Introduction
Psoriasis is a chronic T cell-mediated inflammatory
skin disease that is driven by overproduction of pro-inflammatory
cytokines and excessive proliferation of keratinocytes (1). Interleukin (IL)-17 acts as the
primary pathological effector of psoriasis (1–3) by
regulating keratinocyte proliferation through signal transducer and
activator of transcription 3 (STAT3) (4–6).
STAT3 directly binds to IL-17 (7,8) and
is also a key regulator of keratinocyte function (5). Consistent with this, STA-21, a small
molecule inhibitor of STAT3, significantly alleviates the severity
of lesions in patients with psoriasis (6,9,10).
Keratin 17 (K17) is a marker of psoriasis that is undetectable in
normal epidermis but markedly upregulated in psoriatic lesions
(11,12). A recent study has suggested that
IL-17 induces K17 in the epidermis by promoting the nuclear
translocation of STAT3, which contributes to the pathogenesis of
psoriasis (13).
Topical application of corticosteroids is currently
the standard treatment option for psoriasis (14). Nevertheless, natural, plant-derived
extracts are a promising therapeutic option owing to their fewer
side effects. Shikonin is a 288-kDa liposoluble naphthoquinone
derived from Lithospermum erythrorhizon, which is used in
Traditional Chinese Medicine for the treatment of various diseases,
such as breast and prostate cancer, as well as lung adenocarcinoma
and hepatocellular carcinoma (15,16).
Shikonin displays anti-inflammatory, immunosuppressive and
anti-tumor properties (17–20)
and inhibits the proliferation of skin cancer cells by blocking
STAT3 (21,22). Our previous study demonstrated that
shikonin could suppress IL-17-induced production of cytokines
associated with psoriasis by inhibiting the JAK/STAT3 signaling
pathway (23). Moreover, Liu et
al (24) found that shikonin
could downregulate K17 in proliferating keratinocytes by
interfering with STAT3 signaling. More recently, Yu et al
(25) also demonstrated that
shikonin induced G0/G1 cell cycle arrest in
the human HaCaT keratinocyte cell line. However, the potential
effects of shikonin on the downstream mediators of the JAK/STAT3
signaling pathway are unclear.
The CCAAT/enhancer-binding protein δ (CEBPD) has
been implicated in physiological processes, such as cell
differentiation, metabolism, inflammation, cell cycle arrest and
apoptosis (26). It is regulated
by inflammatory cytokines, such as IL-6, and its protein
overexpression can induce cell cycle arrest and apoptosis in
several types of cancer, such as prostate cancer, neuroblastoma and
acute myeloid leukemia (27–31).
Li et al (32) demonstrated
that ρ-associated kinase 2 knockdown could upregulate CEBPD mRNA
and protein expression levels and activity, resulting in increased
proliferation of hepatocellular carcinoma cells in vitro and
in vivo. In addition, CEBPD is also activated by IL-6 and
IL-17 in response to chemotherapeutic anti-cancer drugs (33,34).
Furthermore, CEBPD is also critical for IL-17 and tumor necrosis
factor-α (TNF-α)-induced expression of lipocalin 2, serum amyloid
A3 pseudogene and IL-6 during psoriasis progression (35). However, to the best of our
knowledge, the mechanisms underlying the effect of CEBPD in
keratinocytes remains largely unknown.
The aim of the present study was to analyze the
effect of shikonin on the IL-6/STAT3 signaling pathway in
IL-17-treated HaCaT cells using the RT2 Profiler™ PCR
Array system. In addition, the effect of shikonin on the imiquimod
(IMQ)-induced murine psoriasis model was also evaluated.
Materials and methods
Cell culture
HaCaT cells (GCC-AO0003CS; Shanghai Jikai Gene
Medical Technology Co., Ltd.) were cultured in DMEM (HyClone; GE
Healthcare Life Sciences) supplemented with 10% heat-inactivated
FBS (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin
(HyClone; GE Healthcare Life Sciences), and 100 µg/ml streptomycin
(HyClone; GE Healthcare Life Sciences). The HaCaT cell line was
authenticated by the supplier using STR profiling. The cells were
maintained in a humidified incubator at 37°C with 5%
CO2.
MTS assay
The proliferation of HaCaT cells was assessed using
a MTS assay (Promega Corporation) according to the manufacturer's
instructions. Briefly, the cells were seeded into 96-well plates,
at a density of 5,000 cells/well and cultured for 24, 48 and 72 h
in the presence of varying concentrations of shikonin (0.00, 0.01,
0.05, 0.25, 0.50, 1.00, 2.50 or 5.00 µM) (Sigma-Aldrich; Merck
KGaA) in a humidified incubator at 37°C with 5% CO2. In
order to verify the effect of shikonin on IL-17 induced HaCaT cell
proliferation, the HaCaT cells were seeded into 96-well plates, at
a density of 5,000 cells/well and cultured in a humidified
incubator at 37°C for 24, 48 and 72 h with IL-17 (PeproTech, Inc.),
shikonin, IL-17 + shikonin, or no treatment. In order to verify the
effect of shikonin on IL-17-induced HaCaT cell proliferation when
CEBPD is silenced, the assay was performed with scrambled siRNA
(NC), RNAi + IL-17, RNAi + shikonin, RNAi + IL-17 + shikonin, and
untreated cells. The number of cells and the duration of treatment
are consistent with the previous description. The absorbance was
measured in each well at 490 nm using a microplate reader (Bio-Rad
Laboratories, Inc.). Each sample was analyzed in six replicates,
and the assay was repeated three times.
iCELLigence system
The proliferation, viability, and morphology of the
cells can be ascertained on the basis of electrical impedance when
cultured on micro-electrodes (36). The HaCaT cells were seeded in two
iCELLigence system 8-well plates (ACEA Bioscience, Inc.) at a
density of 5,000 cells/well in 300 µl medium and cultured for 14 h.
Then, different concentrations of IL-17 (0, 10, 20, 30, 40, 50 or
60 ng/ml) were added and cultured for 56 h in a humidified
incubator at 37°C. Each sample was tested in duplicate. In
addition, the cells were cultured in quadruplicate with 40 ng/ml
IL-17, 1 µM shikonin or both for 96.8 h. The cell index (CI), a
measure of the number of cells, was monitored in real-time to
assess proliferation using RTCA software (37). Each assay was repeated three
times.
Reverse transcription quantitative-PCR
(RT-qPCR)
The HaCaT cells were cultured with 40 ng/ml IL-17
for 0, 2, 4, 6, 8 and 10 h and harvested. Total RNA was isolated
using the miRNeasy Mini kit and cDNA was synthesized from RNA using
a GoScript™ Reverse Transcription System (Promega Corporation)
according to the manufacturer's instructions. RNA concentrations
were obtained using the Nanodrop ND-1000 spectrophotometer
(NanoDrop Technologies; Thermo Fisher Scientific, Inc.). The
sequences of the primers used were as follows: STAT3 forward,
5′-CACCAAGCGAGGACTGAGCAT-3′ and reverse,
5′-GCCAGACCCAGAAGGAGAAGC-3′; CEBPD forward,
5′-ACTTACCACCACTAAACTGCGAG-3′ and reverse,
5′-CTGCATCAACAGGAGTAAGATGTAG-3′; K17 forward,
5′-CCACCCAGAAGACTGTGGAT-3′ and reverse, 5′-TTCTAGACGGCAGGTCAGGT-3′;
and GAPDH forward, 5′-TGGAGTCTACTGGCGTCTT-3′; and reverse,
5′-TGTCATATTTCTCGTGGTTCA-3′ (all Invitrogen; Thermo Fisher
Scientific, Inc.). RT-qPCR was performed in 96-well plates using a
7900HT Fast Real-Time PCR system (Thermo Fisher Scientific, Inc.).
The RT2 SYBR Green qPCR Master mix (Promega Corporation)
was employed for amplification on a reaction mixture containing
primers (0.4 µl each), 2× qPCR Master Mix (10 µl), cDNA (2 µl), and
nuclease free water. Amplification was performed as follows: 95°C
(2 min); 40 cycles at 95°C (15 sec) and 60°C (1 min). The relative
mRNA expression levels were calculated via the 2−ΔΔCq
method (38). Each sample was
analyzed in six replicates, and the assay was repeated three
times.
RT2Profiler™ PCR array
analysis
Cells were seeded in 6-well plates at a density of
5×105 cells/well and treated with 40 ng/ml IL-17, 1 µM
shikonin or both for 8 h. Untreated cells were used as blank
control. Total RNA was extracted, and cDNA was synthesized using
the RT2 First Strand kit (Qiagen GmbH) according to the
manufacturers' recommendations. The RT2 Profiler™ PCR
Array kit 384 (4×96; Qiagen GmbH) specific for the human IL-6/STAT3
signaling pathway was used to analyze the mRNA expression levels of
84 associated genes, which included receptors, up and downstream
signaling, cytokines and pathway activity in response to the
IL-6/STAT3 signaling pathway. The reaction mixture consisted of 650
µl 2× RT2 SYBR Green Mastermix (Qiagen GmbH), 102 µl
cDNA and 548 µl nuclease-free water to a final volume of 1,300 µl,
and 10 µl of this mixture was dispensed in each well of a 96-well
plate. RT-qPCR was performed using the 7900HT Fast Real-Time PCR
system (Thermo Fisher Scientific, Inc.). The thermocycling
conditions consisted of a 10-min, 95°C hot-start, followed by 40
amplification cycles at 95°C for 15 sec and 60°C for 1 min. The PCR
array data was analyzed using the Microsoft Excel 2005 macro
program (Microsoft Corporation) and the SABiosciences PCR Array
Data Analysis Web Portal (http://SABiosciences.com/pcrarrydataanalysis.php).
Each treated group was compared to the blank control. P-values were
calculated using Student's t-test on replicate 2−ΔΔCq
values for each gene (38). A
total of 5 housekeeping genes, including β-actin, β2-microglobulin,
GAPDH, hypoxanthine and ribosomal protein large P0 were used for
normalization.
Small interfering (si) RNA
transfection
Cells were seeded at a density of 5,000 cells/well
in antibiotics-free medium the day before transfection.
Lipofectamine® RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.) was used as siRNA transfection reagent according
to the manufacturer's instructions. Cells grew in medium only and
treated with CEBPD-siRNA or Silencer® Select negative
control siRNA (5′-UUCUCCGAACGUGUCACGUTT-3′) for 24 h at 37°C in a
CO2 incubator. A final concentration of 10 nmol siRNA
was used after optimization. The siRNA targeting human CEBPD and
negative control were designed and synthesized by Shanghai
GenePharma Co. Ltd., and the siRNA sequence used in the study was
5′-CCUGGACUUACCACCACUATT-3′.
Establishment of a murine psoriasis
model and treatment regimen
A total of 20 male BALB/c mice (20–25 g;
8-weeks-old) were purchased from the Center of Experimental Animals
of China Medical University. All the animals were randomly divided
into four groups: i) IMQ group; ii) medium oil (MO) group; iii)
shikonin oil (SO) group; and iv) control (CON) group with five mice
in each group. All animals were housed in an animal facility with a
12/12 h light/dark cycle at 25±2°C with free access to food and
water. All animal experiments were performed in accordance with the
National Institutes of Health Guide for the Care and Use of
Laboratory Animals and approved by the Animal Care Committee at
China Medical University (IACUC no. 16088M)
To induce psoriasis, 50 mg 5% IMQ (Aldara; iNova
Pharmaceuticals Australia Pty Limited) was applied topically on a
2×3 cm shaved area on the back for 8 consecutive days (39,40).
Shikonin (Sigma-Aldrich; Merck KGaA) was dissolved in DMSO and
diluted with MO (Fulinmen; COFCO Corporation) at 60°C to produce SO
at a final concentration of 0.5766 mg/ml. A 0.5 ml volume SO (1 µM)
or MO was applied on the affected area 2 h after each IMQ
administration. The severity of the inflamed lesions was evaluated
by the Psoriasis Area Severity Index (PASI) (24) that independently scores erythema,
scaling and thickening on a scale of 0 to 4: i) 0, none; ii) 1,
slight; iii) 2, moderate; iv) 3, marked; and v) 4, very marked. All
animals were healthy during the experimental period and sacrificed
by cervical dislocation after 8 consecutive days.
Western blot analysis
Mice were sacrificed by cervical dislocation and the
lesion areas were shaved. After sterilizing with povidone iodine
and 70% ethanol, skin pieces measuring 2×3 cm were cut, and the
subcutaneous fat and muscle were excised. The tissues were then cut
into smaller 0.5×1 cm sections and digested with 0.25% trypsin
(HyClone; GE Healthcare Life Sciences) for 2 h at 32°C. The
epidermal layer was detached from the dermis using a sterile
scalpel (41). Cells were seeded
in 6-well plates, at a density of 5×105 cells/well and
treated with IL-17, and/or shikonin for 8 h. Untreated cells were
used as blank control. Total protein was extracted from cultured
cells and epidermal samples using RIPA lysis buffer and quantified
with the bicinchoninic acid Protein Assay kit (both Beyotime
Institute of Biotechnology). Equal amount of proteins per sample
(20 µg and 30 µg for cells and tissue, respectively) were separated
using 10% SDS-PAGE, then electro-transferred to PVDF membranes (EMD
Millipore). After blocking with 5% skimmed milk or BSA in TBS +
Tween-20 for 1 h at room temperature, the blots were incubated
overnight with rabbit anti-CEBPD (cat. no. ab198230; 1:500),
anti-K17 (cat. no. ab109725; 1:1,000) and β-actin (cat. no. ab8226;
1:8,000) (all Abcam) primary antibodies at 4°C. The membranes were
then incubated with horseradish peroxidase-conjugated goat
anti-rabbit IgG (cat. no. ZB-2306; 1:5;000 OriGene Technologies,
Inc.) for 1 h at room temperature. Bands were visualized using an
ECL kit (Bio-Rad Laboratories, Inc.) on a MicroChemi™
Chemiluminescent Imaging System (DNR Bio-Imaging Systems, Ltd.).
Band densities were analyzed by ImageJ software (version 1.52a;
National Institutes of Health).
Histopathology and
immunohistochemistry (IHC)
Skin samples from the mice were excised, washed with
PBS and fixed in formalin, embedded in paraffin and sectioned as 5
µm slices. Then, staining with hematoxylin and eosin (H&E) was
performed, followed by assessment under a light microscope (Olympus
Corporation) at a magnification of ×200. For IHC, skin samples from
the mice were harvested, fixed in 4% paraformaldehyde at room
temperature for 48 h, dehydrated, embedded in paraffin and
sectioned (thickness, 5-µm). Gradient ethanol was used to dewax and
hydrate the samples, and antigen retrieval was performed in 0.01 M
sodium citrate buffer solution (pH, 6.0) for 25 min in a water bath
at 95°C. Sections were incubated with anti-CEBPD antibody (1:100;
cat. no. ab198230; Abcam) overnight at 4°C. An IHC kit (Beijing
Zhongshan Jinqiao Biotechnology Co., Ltd.) was employed for
detection according to the manufacturer's instructions. The
sections were incubated with secondary antibody (provided by the
kit) at room temperature for 1 h. The sections were incubated with
DAB (Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.) for color
development and counterstained with hematoxylin at room temperature
for 1 min. Three randomly selected regions in the center of each
section were assessed under a light microscope (magnification,
×400; Olympus Corporation). The mean optical density was obtained
by dividing integral optic density by the corresponding area using
Image-Pro Plus software (version 6.0; Media Cybernetics, Inc.).
Statistical analysis
Statistical analysis was performed using SPSS v16.0
software (SPSS, Inc.). Grouped pairs were compared using student's
t-test. One-way ANOVA was used to compare multiple groups. Fisher's
Least Significant Difference (for ≤4 groups) or Tukey's post hoc
test (>4) were used to perform inter-group comparisons.
P<0.05 was considered to indicate a statistically significant
difference. Data are presented as the mean ± SEM. All assays were
repeated three times.
Results
Shikonin inhibits IL-17-induced
proliferation of keratinocytes
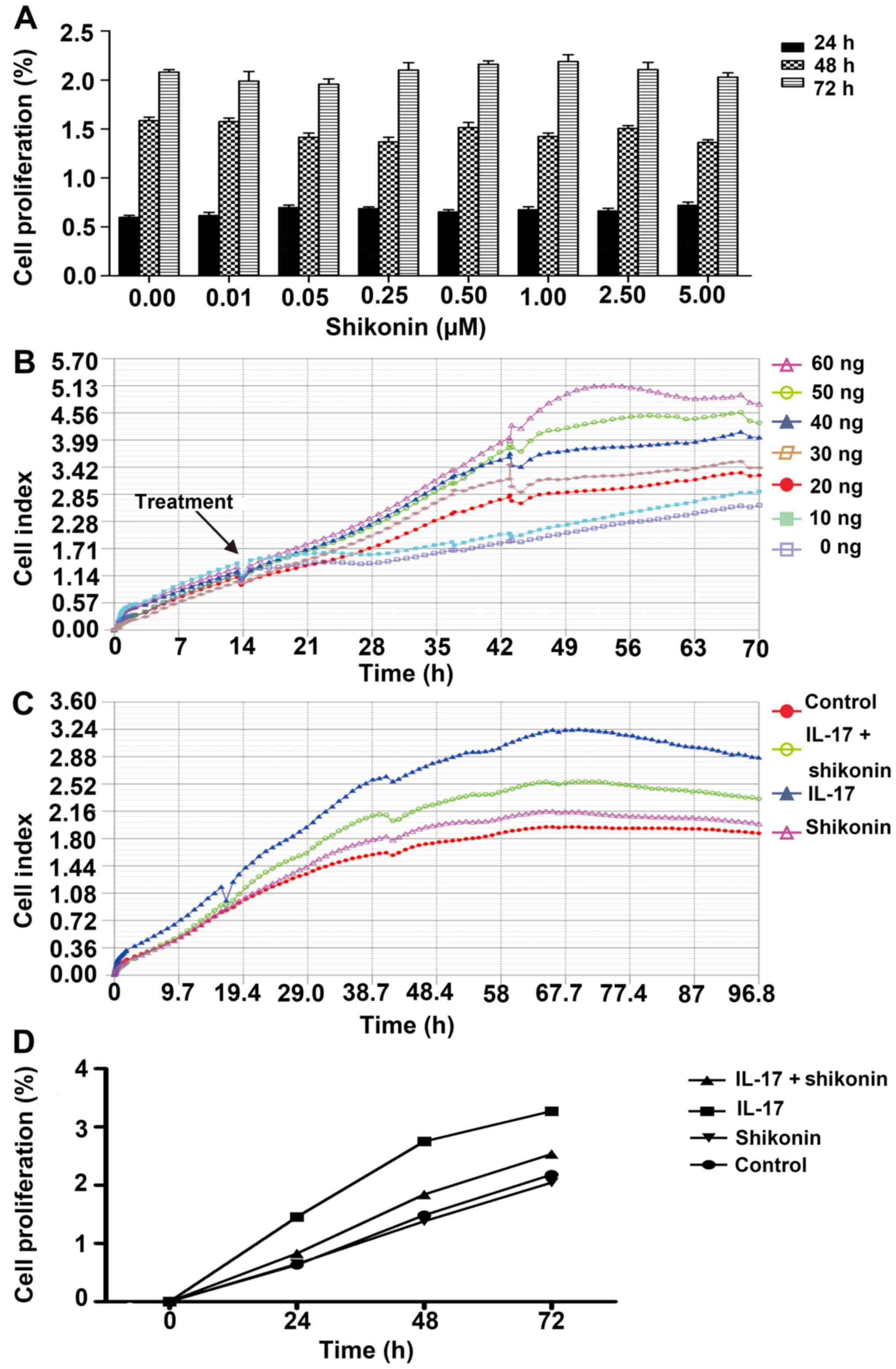
MTS and iCELLigence assays were used to determine
the optimal concentrations of shikonin and IL-17. Shikonin had no
significant cytotoxic effect on HaCaT cell proliferation across
concentrations ranging from 0 to 5 µM. Therefore 1 µM was used in
subsequent experiments (Fig. 1A).
The effect of IL-17 on cell proliferation was assessed in real time
using the iCELLigence system. IL-17 markedly increased the CI of
HaCaT cells in a concentration-dependent manner, compared with the
untreated control (Fig. 1B).
However, preliminary experiments demonstrated that 1 µM shikonin
had a good inhibitory effect on IL-17 induced cell proliferation at
≤40 ng/ml (data not shown). Thus, a concentration of 40 ng/ml was
used in subsequent experiments. Shikonin markedly reversed the
proliferative effects of IL-17, nearly to same levels as the
control group (Fig. 1C). The MTS
assay also confirmed the inhibitory effects of shikonin on
IL-17-stimulated HaCaT cells (Fig.
1D). Altogether, these results suggested that IL-17 could
promote keratinocyte proliferation and that this effect could be
inhibited by shikonin.
Shikonin antagonizes IL-17 by
upregulating CEBPD
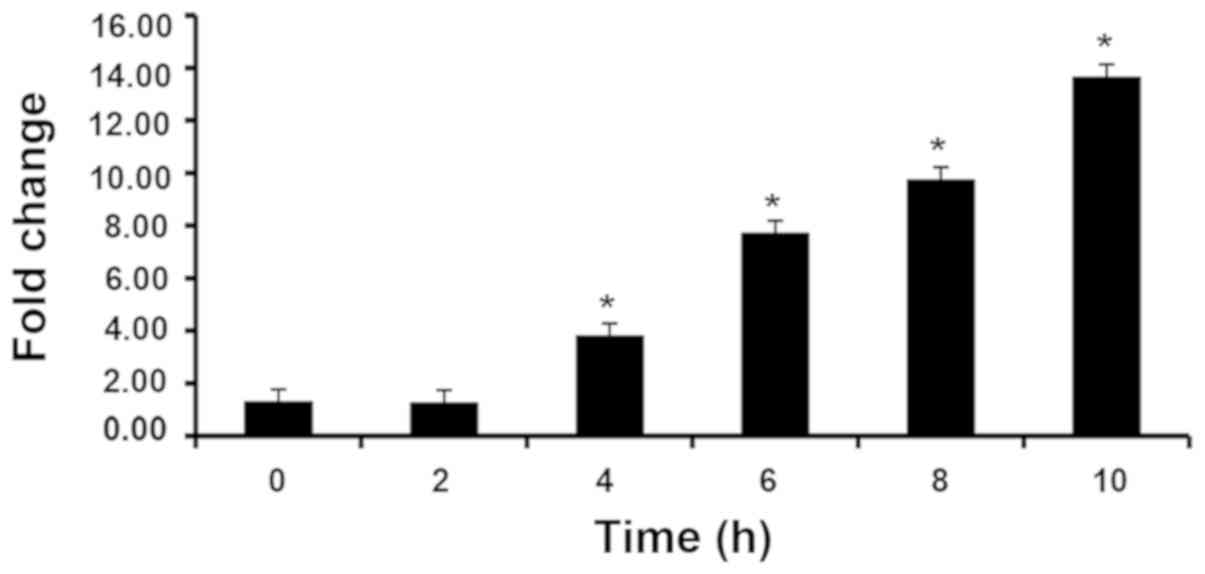
HaCaT cells stimulated with IL-17 caused a
time-dependent significant increase after 4 h in STAT3 expression.
In subsequent experiments, 8 h was randomly selected as the
duration of incubation. (Fig. 2).
Therefore, the changes in the expression levels of genes associated
with the IL-6/STAT3 pathway in the differentially treated cells
using the RT2Profiler™ PCR Array were analyzed. As
presented in Table I, there were
17 genes, including CCL2, CCL4, CEBPD, CSF1, CSF2, CSF3R, IL11,
IL18R1, IL23A, IL4, IL5, IL6, IL6R, JAK2, TLR4, TNFRSF1B and
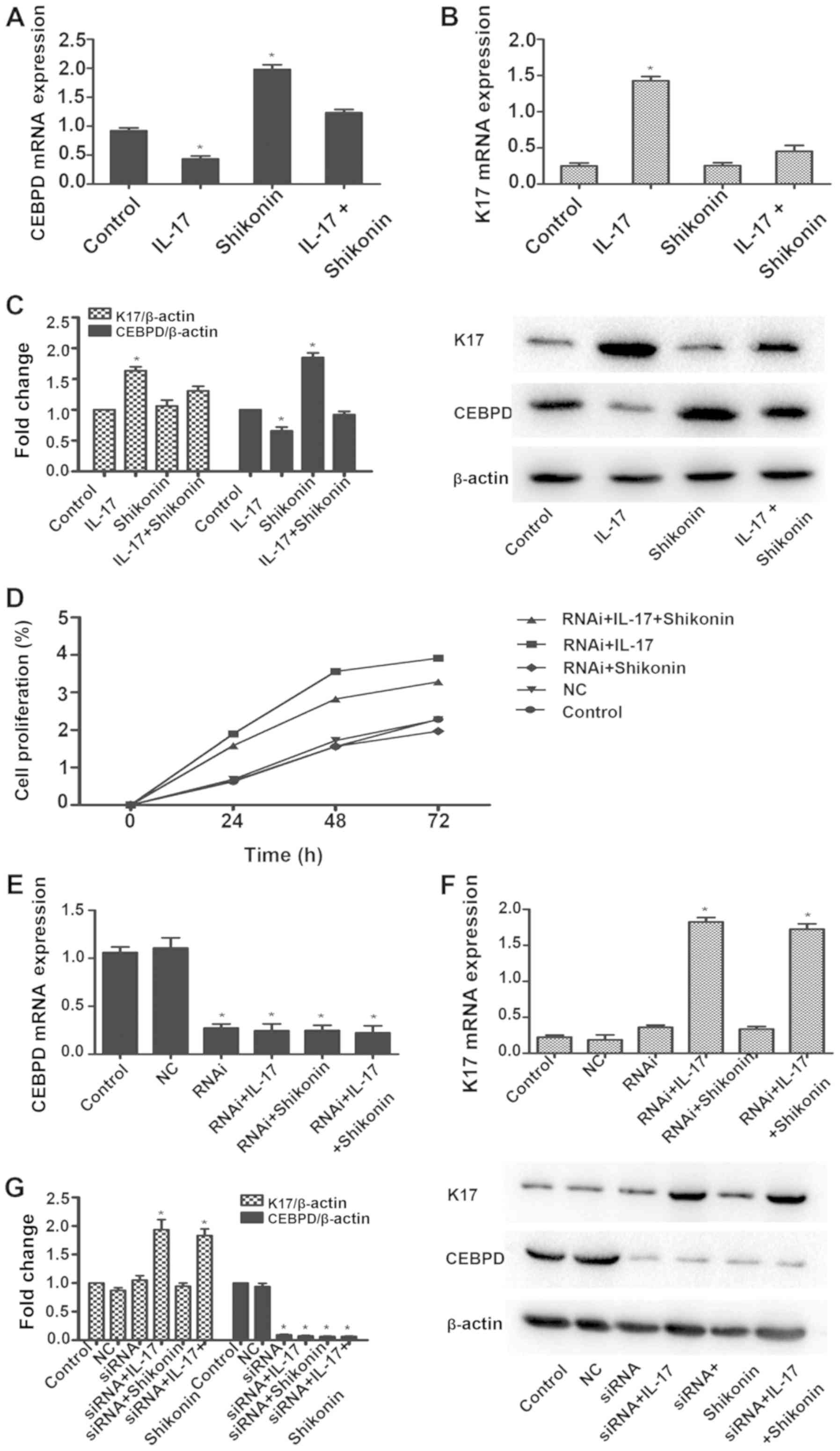
TNFSF10, whose expression levels changed markedly. CEBPD was
downregulated in HaCaT cells in response to IL-17 and upregulated
in the presence of shikonin. The fold change in expression levels
of CEBPD in shikonin + IL-17 was 2.2. There was no notable
difference compared with the blank control group. Similarly, a
significant reduction in CEBPD mRNA and protein levels was observed
following IL-17 treatment, which was restored in the presence of
shikonin (Fig. 3A and C). Thus, it
was hypothesized that CEBPD downregulation might represent a
proliferative signature in IL-17-stimulated HaCaT cells. Consistent
with this hypothesis, the MTS assay confirmed that when CEBPD is
silenced, shikonin does not markedly reverse the proliferative
effects of IL-17 (Fig. 3D).
 | Table I.RT2 Profiler PCR array
genes expression analysis. |
Table I.
RT2 Profiler PCR array
genes expression analysis.
|
|
| Fold change in
expression |
|---|
|
|
|
|
|---|
| Gene symbol | Gene name | IL-17 | Shikonin | IL-17 +
Shikonin |
|---|
| CCL2 | C-C motif chemokine
ligand 2 |
−3.34 | / | / |
| CCL4 | C-C motif chemokine
ligand 4 |
8.64 | / | / |
| CEBPD | CCAAT/enhancer
binding protein δ | −31.95 |
9.62 |
2.2 |
| CSF1 | Colony stimulating
factor 1 |
6.4 |
7.7 |
6.33 |
| CSF2 | Colony stimulating
factor 2 | / |
3.4 | / |
| CSF3R | Colony stimulating
factor 3 receptor |
−5.26 | −14.68 | −23.53 |
| IL11 | Interleukin 11 | / | / |
−3.05 |
| IL18R1 | Interleukin 18
receptor 1 | −17.48 | / |
−3.34 |
| IL23A | Interleukin 23
subunit α |
−5.26 | / | / |
| IL4 | Interleukin 4 | / |
3.25 |
4.07 |
| IL5 | Interleukin 5 | / |
16.51 | / |
| IL6 | Interleukin 6 | / | −11.05 |
−3.16 |
| IL6R | Interleukin 6
receptor | / |
5.91 |
5.29 |
| JAK2 | Janus kinase 2 | / | / | −11.07 |
| TLR4 | Toll-like receptor
4 | / |
3.47 | / |
| TNFRSF1B | Tumor necrosis
factor receptor superfamily member 1B | / |
9.42 |
8.02 |
| TNFSF10 | Tumor necrosis
factor superfamily member 10 | / | / |
−3.02 |
The psoriasis marker, K17 was upregulated in
response to IL-17. However, when IL-17 and shikonin were present,
there was no notable difference in the mRNA and protein expression
levels of K17 compared with the blank control (Fig. 3B and C). When CEBPD was silenced
(Fig. 3E and G), K17 was also
upregulated in response to IL-17 (Fig.
3F and G). When IL-17 and shikonin were present, the expression
levels of K17 in mRNA and protein were upregulated as well
(Fig. 3F and G). Collectively,
these results suggested that IL-17 could induce proliferation of
keratinocytes by downregulating CEBPD, and that the inhibitory
effect of shikonin was mediated through CEBPD.
Shikonin alleviates IMQ-induced
psoriasis in vivo
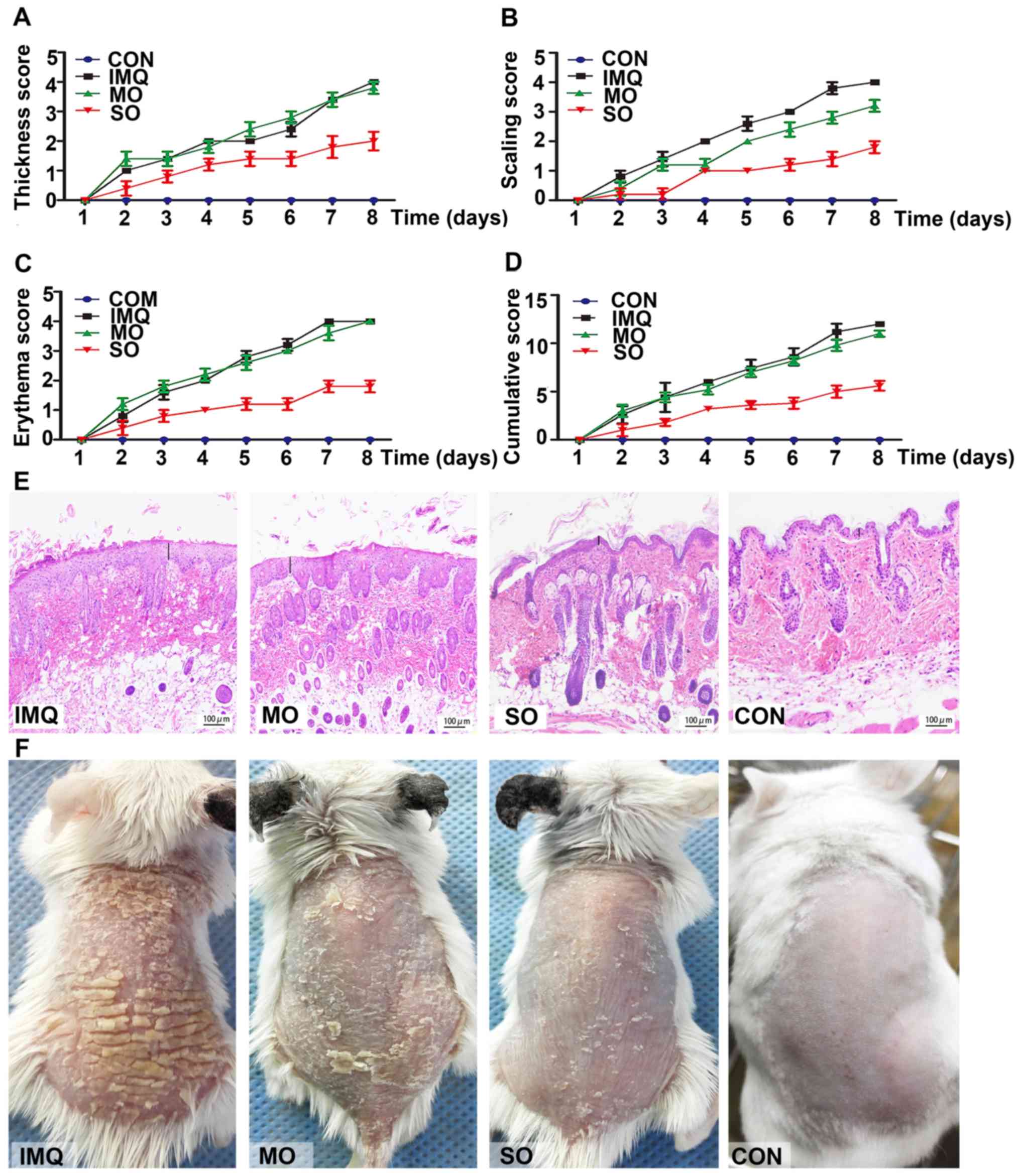
The potential therapeutic effects of shikonin were
investigated in an IMQ-induced model of psoriasis. Topical
application of IMQ resulted in time-dependent thickening, scaling,
erythema and inflammation of the affected skin. However, SO
treatment markedly lowered PASI scores (Fig. 4A-D), resulting in smoother skin and
fewer scales (Fig. 4F).
Shikonin-treated mice did not present any changes in their body
weight compared to those in the untreated control group (data not
shown). MO had no effect on erythema, skin thickening and
cumulative PASI scores compared to the untreated psoriatic mice;
however, it slightly reduced scaling due to its moisturizing
properties. IMQ led to hyperplasia, elongated rete-like ridges and
acanthosis in the epidermis, as well as perivascular infiltration
of inflammatory cells in the upper dermis similar to that seen in
human psoriatic lesions. SO treatment markedly improved these
psoriasis-like lesions (Fig. 4E).
Consistent with previous studies, which reported aberrantly high
K17 levels in early and advanced psoriasis lesions (12), increased in situ K17
expression was detected in the IMQ-induced psoriatic lesions, which
was alleviated by shikonin (Fig.
5C). In addition, CEBPD was undetectable in the epidermal
keratinocytes and the dermal layer of the IMQ-treated skin
(Fig. 5A and B). However, SO
restored CEBPD in the epidermis, especially the basal cell layer,
to levels similar in the untreated controls (Fig. 5C). These results indicated that
shikonin could alleviate IMQ-induced psoriatic lesions in a mouse
model, likely through the upregulation of CEBPD.
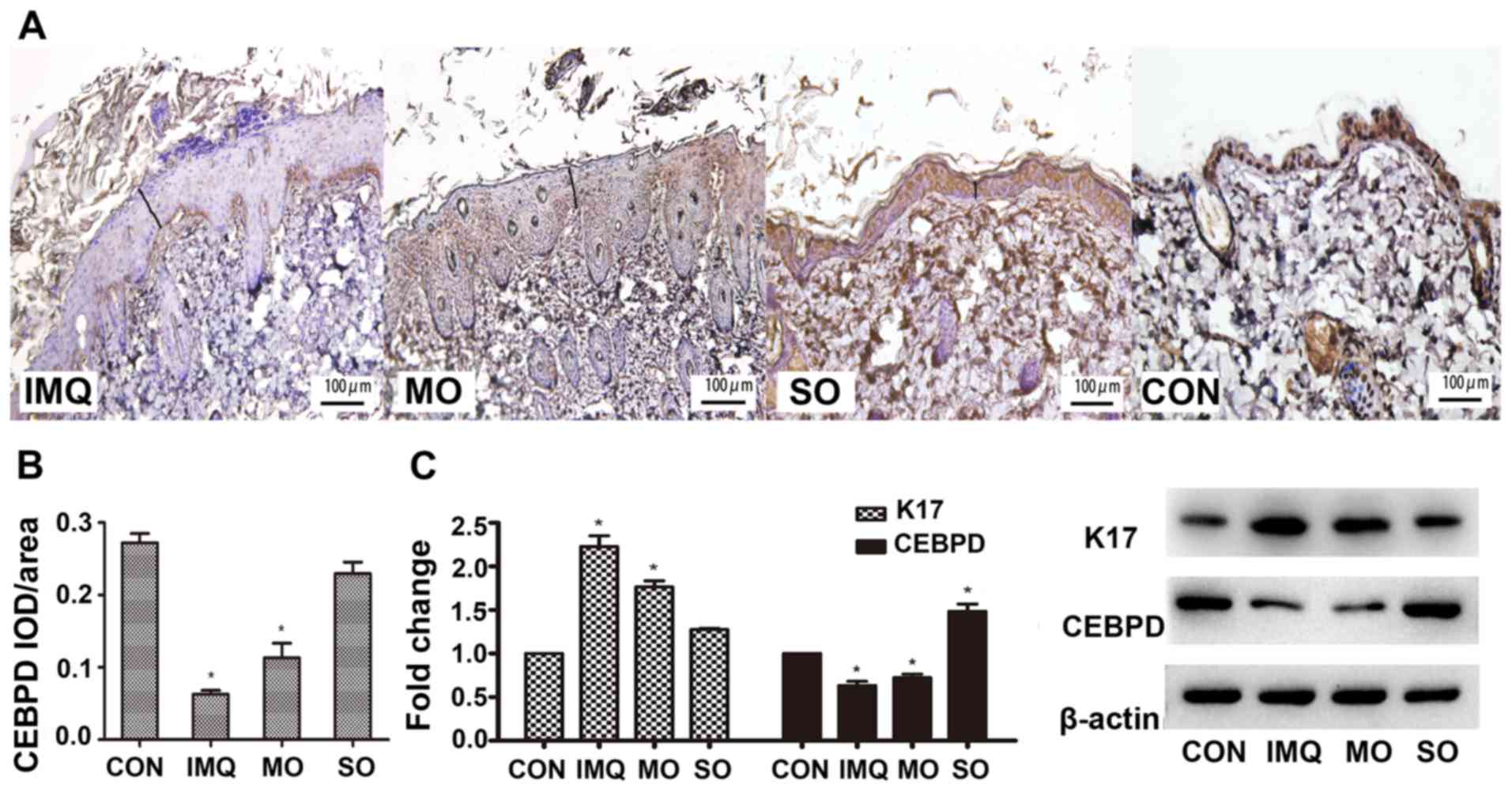 | Figure 5.Effects of shikonin on CEBPD
expression in an IMQ-induced psoriatic mouse model. (A)
Representative IHC images showing in situ expression of
CEBPD in the mouse skin. Magnification, ×400. (B) Mean optical
density of CEBPD. (C) CEBPD and K17 protein expression levels in
psoriatic lesions. *P<0.05, vs. CON. CEBPD,
CCAAT/enhancer-binding protein δ; K17, keratin 17; IMQ, imiquimod,
SO, shikonin oil; MO, medium oil; CON, control; IOD, integrated
optical density. |
Discussion
In the present study, shikonin inhibited the
proliferative effects of IL-17 on keratinocytes both in
vitro and in vivo by targeting the IL-6/STAT3 signaling
pathway. Contradictory to previous reports (33,42),
in the present study, IL-17 downregulated CEBPD in the
hyper-proliferative HaCaT cells, which was reversed by
shikonin.
There are six distinct isoforms in the CEBP family,
including CEBPα, CEBPβ, CEBPγ, CEBPδ and CEBPε as well as CEBP
homologous protein (26). These
are involved in the regulation of growth and differentiation of
various cells, such as hepatocytes, pneumocytes and hematopoietic
cells (26,43). Studies have demonstrated that CEBPD
is implicated in cell cycle control: CEBPD mRNA and protein levels
are markedly induced in cultured mouse mammary epithelial cells
during G0 growth arrest (44). It also plays an important role in
promoting prostate epithelial cell growth arrest and/or apoptosis
after androgen withdrawal (43).
It has been reported that CEBPD may be induced by many
extracellular stimuli, such as IL-1, lipopolysaccharide, interferon
(IFN)-α, IFN-γ, and IL-6 (34,43).
In addition, Wang et al (33) reported that although CEBPD has long
been considered a tumor suppressor gene, CEBPD serves dual roles in
pro- and antitumor processes under conditions such as hypoxia and
inflammation (34). Furthermore,
in human prostate adenocarcinoma LNCaP 104-S and 104-R1 cells,
Chuang et al (34)
demonstrated that DNA- and histone-mediated epigenetic regulation
of CEBPD transcriptional attenuation can occur in a cell type- or
tissue-dependent manner. In lesions of patients with psoriasis,
keratinocytes are characterized by hyperproliferation and aberrant
terminal differentiation and result in the formation of plaque. It
is essential for keratinocytes to intrinsic alterations in the
response to T cell-derived signals in psoriasis (45,46).
It was hypothesized that the latter would also affect the
expression of CEBPD. Thus, lower CEBPD expression induced by IL-17
could lead to excessive proliferation of the HaCaT cells.
CEBPD is a downstream target of p38 (43). A number of studies have suggested
that CEBPD transcriptional activation responds to the activation of
either STAT3 or p38/CREB (cAMP responsive element binding protein)
(33,47). Shan et al (48) demonstrated that shikonin could
inhibit cell proliferation and induce apoptosis by modulating
phosphorylated (p)-p38/mitogen-activated protein kinase (MAPK),
p-JNK and c-Myc. In addition, ERK, JNK and p38 play important roles
in shikonin-induced apoptosis (21,48,49).
The JAK/STAT3 signaling pathway is involved in psoriasis
progression and is also targeted by shikonin to reduce tumor growth
and metastasis (22,50). Our previous study demonstrated that
shikonin suppressed IL-17-induced, psoriasis-associated cytokines
by inhibiting the JAK/STAT3 signaling pathway (23). Several studies suggest that
shikonin and its derivatives are effective inhibitors of STAT3,
which could be the possible mechanistic basis of the upregulation
of CEBPD (21–25). IMQ can induce psoriatic-like
plaques in mice by triggering the IL-23/IL-17 axis (46). In the present study, consistent
with the in vitro findings, CEBPD was downregulated in
IMQ-treated mice and restored by shikonin. Shikonin also alleviated
keratinocyte hyperproliferation, inflammatory infiltration and
other tissue damage. Thus, shikonin inhibited keratinocyte
proliferation and prevented the development of IMQ-induced
psoriatic lesions.
K17 is a widely used marker of psoriasis
pathogenesis. It is rarely expressed in normal cells but is highly
expressed in psoriatic lesions and is upregulated by IFN-γ, IL-22
and IL-17 (11–13). Yang et al (13) confirmed that under a psoriatic
microenvironment with proinflammatory cytokines such as IFN-γ, the
mRNA and protein expression levels of K17 further potentiate the
interaction between K17 and STAT3, which subsequently promotes
STAT3 phosphorylation, nuclear transport and downstream gene
expression levels of cyclin D1. In addition, K17 promotes the
proliferation of psoriatic T cells and production of cytokines such
as IFN-γ (51). Our previous study
suggested that shikonin downregulated K17 by interfering with STAT3
signaling (24). Similarly, in the
present study, shikonin decreased K17 levels in both
IL-17-stimulated HaCaT cells and IMQ-induced psoriasis lesions. In
the absence of CEBPD, however, the inhibitory effect of shikonin
was impaired, these findings may constitute a potential therapeutic
target for psoriasis.
Psoriasis is a complex disease involving
keratinocytes, endothelial cells and immune cells, such as
macrophages and T lymphocytes (1,6,10,13).
Therefore, the effect of shikonin on immune cell infiltration and
psoriasis-related cytokine expression requires further
investigation. Furthermore, the mechanism of CEBPD regulation in
the pathogenesis of psoriasis and the therapeutic effects of
shikonin remain to be determined. In summary, shikonin can protect
against psoriatic progression and CEBPD might represent a promising
therapeutic target in this context.
Acknowledgements
Not applicable.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant nos. 81673055 and
81402595).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
XOL, YYX and YJY performed the experiments. HXW, YY
and HG analyzed and interpreted the data. XOL drafted the
manuscript. RQQ was involved in statistical analysis and data
interpretation. XHG and LG were involved in study conceptualization
and obtained funding. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal Care
Committee at China Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Di Cesare A, Di Meglio P and Nestle FO:
The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J
Invest Dermatol. 129:1339–1350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ramirez-Carrozzi V, Sambandam A, Luis E,
Lin Z, Jeet S, Lesch J, Hackney J, Kim J, Zhou M, Lai J, et al:
IL-17C regulates the innate immune function of epithelial cells in
an autocrine manner. Nat Immunol. 12:1159–1166. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Clark RA: Skin-resident T cells: The ups
and downs of on site immunity. J Invest Dermatol. 130:362–370.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li Y, Yu C, Zhu WM, Xie Y, Qi X, Li N and
Li JS: Triptolide ameliorates IL-10-deficient mice colitis by
mechanisms involving suppression of IL-6/STAT3 signaling pathway
and down-regulation of IL-17. Mol Immunol. 47:2467–2474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Simanski M, Rademacher F, Schröder L,
Schumacher HM, Gläser R and Harder J: IL-17A and IFN-γ
synergistically induce RNase 7 expression via STAT3 in primary
keratinocytes. PLoS One. 8:e595312013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang L, Li B, Dang E, Jin L, Fan X and
Wang G: Impaired function of regulatory T cells in patients with
psoriasis is mediated by phosphorylation of STAT3. J Dermatol Sci.
81:85–92. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang XO, Panopoulos AD, Nurieva R, Chang
SH, Wang D, Watowich SS and Dong C: STAT3 regulates
cytokine-mediated generation of inflammatory helper T cells. J Biol
Chem. 282:9358–9363. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou L, Ivanov II, Spolski R, Min R,
Shenderov K, Egawa T, Levy DE, Leonard WJ and Littman DR: IL-6
programs T(H)-17 cell differentiation by promoting sequential
engagement of the IL-21 and IL-23 pathways. Nat Immunol. 8:967–974.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu L, Kitani A, Stuelten C, McGrady G,
Fuss I and Strober W: Positive and negative transcriptional
regulation of the Foxp3 gene is mediated by access and binding of
the Smad3 protein to enhancer I. Immunity. 33:313–325. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miyoshi K, Takaishi M, Nakajima K, Ikeda
M, Kanda T, Tarutani M, Iiyama T, Asao N, DiGiovanni J and Sano S:
Stat3 as a therapeutic target for the treatment of psoriasis: A
clinical feasibility study with STA-21, a Stat3 inhibitor. J Invest
Dermatol. 131:108–117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fu M and Wang G: Keratin 17 as a
therapeutic target for the treatment of psoriasis. J Dermatol Sci.
67:161–165. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin L and Wang G: Keratin 17: A critical
player in the pathogenesis of psoriasis. Med Res Rev. 34:438–454.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang L, Jin L, Ke Y, Fan X, Zhang T, Zhang
C, Bian H and Wang G: E3 Ligase Trim21 ubiquitylates and stabilizes
Keratin 17 to induce STAT3 activation in psoriasis. J Invest
Dermatol. 138:2568–2577. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
William DJ, Timothy GB and Dirk ME:
Andrews' diseases of the skin clinical dermatology. China Science
Publishing. (Beijing). 195–198. 2015.
|
|
15
|
Lan W, Wan S, Gu W, Wang H and Zhou S:
Mechanisms behind the inhibition of lung adenocarcinoma cell by
shikonin. Cell Biochem Biophys. 70:1459–1467. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gong K, Zhang Z, Chen Y, Shu HB and Li W:
Extracellular signal-regulated kinase, receptor interacting
protein, and reactive oxygen species regulate shikonin-induced
autophagy in human hepatocellular carcinoma. Eur J Pharmacol.
738:142–152. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gaddipati JP, Mani H, Shefal i, Raj K,
Mathad VT, Bhaduri AP and Maheshwari RK: Inhibition of growth and
regulation of IGFs and VEGF in human prostate cancer cell lines by
shikonin analogue 93/637 (SA). Anticancer Res. 20:2547–2552.
2000.PubMed/NCBI
|
|
18
|
Jang SY, Lee JK, Jang EH, Jeong SY and Kim
JH: Shikonin blocks migration and invasion of human breast cancer
cells through inhibition of matrix metalloproteinase-9 activation.
Oncol Rep. 31:2827–2833. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lu L, Qin A, Huang H, Zhou P, Zhang C, Liu
N, Li S, Wen G, Zhang C, Dong W, et al: Shikonin extracted from
medicinal Chinese herbs exerts anti-inflammatory effect via
proteasome inhibition. Eur J Pharmacol. 658:242–247. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen X, Yang L, Oppenheim JJ and Howard
MZ: Cellular pharmacology studies of shikonin derivatives.
Phytother Res. 16:199–209. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li W, Zhang C, Ren A, Li T, Jin R, Li G,
Gu X, Shi R and Zhao Y: Shikonin suppresses skin carcinogenesis via
inhibiting cell proliferation. PLoS One. 10:e01264592015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qiu HY, Fu JY, Yang MK, Han HW, Wang PF,
Zhang YH, Lin HY, Tang CY, Qi JL, Yang RW, et al: Identification of
new shikonin derivatives as STAT3 inhibitors. Biochem Pharmacol.
146:74–86. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu Y, Xu X, Gao X, Chen H and Geng L:
Shikonin suppresses IL-17-induced VEGF expression via blockage of
JAK2/STAT3 pathway. Int Immunopharmacol. 19:327–333. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu L, Wu Y, Cao K, Xu YY, Gao XH, Chen HD
and Geng L: Shikonin inhibits IFN-γ-induced K17 over-expression of
HaCaT cells by interfering with STAT3 signaling. Int J Clin Exp
Pathol. 8:9202–9207. 2015.PubMed/NCBI
|
|
25
|
Yu YJ, Xu YY, Lan XO, Liu XY, Zhang XL,
Gao XH and Geng L: Shikonin induces apoptosis and suppresses growth
in keratinocytes via CEBP-δ upregulation. Int Immunopharmacol.
72:511–521. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ikezoe T, Gery S, Yin D, O'Kelly J,
Binderup L, Lemp N, Taguchi H and Koeffler HP:
CCAAT/enhancer-binding protein delta: A molecular target of
1,25-dihydroxyvitamin D3 in androgen-responsive prostate cancer
LNCaP cells. Cancer Res. 65:4762–4768. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Alonzi T, Maritano D, Gorgoni B, Rizzuto
G, Libert C and Poli V: Essential role of STAT3 in the control of
the acute-phase response as revealed by inducible gene inactivation
[correction of activation] in the liver. Mol Cell Biol.
21:1621–1632. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang JM, Ko CY, Chen LC, Wang WL and Chang
WC: Functional role of NF-IL6β and its sumoylation and acetylation
modifications in promoter activation of cyclooxygenase 2 gene.
Nucleic Acids Res. 34:217–231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ko CY, Chang LH, Lee YC, Sterneck E, Cheng
CP, Chen SH, Huang AM, Tseng JT and Wang JM: CCAAT/enhancer binding
protein delta (CEBPD) elevating PTX3 expression inhibits
macrophage-mediated phagocytosis of dying neuron cells. Neurobiol
Aging. 33:422.e11–422.e25. 2012. View Article : Google Scholar
|
|
30
|
Tang D, Sivko GS and DeWille JW: Promoter
methylation reduces C/EBPdelta (CEBPD) gene expression in the
SUM-52PE human breast cancer cell line and in primary breast
tumors. Breast Cancer Res Treat. 95:161–170. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Agrawal S, Hofmann WK, Tidow N, Ehrich M,
van den Boom D, Koschmieder S, Berdel WE, Serve H and Müller-Tidow
C: The C/EBPδ tumor suppressor is silenced by hypermethylation in
acute myeloid leukemia. Blood. 109:3895–3905. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li M, Zhou W, Yuan R, Chen L, Liu T, Huang
D, Hao L, Xie Y and Shao J: ROCK2 promotes HCC proliferation by
CEBPD inhibition through phospho-GSK3β/β-catenin signaling. FEBS
Lett. 589:5892015. View Article : Google Scholar
|
|
33
|
Wang WJ, Li CF, Chu YY, Wang YH, Hour TC,
Yen CJ, Chang WC and Wang JM: Inhibition of the EGFR/STAT3/CEBPD
axis reverses cisplatin cross-resistance with paclitaxel in the
urothelial carcinoma of the urinary bladder. Clin Cancer Res.
23:503–513. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chuang CH, Wang WJ, Li CF, Ko CY, Chou YH,
Chuu CP, Cheng TL and Wang JM: The combination of the prodrugs
perforin-CEBPD and perforin-granzyme B efficiently enhances the
activation of caspase signaling and kills prostate cancer. Cell
Death Dis. 5:e12202014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sønder SU, Paun A, Ha HL, Johnson PF and
Siebenlist U: CIKS/Act1-mediated signaling by IL-17 cytokines in
context: Implications for how a CIKS gene variant may predispose to
psoriasis. J Immunol. 188:5906–5914. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yerlikaya A, Erdoğan E, Okur E, Yerlikaya
Ş and Savran B: A novel combination treatment for breast cancer
cells involving BAPTA-AM and proteasome inhibitor bortezomib. Oncol
Lett. 12:323–330. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Türker Şener L, Albeniz G, Dinç B and
Albeniz I: iCELLigence real-time cell analysis system for examining
the cytotoxicity of drugs to cancer cell lines. Exp Ther Med.
14:1866–1870. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin YK, Yang SH, Chen CC, Kao HC and Fang
JY: Using imiquimod-induced psoriasis-like skin as a model to
measure the skin penetration of anti-psoriatic drugs. PLoS One.
10:e01378902015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Van Der Fits L, Mourits S, Voerman JS,
Kant M, Boon L, Laman JD, Cornelissen F, Mus AM, Florencia E, Prens
EP and Lubberts E: Imiquimod-induced psoriasis-like skin
inflammation in mice is mediated via the IL-23/IL-17 axis. J.
Immunol. 182:5836–5845. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Morris RJ, Readio N, Boland K, Johnson K,
Lad S, Singh A, Singh A, Holtorf S and Skaar S: Isolation of mouse
epidermal keratinocytes and their in vitro clonogenic culture. J
Vis Exp. 10:e587012019.
|
|
42
|
Hu Y, Shen F, Crellin NK and Ouyang W: The
IL-17 pathway as a major therapeutic target in autoimmune diseases.
Ann N Y Acad Sci. 1217:60–76. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang JM, Tseng JT and Chang WC: Induction
of human NF-IL6β by epidermal growth factor is mediated through the
p38 signaling pathway and cAMP response element-binding protein
activation in A431 cells. Mol Biol Cell. 16:3365–3376. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
O'Rourke J and Yuan R, DeWille J, O'
Rourke JP and Yuan R: CCAAT/enhancer-binding protein-delta
(C/EBP-δ) is induced in growth-arrested mouse mammary epithelial
cells. J Biol Chem. 272:6291–6296. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Albanesi C, De Pità O and Girolomoni G:
Resident skin cells in psoriasis: A special look at the
pathogenetic functions of keratinocytes. Clin Dermatol. 25:581–588.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen Y, Yan H, Song Z, Chen F, Wang H, Niu
J, Shi X, Zhang D, Zhang N, Zhai Z, et al: Downregulation of TNIP1
expression leads to increased proliferation of human keratinocytes
and severer psoriasis-like conditions in an imiquimod-induced mouse
model of dermatitis. PLoS One. 10:e01279572015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pan YC, Li CF, Ko CY, Pan MH, Chen PJ,
Tseng JT, Wu WC, Chang WC, Huang AM, Sterneck E, et al: CEBPD
reverses RB/E2F1-mediated gene repression and participates in
HMDB-induced apoptosis of cancer cells. Clin Cancer Res.
16:5770–5780. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shan ZL, Zhong L, Xiao CL, Gan LG, Xu T,
Song H, Yang R, Li L and Liu BZ: Shikonin suppresses proliferation
and induces apoptosis in human leukemia NB4 cells through
modulation of MAPKs and c Myc. Mol Med Rep. 16:3055–3060. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Huang WR, Zhang Y and Tang X: Shikonin
inhibits the proliferation of human lens epithelial cells by
inducing apoptosis through ROS and caspase-dependent pathway.
Molecules. 19:7785–7797. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Qiu HY, Zhu X, Luo YL, Lin HY, Tang CY, Qi
JL, Pang YJ, Yang RW, Lu GH, Wang XM, et al: Identification of new
shikonin derivatives as antitumor agents targeting STAT3 SH2
domain. Sci Rep. 7:28632017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yang L, Fan X, Cui T, Dang E and Wang G:
Nrf2 promotes keratinocyte proliferation in psoriasis through
up-regulation of Keratin 6, Keratin 16, and Keratin 17. J Invest
Dermatol. 137:2168–2176. 2017. View Article : Google Scholar : PubMed/NCBI
|