos G and Georgatos SD: The lamin B
receptor-associated protein p34 shares sequence homology and
antigenic determinants with the splicing factor 2-associated
protein p32. FEBS Lett. 346:225–228. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Deb TB and Datta K: Molecular cloning of
human fibroblast hyaluronic acid-binding protein confirms its
identity with P-32, a protein co-purified with splicing factor SF2.
Hyaluronic acid-binding protein as P-32 protein, co-purified with
splicing factor SF2. J Biol Chem. 271:2206–2212. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lim BL, Reid KB, Ghebrehiwet B, Peerschke
EI, Leigh LA and Preissner KT: The binding protein for globular
heads of complement C1q, gC1qR. Functional expression and
characterization as a novel vitronectin binding factor. J Biol
Chem. 271:26739–26744. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu L, Zhang Z, Loewenstein PM, Desai K,
Tang Q, Mao D, Symington JS and Green M: Molecular cloning and
characterization of a cellular protein that interacts with the
human immunodeficiency virus type 1 Tat transactivator and encodes
a strong transcriptional activation domain. J Virol. 69:3007–3016.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Y, Finan JE, Middeldorp JM and
Hayward SD: P32/TAP, a cellular protein that interacts with EBNA-1
of Epstein-Barr virus. Virology. 236:18–29. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Braun L, Ghebrehiwet B and Cossart P:
gC1q-R/p32, a C1q-binding protein, is a receptor for the InlB
invasion protein of Listeria monocytogenes. EMBO J.
19:1458–1466. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fleming I, Fisslthaler B, Dimmeler S, Kemp
BE and Busse R: Phosphorylation of Thr(495) regulates
Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase
activity. Circ Res. 88:E68–E75. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Salerno JC, Harris DE, Irizarry K, Patel
B, Morales AJ, Smith SM, Martasek P, Roman LJ, Masters BS, Jones
CL, et al: An autoinhibitory control element defines
calcium-regulated isoforms of nitric oxide synthase. J Biol Chem.
272:29769–29777. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lobatón CD, Vay L, Hernández-Sanmiguel E,
Santodomingo J, Moreno A, Montero M and Alvarez J: Modulation of
mitochondrial Ca(2+) uptake by estrogen receptor agonists and
antagonists. Br J Pharmacol. 145:862–871. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koo BH, Hwang HM, Yi BG, Lim HK, Jeon BH,
Hoe KL, Kwon YG, Won MH, Kim YM, Berkowitz DE, et al: Arginase II
Contributes to the Ca2+/CaMKII/eNOS Axis by Regulating
Ca2+ Concentration Between the Cytosol and Mitochondria
in a p32-Dependent Manner. J Am Heart Assoc. 7:e0095792018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ryoo S, Gupta G, Benjo A, Lim HK, Camara
A, Sikka G, Lim HK, Sohi J, Santhanam L, Soucy K, et al:
Endothelial arginase II: A novel target for the treatment of
atherosclerosis. Circ Res. 102:923–932. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Peerschke EI and Ghebrehiwet B: The
contribution of gC1qR/p33 in infection and inflammation.
Immunobiology. 212:333–342. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
24. Koo BH, Hong D, Hong HD, Lim HK, Hoe
KL, Won M-H, Kim YM, Berkowitz DE and Ryoo S: Arginase II activity
regulates cytosolic Ca(2+) level in a p32-dependent manner that
contributes to Ca(2+)-dependent vasoconstriction in native
low-density lipoprotein-stimulated vascular smooth muscle cells.
Exp Mol Med. 51:1–12. 2019. View Article : Google Scholar
|
|
25
|
Sessa WC: eNOS at a glance. J Cell Sci.
117:2427–2429. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ignarro LJ, Buga GM, Wei LH, Bauer PM, Wu
G and del Soldato P: Role of the arginine-nitric oxide pathway in
the regulation of vascular smooth muscle cell proliferation. Proc
Natl Acad Sci USA. 98:4202–4208. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li H, Meininger CJ, Hawker JR Jr, Haynes
TE, Kepka-Lenhart D, Mistry SK, Morris SM Jr and Wu G: Regulatory
role of arginase I and II in nitric oxide, polyamine, and proline
syntheses in endothelial cells. Am J Physiol Endocrinol Metab.
280:E75–E82. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Morris SM Jr, Kepka-Lenhart D and Chen LC:
Differential regulation of arginases and inducible nitric oxide
synthase in murine macrophage cells. Am J Physiol. 275:E740–E747.
1998.PubMed/NCBI
|
|
29
|
Louis CA, Reichner JS, Henry WL Jr,
Mastrofrancesco B, Gotoh T, Mori M and Albina JE: Distinct arginase
isoforms expressed in primary and transformed macrophages:
Regulation by oxygen tension. Am J Physiol. 274:R775–R782.
1998.PubMed/NCBI
|
|
30
|
Collado B, Sánchez-Chapado M, Prieto JC
and Carmena MJ: Hypoxia regulation of expression and angiogenic
effects of vasoactive intestinal peptide (VIP) and VIP receptors in
LNCaP prostate cancer cells. Mol Cell Endocrinol. 249:116–122.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Berkowitz DE, White R, Li D, Minhas KM,
Cernetich A, Kim S, Burke S, Shoukas AA, Nyhan D, Champion HC, et
al: Arginase reciprocally regulates nitric oxide synthase activity
and contributes to endothelial dysfunction in aging blood vessels.
Circulation. 108:2000–2006. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hein TW, Zhang C, Wang W, Chang CI,
Thengchaisri N and Kuo L: Ischemia-reperfusion selectively impairs
nitric oxide-mediated dilation in coronary arterioles:
Counteracting role of arginase. FASEB J. 17:2328–2330. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jung C, Gonon AT, Sjöquist PO, Lundberg JO
and Pernow J: Arginase inhibition mediates cardioprotection during
ischaemia-reperfusion. Cardiovasc Res. 85:147–154. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang C, Hein TW, Wang W, Miller MW,
Fossum TW, McDonald MM, Humphrey JD and Kuo L: Upregulation of
vascular arginase in hypertension decreases nitric oxide-mediated
dilation of coronary arterioles. Hypertension. 44:935–943. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Johnson FK, Johnson RA, Peyton KJ and
Durante W: Arginase inhibition restores arteriolar endothelial
function in Dahl rats with salt-induced hypertension. Am J Physiol
Regul Integr Comp Physiol. 288:R1057–R1062. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Peyton KJ, Ensenat D, Azam MA, Keswani AN,
Kannan S, Liu XM, Wang H, Tulis DA and Durante W: Arginase promotes
neointima formation in rat injured carotid arteries. Arterioscler
Thromb Vasc Biol. 29:488–494. 2009. View Article : Google Scholar : PubMed/NCBI
|