Introduction
Acute lung injury (ALI) is a common critical illness
in clinical anesthesia and the intensive care unit (ICU) (1–3). ALI
refers to the injury of pulmonary capillary endothelial cells and
alveolar epithelial cells during non-cardiac diseases, such as
severe infection, shock, trauma and burns, and can cause diffuse
pulmonary interstitial and alveolar edema, which can lead to acute
hypoxic respiratory insufficiency or failure (4). Moreover, the development of ALI to
the severe stages can result in acute respiratory distress syndrome
(ARDS) (5,6); ALI/ARDS seriously threaten the lives
of patients, affecting quality of life and increasing the economic
burden. Compared with older children and adults, infants have
smaller airways, increased incomplete alveolar vascular bed
development, improved chest wall elasticity and lower functional
residual capacity, and thus are a high-risk group of ALI/ARDS
(7). Children have high chest wall
compliance, low rib support to the lungs, and difficulty in
maintaining negative intrathoracic pressure. As a result, the
residual volume of lung function is reduced, which is a
disadvantageous factor in acute lung injury. Furthermore, the
overall mortality rate of ALI in pediatrics is 18–27%, and the
mortality rate of ARDS is 29–50% (8,9). Any
response of the lung to the injurious stimulus may be mediated by
several pathways, depending on the cause of injury. For instance,
bacterial antigens trigger the inflammatory response by activating
Toll-like receptors (TLR) (10),
and chemical injury may induce damage to cell membrane and
oxidative stress, leading to the activation of intracellular
kinases (11,12). Although the development of
intensive care medicine has made progress worldwide, the
understanding and treatment status of ALI/ARDS remains
unsatisfactory. Therefore, it is of great clinical significance to
elucidate the pathogenesis of ALI/ARDS and develop targeted drugs
to effectively prevent or treat the disease. As an α2-adrenergic
receptor agonist, dexmedetomidine (Dex) has an analgesic effect and
is widely used for sedation and anesthesia in patients with ICU
ventilator support; however, there are few studies related to the
progression of Dex and ALI. Our previous clinical study reported
that Dex can reduce the occurrence of delirium during postoperative
recovery by reducing the level of inflammatory response (13); however, the detailed underlying
signaling mechanisms remain to be elucidated.
The NOD-like receptor protein 3 (NLRP3) signaling
pathway plays an important role in the inflammatory response and is
involved in the pathogenesis of ALI/ARDS (14–17).
Moreover, NLRP3 is composed of NLRs and caspase-1 and is widely
present in T cells, B cells, monocytes, macrophages, dendritic
cells and granulocytes (18,19).
In the absence of an activating substance, the leucine-rich repeat
domain of NLRP3 binds to the NACHT domain, inhibiting
self-oligomerization and destabilizing the inactive state (20,21).
There are numerous substances that activate NLRP3, including
lipopolysaccharide (LPS) bacterial toxin, extracellular ATP or
necrotic cellular components (22). Furthermore, the activated NLRP3
inflammasome provides a platform for cytokines, including
interleukin (IL)-1, IL-6, IL-18 and IL-33, which are subsequently
converted into activated IL-1β, IL-6, IL-18 and IL-33 states
(23,24).
Heme oxygenase 1 (HO-1) plays a key role in
regulating organ protection from ischemia-reperfusion injury
(25). In addition, the nuclear
factor erythroid 2-related factor 2 (Nrf2)/HO-1 signaling pathway
plays an important role in preventing the occurrence of ALI/ARDS,
and activation of Nrf2 reduces the severity of ALI/ARDS (26). The activation of the Nrf2/HO-1
signaling pathway also inhibits the activation of NLRP3
inflammasome, thus reducing IL-1β expression and exerting
anti-inflammatory and cytoprotective effects (27,28).
p38 kinase, which is a member of the mitogen-activated protein
kinase (MAPK) family, plays a central role in inflammatory
responses in a variety of disease models, such as Parkinson's
disease, cancer and inflammatory disease (29–31),
and has been the subject of basic research and drug discovery
(32). Furthermore, inhibition of
p38 activation has been revealed to suppress LPS-induced
inflammation in different situations, such as in LPS-induced
inflammation in IPEC-J2 cells, bronchial epithelial cells,
macrophages and rats (29,33–35).
In the present study, it was hypothesized that Dex
may activate the HO-1 signaling pathway by suppressing p38 to
mediate the anti-inflammatory effect in LPS induced ALI. Therefore,
the activity of NLRP3 may be inhibited by Dex, which could reduce
IL-1β secretion and the level of inflammatory response in
Sprague-Dawley rats with ALI. Therefore, the present study may
provide a theoretical basis and novel evidence for the discovery of
new targets for clinical treatment of ALI.
Materials and methods
Animal model of ALI and experimental
groups
Newborn male specific pathogen-free Sprague-Dawley
rats (age, 7 days; weight, 250–300 g) were provided by Experimental
Animal Center of Anhui Medical University (no. SCXK-2017-023). Rats
were housed in standard laboratory cages under standard housing
conditions of 12 h light/12 h dark cycles at a temperature of
22±2°C, along with free access to food and water, and all animal
experiments were performed in accordance with the international
standards on animal welfare, as well as being compliant with the
committee of Anhui Medical University.
The rats (n=48) were randomly divided into four
groups (n=12 rats each) (36,37):
i) Saline control group; ii) LPS (cat. no. L2630-100MG; Sigma
Aldrich; Merck KGaA) group; iii) LPS and Dex (cat. no.
MB1434-S-100MG; Dalian Meilunbio Biology Technology Co., Ltd.)
group; and iv) Dex and SB203580 [cat. no. 5633S; Cell Signaling
Technology, Inc. (CST)] group. The rats were anesthetized with an
intraperitoneal injection of 40 mg/kg pentobarbital sodium (cat.
no. 1063180500; Merck KGaA). The rats were tracheal intubated with
a micro-atomizer and the control group was given 300 µl
physiological saline, the experimental group was administered LPS
(5 mg/kg) in 300 µl physiological saline, which was fully
administered via a nebulizer at an oxygen flow-rate of 4 L/min for
25 min. The rats were normally fed without intubation for 30 min
after each 6 h intubation. Then, 12 h later, rats were sacrificed
by decapitation, bronchoalveolar lavage and lung tissue were
assessed, after which they underwent thoracotomy. The right
bronchus was ligated, and the right lung tissue specimen was
isolated for preparation for subsequent experiments.
Morphological analysis of the
lungs
The fresh lung samples were weighed for wet mass
after harvesting or dried overnight at 75°C for dry mass
measurement (38). Haematoxylin
and eosin (H&E) staining was used to observe histological
pulmonary structures and the condition of inflammation following
exposure to LPS. After harvesting at the assigned time points, the
lungs were imaged and fixed in 4% paraformaldehyde at room
temperature for 12 h, dehydrated, embedded in paraffin wax and
serially sectioned at 5-µm. The sections were stained with
haematoxylin for 20 min and eosin for 15 min at room temperature.
The sections were imaged using a fluorescence microscope (Olympus
IX50; Olympus Corporation) linked to the NIS-Elements F3.2 software
(Nikon Corporation). The airspace volume density was measured by
dividing the sum of the airspace area by the total area (39). In total, ≥3 randomly selected
images from five samples were assessed per group at the assigned
time point.
Immunohistochemistry
Immunostaining was performed on paraffin transverse
sections against IL-1β and NLRP3 (40). Transverse sections of rat lungs
were fixed by carnoy solution (1:500) at room temperature for 3 h,
de-waxed in xylene, rehydrated (100, 90 and 70% ethanol), heated in
a microwave at 92–98°C for 15 min for antigen retrieval before
exposure to the primary antibody with citrate buffer (pH=6.0) and
serially sectioned at 5 µm. Next, the sections were immersed in 3%
hydrogen peroxide for 10 min to block endogenous peroxidase.
Non-specific immunoreactions were blocked using 5% inactivated goat
serum (Gibco) in PBS for 30 min at room temperature. The sections
were washed in PBS and incubated with NLRP3 (1:250; cat. no.
ab263899; Abcam) and IL-1β (1:200; cat. no. ab9722; Abcam)
antibodies overnight with shaking at 4°C. For immunohistochemistry,
following extensive washing, the sections were incubated in
horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG
secondary antibody (1:400; cat. no. E030120; EarthOx Life Sciences)
for 2 h at room temperature in a dark box, and were then
subsequently stained with 3′3-diaminobenzidine at room temperature
for 5 min (Fuzhou Maixin Biotech Co., Ltd.). After immunostaining,
the sections were counterstained with haematoxylin at room
temperature for 10 min and observed using an IX50 confocal
microscope (Olympus Corporation; magnification, ×200).
TUNEL analysis
TUNEL staining was performed using an In Situ
Cell Death Detection kit (Roche Diagnostics) according to the
manufacturer's instructions. Sections were fixed with 4%
paraformaldehyde for 20 min at 20°C. Sections (thickness, 4 µm)
were deparaffinized in xylene by heating at 60°C, rehydrated in
decreasing concentrations of ethanol (100, 95, 90, 80 and 70%) and
heated for antigen retrieval at 37°C. Endogenous peroxidase was
blocked in 3% hydrogen peroxide. Then, three different dilutions
(1:7, 1:11 and 1:16) of terminal deoxynucleotidyl transferase in
reaction buffer (containing a fixed concentration of
digoxigenin-labelled nucleotides) were applied to serial sections
at 37°C for 1 h before the slides were placed in stop/wash buffer
for 10 min. Following intensive washing, a pre-diluted
anti-digoxigenin peroxidase-conjugated antibody (1:2; cat. no.
11207733910; Roche Diagnostics) was applied for 30 min at room
temperature. For immunofluorescent staining, the sections were
incubated with the corresponding Alexa Fluor 488 secondary antibody
(1:1,000; cat. no. A-11029; Invitrogen; Thermo Fisher Scientific,
Inc.) at room temperature for 2 h in a dark box. All sections were
then counterstained with DAPI (1:1,000; Invitrogen; Thermo Fisher
Scientific, Inc.) at room temperature for 30 min. Sections were
mounted with Prolong Gold Antifade mounting media containing DAPI
(Invitrogen; Thermo Fisher Scientific, Inc.). Stained cells were
observed in five randomly selected fields of view. The presence of
TUNEL+ cells was determined using Image Analysis
Software V.2.4.2 (Olympus Corporation). The percentage of
TUNEL+ cells relative to the total cells in the same
area between the control and experimental groups was evaluated (n=8
lungs for each group).
Western blotting
Western blotting was performed in accordance with a
standard procedure using polyclonal antibodies that specifically
recognized phosphorylated (p)-p38, p38, HO-1 and NLRP3. The methods
for protein extraction and immunoblotting used in this study have
been described previously (40).
Protein samples were extracted from lung tissue homogenate using a
RIPA buffer (Sigma-Aldrich; Merck KGaA) supplemented with protease
and phosphatase inhibitors, and the protein concentrations were
quantified using the bicinchoninic acid assay. The extracted
protein samples were separated by SDS-PAGE on a 10% gel, and
subsequently transferred onto a PVDF membrane (EMD Millipore). The
membrane was blocked with 5% non-fat milk at room temperature for 1
h and incubated with antibodies against p38 (1:500; cat. no. 8690;
CST), p-p38 (1:500; cat. no. ab4822; Abcam), HO-1 (1:500; cat. no.
82206; CST) and NLRP3 (1:500; cat. no. ab214185; Abcam) in TBS
buffer at 4°C overnight. GAPDH was used as a loading control
(1:1,000; cat. no. 5174; CST). After incubation with the secondary
antibodies at room temperature for 1 h, which were either
HRP-conjugated goat anti-rabbit IgG (1:3,000; cat. no. E030120;
EarthOx Life Sciences) or HRP-conjugated goat anti-mouse IgG
(1:3,000; cat. no. E030110; EarthOx Life Sciences), the blots were
developed with the SuperSignal™ West Femto Chemiluminescent
Substrate (Thermo Fisher Scientific, Inc.) and Gel Doc™ XR+ System
(Bio-Rad Laboratories, Inc.). The intensity of the bands was
analyzed using the Quantity One software V.4.6.7 (Bio-Rad
Laboratories, Inc.), according to the manufacturer's instructions.
The western blotting results are representative of three
independent experiments.
ELISA
Briefly, the left lung was lavaged three times with
500 µl saline via tracheal catheter to obtain bronchoalveolar
lavage (BAL) fluid. BAL fluid was centrifuged at 4°C at 600 × g for
10 min. The supernatant was stored at −20°C until further
examination. The level of secreted IL-1β in BAL fluid was detected
using a Rat IL-1β ELISA kit (cat. no. ab100704; Abcam), according
to the manufacturer's instructions.
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from fresh rat lung tissue or
MLE-12 cells (American Type Culture Collection) stored on ice using
the E.Z.N.A® Total RNA kit (Omega Bio-Tek, Inc.),
according to the manufacturer's instructions. Total RNA was reverse
transcribed into cDNA at 42°C for 15 min and 85°C for 5 sec using
the PrimeScript™ RT Reagent kit (Takara Bio, Inc.). Subsequently,
qPCR was performed using SYBR® Green qPCR assay (Thermo
Fisher Scientific, Inc.). All the specific primers used are
described in Table I (41–43).
qPCR was performed in the Bio-Rad S1000TM thermocycler (Bio-Rad
Laboratories, Inc.), with the following qPCR thermocycling
conditions: Initial denaturation at 95°C for a 3 min, followed by
40 PCR cycles (95°C for 5 sec, 60°C for 20 sec and 72°C for 20
sec), using ABI 7000 RT PCR machines. Corresponding relative mRNA
expression was calculated by the 2−ΔΔCq method (44) and normalized to β-actin. The qPCR
results are representative of three independent experiments.
 | Table I.Primers used for reverse
transcription-quantitative PCR. |
Table I.
Primers used for reverse
transcription-quantitative PCR.
| Gene | Primer sequence
(5′-3′) |
|---|
| HO-1 | F:
CGTGCAGAGAATTCTGAGTTC |
|
| R:
AGACGCTTTACGTAGTGCTG |
| p38 | F:
AGGGCGATGTGACGTTT |
|
| R:
CTGGCAGGGTGAAGTTGG |
| NLRP3 | F:
GGGACTCAAGCTCCTCTGTG |
|
| R:
GAGGCTCTGGTTATGGGTCA |
| GAPDH | F:
AGCCACATCGCTCAGACA |
|
| R:
TGGACTCCACGACGTACT |
Data analysis
Data analysis and the construction of statistical
graphs were performed using GraphPad Prism 5 software package
(GraphPad Software, Inc.). Data are presented as the mean ± SEM
from ≥3 independent experiments. ANOVA (followed by Tukey's post
hoc test) or an unpaired Student's t-test were used to analyze
whether there were any significant differences between the control
and treatment groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
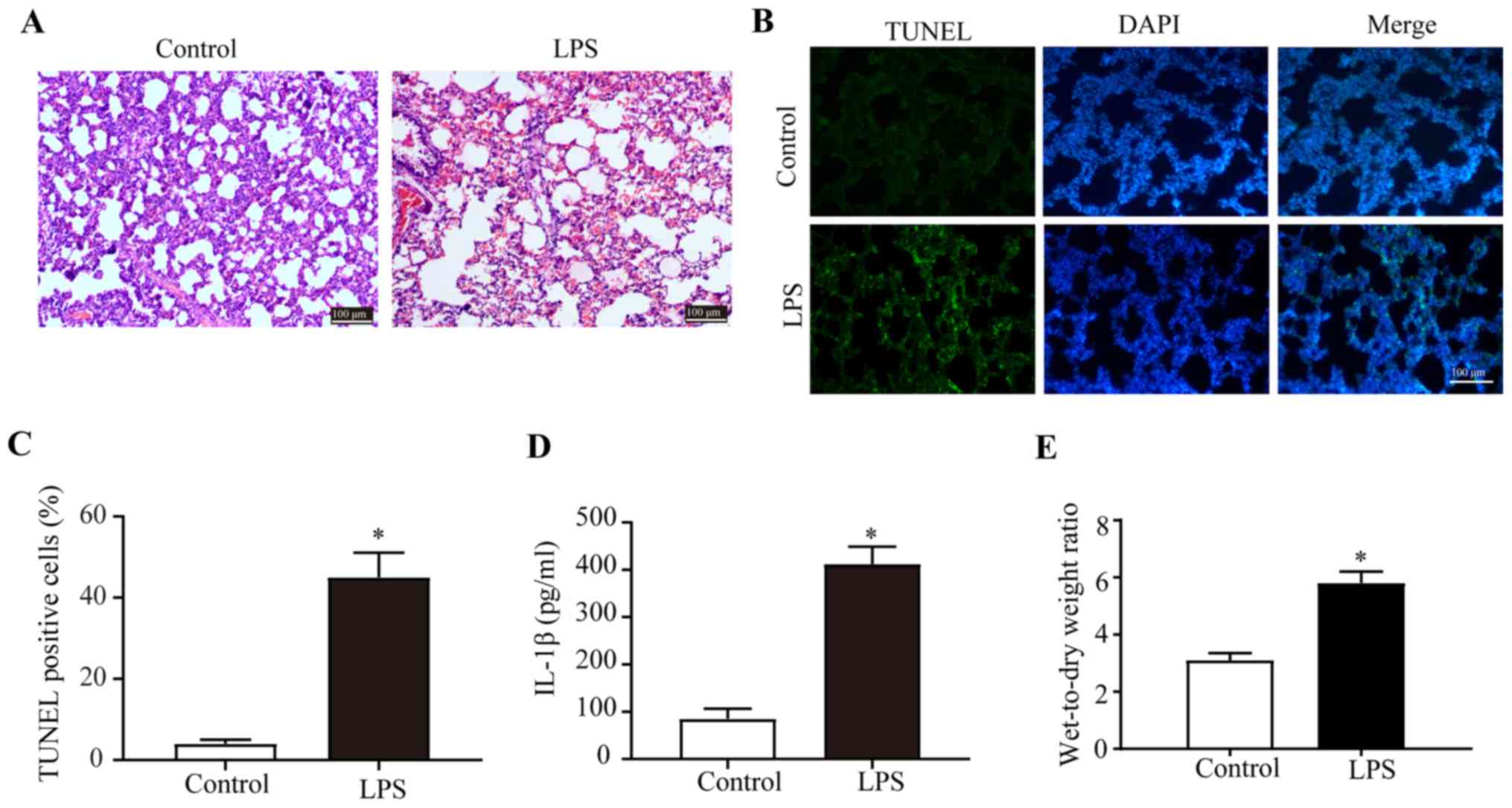
LPS exposure induces ALI
The histological characteristics of the H&E
stained transverse sections of the lungs were compared between the
control and LPS groups (Fig. 1A).
The integrity of the structure of alveoli before LPS exposure was
identified by examination of pathological tissue sections. After
LPS exposure, diffuse damage was observed in the alveoli, alveolar
sacs, alveolar tubes, alveolar septa and bronchi. Moreover, there
were numerous inflammatory cells in the alveolar septa of the LPS
group (Fig. 1A). Furthermore, LPS
treatment significantly increased the number of TUNEL+
pulmonary cells in lungs of the LPS group compared with the control
(Fig. 1B and C), indicating that
LPS promoted apoptosis in the pulmonary cells. IL-1β plays an
important role in the body's inflammatory response and is involved
in the pathogenesis of ALI. With collected rat bronchoalveolar
lavage, using ELISA kits, it was demonstrated that the secretion
levels of IL-1β in the bronchoalveolar lavage fluid were
significantly upregulated upon LPS exposure (Fig. 1D).
In addition, the quantity of the wet lung and dry
lung in the control group and LPS group was weighed. It was
indicated that the wet-to-dry weight ratio of the LPS group was
significantly higher compared with the control group, which
suggested that a large amount of edema fluid was accumulated in the
alveolar and interstitial lungs in the LPS group (Fig. 1E). Collectively, the results
demonstrated the successful establishment of the ALI model in rats
and that LPS induced severe injury in the lungs.
p38/HO-1 signaling pathway is involved
in the regulation of NLRP3
p38 plays a central role in the inflammatory
response, and to further assess the possible underlying mechanisms
of the LPS-induced ALI in rat lungs, the expression levels of
inflammatory factors were measured by immunohistochemistry, RT-qPCR
and western blotting. NLRP3 inflammasome is a critical component of
the innate immune system, and is upregulated when the lungs are
injured. The immunohistochemistry (Fig. 2A and B), western blotting (Fig. 2C and D) and RT-qPCR (Fig. 2E) results suggested that the mRNA
and protein expression levels of NLRP3 were significantly
upregulated upon LPS exposure in lungs. Moreover, western blotting
results demonstrated that LPS induced the upregulation of p-p38
expression (Fig. 2C and D).
However, there were no significant differences (P>0.05) in the
expression of p38 between the two groups (Fig. 2C and D).
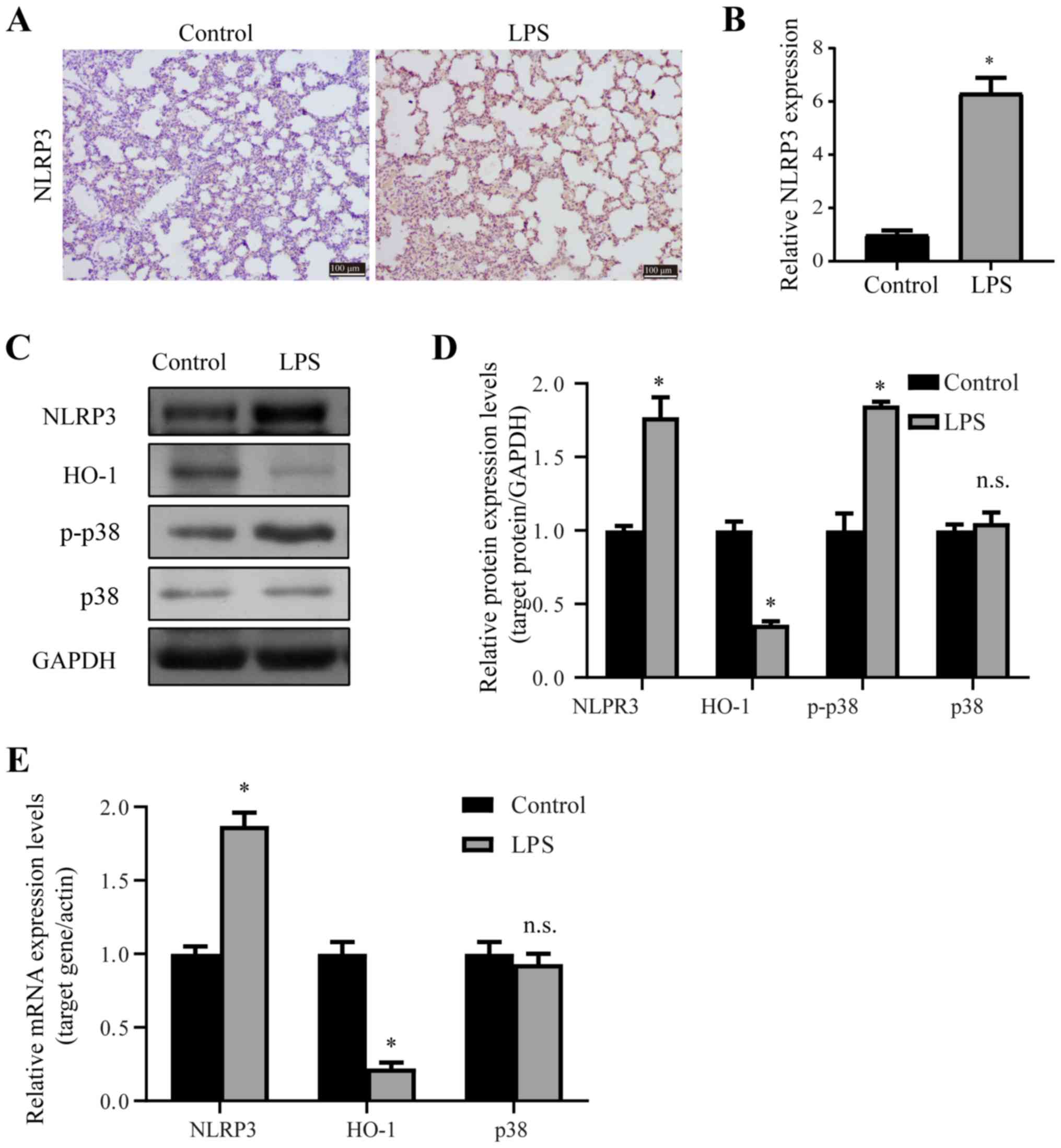 | Figure 2.p38/HO-1 signaling pathway is
involved in the regulation of LPS-induced ALI. (A and B) NLRP3
immunohistochemistry staining was performed on the transverse
sections of rat lungs from the control and LPS-treated groups. (C
and D) Western blotting results of the relative protein expression
levels of NLRP3, HO-1, p-p38 and p38 in rat lungs from the control
and LPS-treated group. (E) Reverse transcription-quantitative PCR
results comparing the relative mRNA expression of NLRP3, HO-1 and
p38 in the lungs of the control or LPS-treated rats. Scale bar, 100
µm. *P<0.05 vs. control. n.s., no significant differences; p-,
phosphorylated; NLRP3, NOD-like receptor protein 3; HO-1, Heme
oxygenase 1; LSP, lipopolysaccharide; ALI, acute lung injury. |
HO-1 is reported to be a downstream effector of p38,
and activation of p38 suppresses the anti-inflammatory effect of
HO-1 (45). Thus, the present
study determined the expression of HO-1 in ALI, and it was found
that both the mRNA and protein expression levels of HO-1 were
significantly downregulated after LPS exposure in lungs (Fig. 2C-E). Thus, LPS exposure induced the
activation of p38, the downregulation of HO-1 and the upregulation
of NLRP3.
Dex and pharmacological inhibition of
p38 co-operate to suppress ALI
Our previous results revealed that Dex can reduce
the level of inflammatory response (13); however, the role of Dex in ALI
remains to be elucidated. Thus, Dex administration was given to
LPS-induced ALI model rats. Moreover, inflammation and
morphological changes were examined by H&E staining, and the
integrity of alveoli structure before LPS exposure was observed by
examination of pathologic tissue sections (Fig. 3A). Furthermore, diffuse damage was
observed in the alveoli, alveolar sacs, alveolar tubes, alveolar
septa and bronchi after LPS exposure, and there was considerable
lymphocyte infiltration in the pulmonary interstitium (Fig. 3A). However, the addition of Dex
reversed LPS-induced inflammation and morphological damage in the
lungs (Fig. 3A). It was
demonstrated that LPS induced p38 activation (Fig. 2), and thus it was further
investigated whether inhibition of p38 attenuated ALI, using
SB203580 (p38 MAPK inhibitor) in the group exposed to LPS and Dex.
Compared with the group exposed to Dex and LPS, the occurrence of
inflammation and morphological damage was further inhibited by
suppressing p38 signaling pathway with SB203580 (Fig. 3A). Furthermore, Dex administration
in the LPS group reversed LPS-induced upregulation of NLRP3
expression in the lungs, and this effect could be further enhanced
by SB203580 (Fig. 3B and C).
Bronchoalveolar lavage was collected and the concentration of
inflammatory factor IL-1β was measured by ELISA, the data showed
the secretion of IL-1β demonstrated the same trend as NLRP3
expression (Fig. 3D). Therefore,
it was speculated that Dex inhibited ALI, which was further
enhanced by p38 suppression.
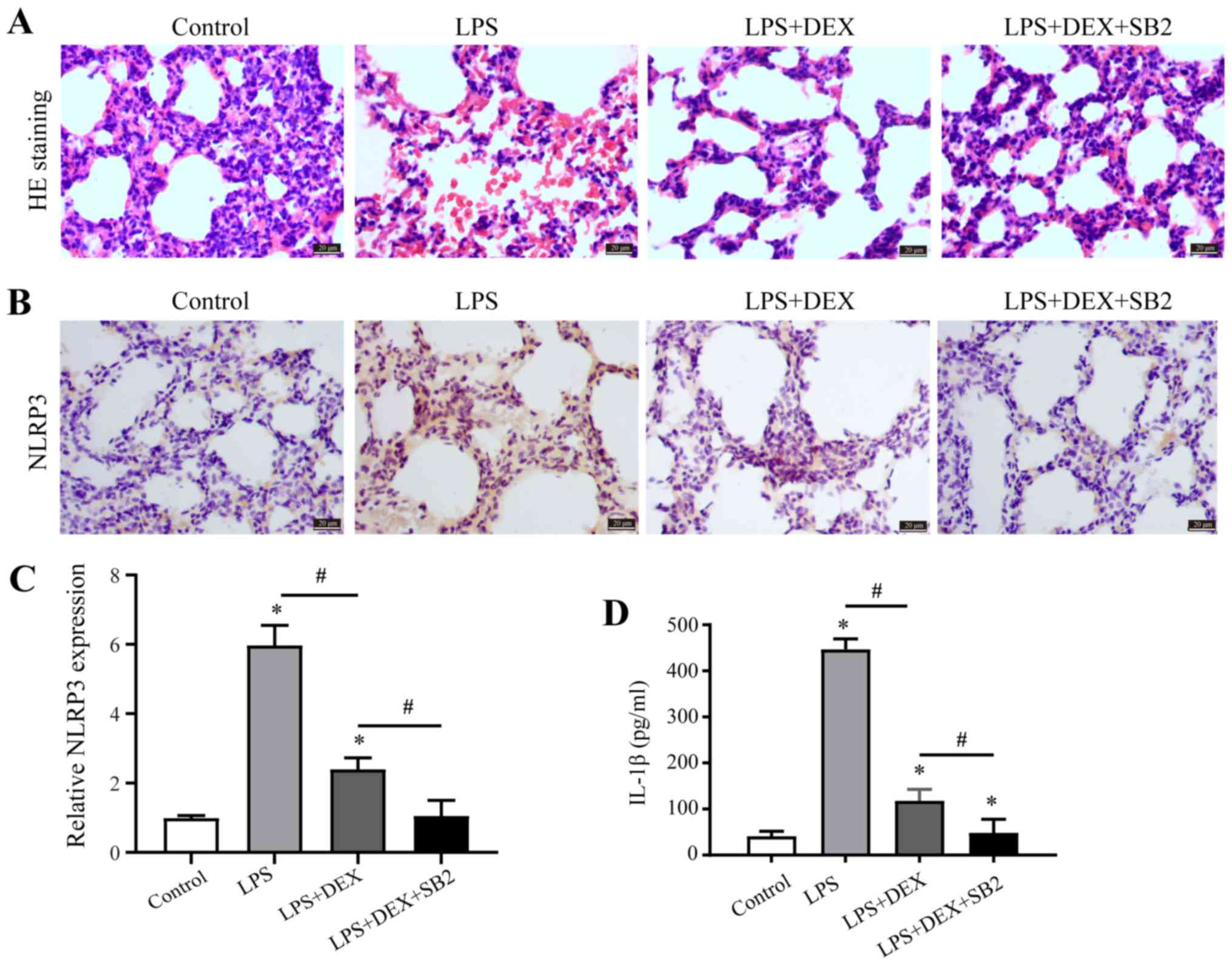 | Figure 3.Dex and p38 inhibition alleviate ALI.
(A) H&E staining was performed on transverse sections of the
rat lungs from the control group, LPS-treated group, LPS +
Dex-treated group, and the LPS + Dex + SB203580-treated group. (B
and C) NLRP3 immunohistochemistry staining was performed on the
transverse sections of rat lungs from the control group,
LPS-treated groups, LPS + Dex-treated group, and the LPS + Dex +
SB203580-treated group. (D) IL-1β immunolabeling intensities
(IL-1β+ cells) in lung tissue from the control group,
LPS-treated groups, LPS + Dex-treated group, and the LPS + Dex +
SB203580-treated group. Scale bar, 50 µm. *P<0.05 vs. control;
#P<0.05 vs. LPS + Dex group. Dex, dexmedetomidine;
IL, interleukin; LSP, lipopolysaccharide; ALI, acute lung injury;
H&E, haematoxylin and eosin. |
Dex alleviates ALI via inhibition of
the p38 signaling pathway
As pharmacological inhibition of p38 enhanced the
effect of Dex in ALI, whether Dex function upstream or parallel to
the p38 pathway was assessed by detecting its mRNA and protein
expression levels by RT-qPCR and western blotting, respectively.
Compared with the control group, the phosphorylation level of p38
was significantly suppressed by Dex, while addition of SB203580
further enhanced this effect. Moreover, the mRNA and protein
expression levels of NLRP3 were suppressed by Dex, and further
reduced by SB203580 treatment (Fig. 4A
and C). The LPS-induced decrease in HO-1 expression was
reversed by Dex, and the addition of SB203580 further increased
HO-1 expression (Fig. 4A-C).
Collectively, the results indicated that Dex alleviated ALI by
suppressing p38 activation and inducing HO-1 expression.
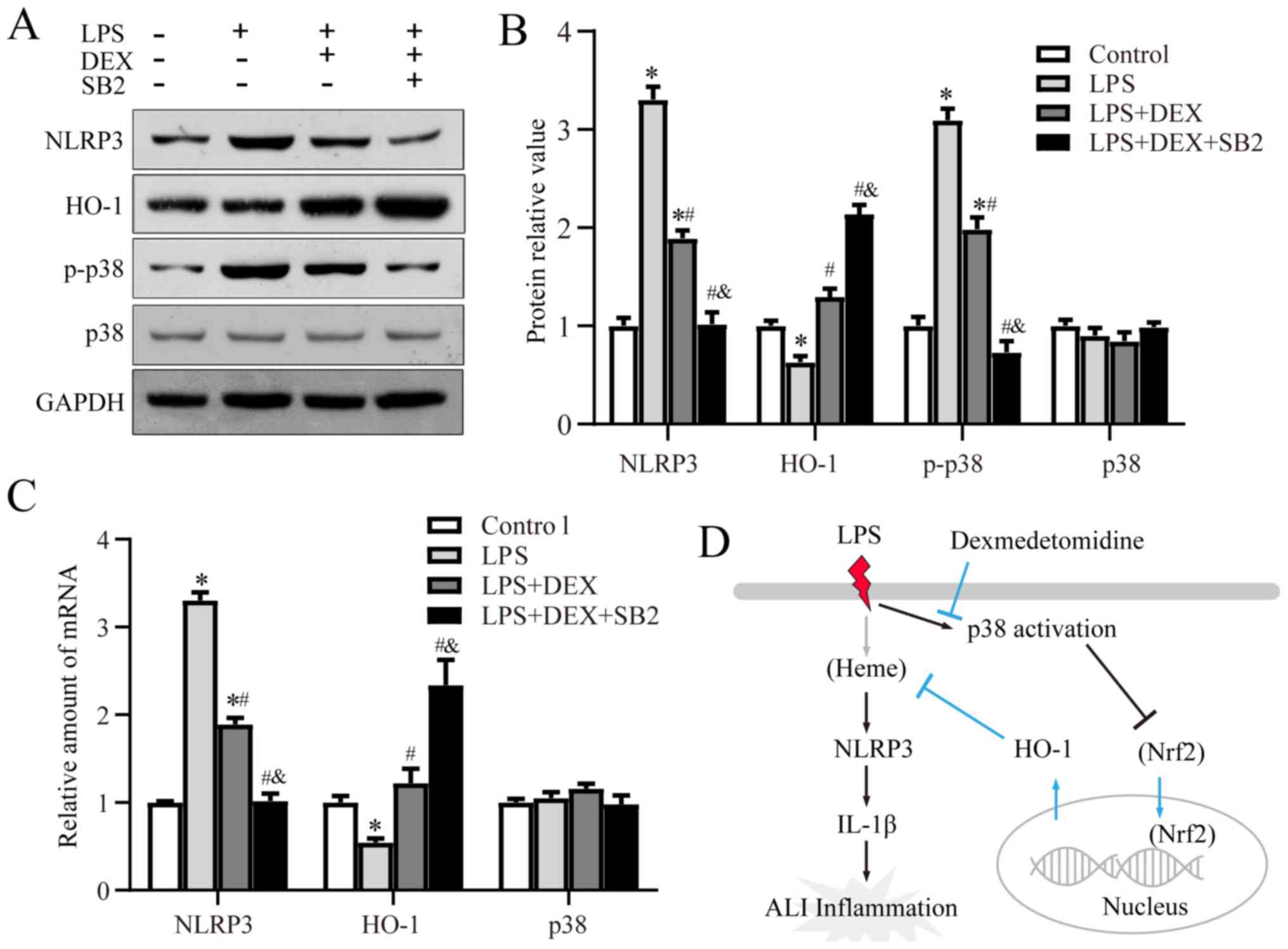 | Figure 4.Dex alleviates ALI via inhibition of
p38. (A and B) Western blotting results of the relative protein
expression levels of NLRP3, HO-1, p-p38 and p38 in rat lungs from
the control, LPS-treated group, LPS + Dex-treated group, and the
LPS + Dex + SB203580-treated group. (C) Reverse
transcription-quantitative PCR results of the relative mRNA
expression levels of NLRP3, HO-1 and p38. (D) Diagram of the
proposed signaling pathway. *P<0.05 vs. control;
#P<0.05 vs. LPS group; &P<0.05 vs.
LPS + Dex group. p-, phosphorylated; NLRP3, NOD-like receptor
protein 3; HO-1, Heme oxygenase 1; LSP, lipopolysaccharide; ALI,
acute lung injury. |
Discussion
ALI is a serious complication of critical illness
and clinical anesthesia, however, the underlying mechanisms remain
to be elucidated. The present results suggested that LPS exposure
could lead to ALI. Furthermore, it was speculated that LPS
activated p38, which subsequently suppressed HO-1 signaling,
promoted the expression of NLRP3 and induction of IL-1β, and
finally resulted in lung damage. Moreover, it was demonstrated that
Dex could prevent ALI by inhibiting p38 activation, rescuing the
expression of HO-1 signaling and decreasing NLRP3; these signal
pathway interactions are summarized in a schematic diagram
(Fig. 4D).
It has been reported that systemic diseases caused
by various pathogenic factors in the lungs and outside the lungs
are the underlying causes of ALI/ARDS in pediatrics. Moreover, the
incidence of ALI/ARDS in sepsis is 25–50%, which remains the main
cause of serious complications and mortality (46–48).
In addition, a large number of clinical blood transfusions,
multiple trauma and aspiration can also cause ALI/ARDS, with an
incidence rate of 40, 11–25 and 9%, respectively (49,50).
Sepsis can induce inflammatory cell activation, which leads to
increased permeability of pulmonary capillary endothelial cells and
alveolar epithelium, and a large amount of edema fluid accumulation
in the alveolar and interstitial lungs (51,52).
Moreover, a variety of inflammatory cells, mainly neutrophils,
adhere to the surface of damaged vascular endothelial cells and
further migrate to the interstitial and alveolar spaces (53). During this process, a large number
of pro-inflammatory mediators, such as IL-1β, IL-6, peroxide,
leukotrienes and proteases, are released, which are involved in
neutrophil-mediated lung injury, alveolar degeneration, necrosis,
apoptosis and alveolar collapse, causing lung dysfunction (54–56).
The Nrf2/HO-1 signaling pathway plays an important
role in preventing the occurrence of ALI/ARDS, and activation of
Nrf2 prevents or reduces the severity of ALI/ARDS (26). Furthermore, activated Nrf2 is
translocated into the nucleus to regulate the expression of
HO-1-related genes. The activated HO-1 gene can inhibit the
inflammatory cascade, thus protecting organ function and reducing
ALI caused by inflammation (57).
Previous studies have also reported that activation of the
Nrf2/HO-1 signaling pathway inhibits the activation of NLRP3
inflammasome, thus reducing IL-1β expression and exerting
anti-inflammatory and cytoprotective effects (58–60).
Furthermore, HO-1 expression attenuates NLRP3 inflammasome
activation (61), and multiple
HO-1 inducers regulate HO-1 gene expression via ≥1 MAPK subtype
signal chains (62). In the MAPK
family, p38 signal transduction pathway can modulate the expression
of HO-1, however, whether it promotes or inhibits the expression of
HO-1 remains controversial. Previous studies have revealed that
there is relationship between p38, Nrf2 and HO-1, which is
characterized by sequential activation (62–64).
Kim et al (65) reported
that glycyrrhizin could activate p38, following which the
expression of HO-1 increased via Nrf2, which could significantly
reduce the secretion of inflammatory factors in sepsis. It has also
been demonstrated that p38 can inhibit Nrf2/HO-1 gene expression.
Furthermore, inhibition or genetic deficiency of p38 upregulates
HO-1 via Nrf2 in monocytic cells (45) and in human hepatoma HepG2 cells
(66,67). The present results also suggested
that LPS stimulated p38 activation to suppress the expression of
HO-1 to induce inflammation.
p38 has attracted increased attention in
inflammation, and p38 kinase plays a central role in inflammatory
responses (29–31). Moreover, inhibition of p38
activation has been demonstrated to suppress LPS-induced
inflammation (29,33–35).
The present results indicated that LPS exposure caused severe
damage in rat lungs by examination of pathological tissue sections.
After LPS exposure, diffuse damage was observed in the alveoli,
alveolar sacs, alveolar tubes, alveolar septa and bronchi, and
there were a considerable number of inflammatory cells in the
alveolar septa of the LPS group. Furthermore, the increase of
TUNEL+ pulmonary cells in lungs and secretion levels of
IL-1β also indicated that the LPS-induced ALI model was successful.
It was also demonstrated that the HO-1 signaling pathway was
involved in the regulation of NLRP3 via p38 in ALI. Dex has an
analgesic effect and is widely used for sedation and anesthesia in
patients on ICU ventilator support (68–70).
However, the role of Dex in the progression of ALI is not fully
understood. The current results suggested that Dex could inhibit
ALI by suppressing p38 activation and activating the HO-1 signaling
pathway to inhibit the LPS-induced inflammatory response.
In conclusion, the present study investigated the
effect of Dex on decreasing lung inflammation in the neonatal rat
LPS-induced ALI model, which may provide a theory for the
pathogenesis of septic lung injury in infants and young pediatric
patients. Furthermore, the results indicated that Dex functions via
the p38/HO-1 and NLRP3/IL-1β signaling pathways, which may be a
possible mechanism of Dex intervention in ALI. Thus, the current
study provides a theoretical basis for the use of Dex in clinical
patients with ALI, providing novel evidence for the discovery of
new targets for clinical treatment of ALI.
Acknowledgements
Not applicable.
Funding
This work was supported by Natural Science
Foundation of Anhui Province, China (grant no. 1808085MH230).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
YS and XY conceived and designed the present study,
and provided administrative support. XY was involved in provision
of study materials. YS, YX, XL and JL collected and assembled data.
YS, WH and HY analyzed and interpreted the data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hughes KT and Beasley MB: Pulmonary
manifestations of acute lung injury: More than just diffuse
alveolar damage. Arch Pathol Lab Med. 141:916–922. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dias-Freitas F, Metelo-Coimbra C and
Roncon-Albuquerque R Jr: Molecular mechanisms underlying hyperoxia
acute lung injury. Respir Med. 119:23–28. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Monsel A, Zhu YG, Gudapati V, Lim H and
Lee JW: Mesenchymal stem cell derived secretome and extracellular
vesicles for acute lung injury and other inflammatory lung
diseases. Expert Opin Biol Ther. 16:859–871. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Walkey AJ, Kirkpatrick AR and Summer RS:
Systemic inflammatory response syndrome criteria for severe sepsis.
N Engl J Med. 373:8802015.PubMed/NCBI
|
|
5
|
Kaukonen KM, Bailey M and Bellomo R:
Systemic inflammatory response syndrome criteria for severe sepsis.
N Engl J Med. 373:8812015.PubMed/NCBI
|
|
6
|
Khemani RG, Smith LS, Zimmerman JJ and
Erickson S; Pediatric Acute Lung Injury Consensus Conference Group,
: Pediatric acute respiratory distress syndrome: Definition,
incidence, and epidemiology: Proceedings from the pediatric acute
lung injury consensus conference. Pediatr Crit Care Med 16 (5 Suppl
1). S23–S40. 2015. View Article : Google Scholar
|
|
7
|
Shi T, McAllister DA, O'Brien KL, Simoes
EAF, Madhi SA, Gessner BD, Polack FP, Balsells E, Acacio S, et al:
Global, regional, and national disease burden estimates of acute
lower respiratory infections due to respiratory syncytial virus in
young children in 2015: a systematic review and modelling study.
Lancet. 390:946–958. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou T and Chen YL: The functional
mechanisms of miR-30b-5p in acute lung injury in children. Med Sci
Monit. 25:40–51. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shein SL, Karam O, Beardsley A, Karsies T,
Prentice E, Tarquinio KM and Willson DF; Pediatric Acute Lung
Injury and Sepsis Investigator (PALISI) Network, : Development of
an antibiotic guideline for children with suspected
ventilator-associated infections. Pediatr Crit Care Med.
20:697–706. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Piñero P, Juanola O, Caparrós E, Zapater
P, Giménez P, González-Navajas JM, Such J and Francés R: Toll-like
receptor polymorphisms compromise the inflammatory response against
bacterial antigen translocation in cirrhosis. Sci Rep. 7:464252017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Byczkowski JZ and Channel SR:
Chemically-induced oxidative stress and tumorigenesis: Effects on
signal transduction and cell proliferation. Toxic Subst Mechan.
15:101–128. 1996.
|
|
12
|
Zhang G, Li J, Xie R, Fan X, Liu Y, Zheng
S, Ge Y and Chen PR: Bioorthogonal chemical activation of kinases
in living systems. ACS Cent Sci. 2:325–331. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun Y, Liu J, Yuan X and Li Y: Effects of
dexmedetomidine on emergence delirium in pediatric cardiac surgery.
Minerva Pediatr. 69:165–173. 2017.PubMed/NCBI
|
|
14
|
Zhang A, Pan W, Lv J and Wu H: Protective
effect of amygdalin on LPS-induced acute lung injury by inhibiting
NF-κB and NLRP3 signaling pathways. Inflammation. 40:745–751. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang B, Wang B, Cao S, Wang Y and Wu D:
Silybin attenuates LPS-induced lung injury in mice by inhibiting
NF-κB signaling and NLRP3 activation. Int J Mol Med. 39:1111–1118.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang A, Wang S, Zhang J and Wu H: Genipin
alleviates LPS-induced acute lung injury by inhibiting NF-κB and
NLRP3 signaling pathways. Int Immunopharmacol. 38:115–119. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li W, Li W, Zang L, Liu F, Yao Q, Zhao J,
Zhi W and Niu X: Fraxin ameliorates lipopolysaccharide-induced
acute lung injury in mice by inhibiting the NF-κB and NLRP3
signalling pathways. Int Immunopharmacol. 67:1–12. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gaidt MM and Hornung V: The NLRP3
inflammasome renders cell death pro-inflammatory. J Mol Biol.
430:133–141. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim JK, Jin HS, Suh HW and Jo EK: Negative
regulators and their mechanisms in NLRP3 inflammasome activation
and signaling. Immunol Cell Biol. 95:584–592. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu X, Pichulik T, Wolz OO, Dang TM, Stutz
A, Dillen C, Delmiro Garcia M, Kraus H, Dickhöfer S, Daiber E, et
al: Human NACHT, LRR, and PYD domain-containing protein 3 (NLRP3)
inflammasome activity is regulated by and potentially targetable
through bruton tyrosine kinase. J Allergy Clin Immunol.
140:1054–1067.e10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hughes FM Jr, Kennis JG, Youssef MN, Lowe
DW, Shaner BE and Purves JT: The NACHT, LRR and PYD
domains-containing protein 3 (NLRP3) inflammasome mediates
inflammation and voiding dysfunction in a
lipopolysaccharide-induced rat model of cystitis. J Clin Cell
Immunol. 7:3962016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martinon F, Burns K and Tschopp J: The
inflammasome: A molecular platform triggering activation of
inflammatory caspases and processing of proIL-beta. Mol Cell.
10:417–426. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee J, Wan J, Lee L, Peng C, Xie H and Lee
C: Study of the NLRP3 inflammasome component genes and downstream
cytokines in patients with type 2 diabetes mellitus with carotid
atherosclerosis. Lipids Health Dis. 16:2172017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Y, Li N, Yan Z, Li H, Chen L, Zhang Z,
Fan G, Xu K and Li Z: Dysregulation of the NLRP3 inflammasome
complex and related cytokines in patients with multiple myeloma.
Hematology. 21:144–151. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ishii T and Mann GE: Redox status in
mammalian cells and stem cells during culture in vitro: Critical
roles of Nrf2 and cystine transporter activity in the maintenance
of redox balance. Redox Biol. 2:786–794. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rojo de la Vega M, Dodson M, Gross C,
Mansour HM, Lantz RC, Chapman E, Wang T, Black SM, Garcia JG and
Zhang DD: Role of Nrf2 and autophagy in acute lung injury. Curr
Pharmacol Rep. 2:91–101. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Luo YP, Jiang L, Kang K, Fei DS, Meng XL,
Nan CC, Pan SH, Zhao MR and Zhao MY: Hemin inhibits NLRP3
inflammasome activation in sepsis-induced acute lung injury,
involving heme oxygenase-1. Int Immunopharmacol. 20:24–32. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao Z, Han Y, Hu Y, Wu X, Wang Y, Zhang X,
Fu J, Zou X, Zhang J, Chen X, et al: Targeting HO-1 by
epigallocatechin-3-gallate reduces contrast-induced renal injury
via anti-oxidative stress and anti-inflammation pathways. PLoS One.
11:e01490322016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dong N, Xu X, Xue C, Wang C, Li X, Bi C
and Shan A: Ethyl pyruvate inhibits LPS induced IPEC-J2
inflammation and apoptosis through p38 and ERK1/2 pathways. Cell
Cycle. 18:2614–2628. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xin L, Che B, Zhai B, Luo Q, Zhang C, Wang
J, Wang S, Fan G, Liu Z, Feng J and Zhang Z: 1,25-Dihydroxy vitamin
D3 attenuates the oxidative stress-mediated inflammation induced by
PM2.5via the p38/NF-κB/NLRP3 pathway. Inflammation. 42:702–713.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bailey KA, Moreno E, Haj FG, Simon SI and
Passerini AG: Mechanoregulation of p38 activity enhances
endoplasmic reticulum stress-mediated inflammation by arterial
endothelium. FASEB J. 33:12888–12899. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schieven GL: The biology of p38 kinase: A
central role in inflammation. Curr Top Med Chem. 5:921–928. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou LF, Chen QZ, Yang CT, Fu ZD, Zhao ST,
Chen Y, Li SN, Liao L, Zhou YB, Huang JR and Li JH: TRPC6
contributes to LPS-induced inflammation through ERK1/2 and p38
pathways in bronchial epithelial cells. Am J Physiol Cell Physiol.
314:C278–C288. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Abarikwu SO: Kolaviron, a natural
flavonoid from the seeds of Garcinia kola, reduces LPS-induced
inflammation in macrophages by combined inhibition of IL-6
secretion, and inflammatory transcription factors, ERK1/2, NF-κB,
p38, Akt, p-c-JUN and JNK. Biochim Biophys Acta. 1840:2373–2381.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Haddad EB, Birrell M, McCluskie K, Ling A,
Webber SE, Foster ML and Belvisi MG: Role of p38 MAP kinase in
LPS-induced airway inflammation in the rat. Br J Pharmacol.
132:1715–1724. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ayala TS, Tessaro FHG, Jannuzzi GP, Bella
LM, Ferreira KS and Martins JO: High glucose environments interfere
with bone marrow-derived macrophage inflammatory mediator release,
the TLR4 pathway and glucose metabolism. Sci Rep. 9:114472019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Perrone M, Moon BS, Park HS, Laquintana V,
Jung JH, Cutrignelli A, Lopedota A, Franco M, Kim SE, Lee BC and
Denora N: A novel PET imaging probe for the detection and
monitoring of translocator protein 18 kDa expression in
pathological disorders. Sci Rep. 6:204222016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Herriges M and Morrisey EE: Lung
development: Orchestrating the generation and regeneration of a
complex organ. Development. 141:502–513. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Plosa EJ, Young LR, Gulleman PM,
Polosukhin VV, Zaynagetdinov R, Benjamin JT, Im AM, van der Meer R,
Gleaves LA, Bulus N, et al: Epithelial β1 integrin is required for
lung branching morphogenesis and alveolarization. Development.
141:4751–4762. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
He MY, Wang G, Han SS, Li K, Jin Y, Liu M,
Si ZP, Wang J, Liu GS and Yang X: Negative impact of hyperglycaemia
on mouse alveolar development. Cell Cycle. 17:80–91. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chi W, Li F, Chen H, Wang Y, Zhu Y, Yang
X, Zhu J, Wu F, Ouyang H, Ge J, et al: Caspase-8 promotes
NLRP1/NLRP3 inflammasome activation and IL-1β production in acute
glaucoma. Proc Natl Acad Sci USA. 111:11181–11186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Suzuki H, Kanamaru K, Tsunoda H, Inada H,
Kuroki M, Sun H, Waga S and Tanaka T: Heme oxygenase-1 gene
induction as an intrinsic regulation against delayed cerebral
vasospasm in rats. J Clin Invest. 104:59–66. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang L and Tang Z: Expression and
regulation of the ERK1/2 and p38 MAPK signaling pathways in
periodontal tissue remodeling of orthodontic tooth movement. Mol
Med Rep. 17:1499–1506. 2018.PubMed/NCBI
|
|
44
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Naidu S, Vijayan V, Santoso S, Kietzmann T
and Immenschuh S: Inhibition and genetic deficiency of p38 MAPK
up-regulates heme oxygenase-1 gene expression via Nrf2. J Immunol.
182:7048–7057. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bosmann M and Ward PA: The inflammatory
response in sepsis. Trends Immunol. 34:129–136. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rice TC, Pugh AM, Caldwell CC and
Schneider BSP: Balance between the proinflammatory and
anti-inflammatory immune responses with blood transfusion in
sepsis. Crit Care Nurs Clin North Am. 29:331–340. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kaukonen KM, Bailey M, Pilcher D, Cooper
DJ and Bellomo R: Systemic inflammatory response syndrome criteria
in defining severe sepsis. N Engl J Med. 372:1629–1638. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chang M, Lu HY, Xiang H and Lan HP:
Clinical effects of different ways of mechanical ventilation
combined with pulmonary surfactant in treatment of acute lung
injury/acute respiratory distress syndrome in neonates: A
comparative analysis. Zhongguo Dang Dai Er Ke Za Zhi. 18:1069–1074.
2016.(In Chinese). PubMed/NCBI
|
|
50
|
Chaiwat O, Chittawatanarat K,
Piriyapathsom A, Pisitsak C, Thawitsri T, Chatmongkolchart S and
Kongsayreepong S: Incidence of and risk factors for acute
respiratory distress syndrome in patients admitted to surgical
intensive care units: The multicenter thai University-based
surgical intensive care unit (THAI-SICU) study. J Med Assoc Thai.
99 (Suppl 6):S118–S127. 2016.PubMed/NCBI
|
|
51
|
Gong Y, Lan H, Yu Z, Wang M, Wang S, Chen
Y, Rao H, Li J, Sheng Z and Shao J: Blockage of glycolysis by
targeting PFKFB3 alleviates sepsis-related acute lung injury via
suppressing inflammation and apoptosis of alveolar epithelial
cells. Biochem Biophys Res Commun. 491:522–529. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang X and Dong S: Protective effects of
erythropoietin towards acute lung injuries in rats with sepsis and
its related mechanisms. Ann Clin Lab Sci. 49:257–264.
2019.PubMed/NCBI
|
|
53
|
Wygrecka M, Jablonska E, Guenther A,
Preissner KT and Markart P: Current view on alveolar coagulation
and fibrinolysis in acute inflammatory and chronic interstitial
lung diseases. Thromb Haemost. 99:494–501. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lim R, Barker G and Lappas M: TLR2, TLR3
and TLR5 regulation of pro-inflammatory and pro-labour mediators in
human primary myometrial cells. J Reprod Immunol. 122:28–36. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Jin Z, Yang YZ, Chen JX and Tang YZ:
Inhibition of pro-inflammatory mediators in RAW264.7 cells by
7-hydroxyflavone and 7,8-dihydroxyflavone. J Pharm Pharmacol.
69:865–874. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lappas M: The IL-1β signalling pathway and
its role in regulating pro-inflammatory and pro-labour mediators in
human primary myometrial cells. Reprod Biol. 17:333–340. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Feng G, Sun B, Liu HX, Liu QH, Zhao L and
Wang TL: EphA2 antagonism alleviates LPS-induced acute lung injury
via Nrf2/HO-1, TLR4/MyD88 and RhoA/ROCK pathways. Int
Immunopharmacol. 72:176–185. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen Z, Zhong H, Wei J, Lin S, Zong Z,
Gong F, Huang X, Sun J, Li P, Lin H, et al: Inhibition of Nrf2/HO-1
signaling leads to increased activation of the NLRP3 inflammasome
in osteoarthritis. Arthritis Res Ther. 21:3002019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mahmoud AM, Hussein OE, Abd El-Twab SM and
Hozayen WG: Ferulic acid protects against methotrexate
nephrotoxicity via activation of Nrf2/ARE/HO-1 signaling and PPARγ,
and suppression of NF-κB/NLRP3 inflammasome axis. Food Funct.
10:4593–4607. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu S, Tian L, Chai G, Wen B and Wang B:
Targeting heme oxygenase-1 by quercetin ameliorates alcohol-induced
acute liver injury via inhibiting NLRP3 inflammasome activation.
Food Funct. 9:4184–4193. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xiaoyu H, Si H, Li S, Wang W, Guo J, Li Y,
Cao Y, Fu Y and Zhang N: Induction of heme oxygenas-1 attenuates
NLRP3 inflammasome activation in lipopolysaccharide-induced
mastitis in mice. Int Immunopharmacol. 52:185–190. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhang X, Xu X, Yu Y, Chen C, Wang J, Cai C
and Guo W: Integration analysis of MKK and MAPK family members
highlights potential MAPK signaling modules in cotton. Sci Rep.
6:297812016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Roux PP and Blenis J: ERK and p38
MAPK-activated protein kinases: A family of protein kinases with
diverse biological functions. Microbiol Mol Biol Rev. 68:320–344.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kim YM, Kim HJ and Chang KC: Glycyrrhizin
reduces HMGB1 secretion in lipopolysaccharide-activated RAW 264.7
cells and endotoxemic mice by p38/Nrf2-dependent induction of HO-1.
Int Immunopharmacol. 26:112–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Keum YS, Yu S, Chang PP, Yuan X, Kim JH,
Xu C, Han J, Agarwal A and Kong AN: Mechanism of action of
sulforaphane: Inhibition of p38 mitogen-activated protein kinase
isoforms contributing to the induction of antioxidant response
element-mediated heme oxygenase-1 in human hepatoma HepG2 cells.
Cancer Res. 66:8804–8813. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yu R, Mandlekar S, Lei W, Fahl WE, Tan TH
and Kong AN: p38 mitogen-activated protein kinase negatively
regulates the induction of phase II drug-metabolizing enzymes that
detoxify carcinogens. J Biol Chem. 275:2322–2327. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Venkatraman R, Hungerford JL, Hall MW,
Moore-Clingenpeel M and Tobias JD: Dexmedetomidine for sedation
during noninvasive ventilation in pediatric patients. Pediatr Crit
Care Med. 18:831–837. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sottas CE and Anderson BJ:
Dexmedetomidine: The new all-in-one drug in paediatric anaesthesia?
Curr Opin Anaesthesiol. 30:441–451. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Devasya A and Sarpangala M:
Dexmedetomidine: A review of a newer sedative in dentistry. J Clin
Pediatr Dent. 39:401–409. 2015. View Article : Google Scholar : PubMed/NCBI
|