Introduction
Retinitis pigmentosa (RP) is a group of inherited
retinal dystrophies, which is characterized by the progressive loss
of function in rod and cone photoreceptors and pigment epithelial
cells (1,2). RP affects approximately 1 in 4,000
individuals worldwide (3).
Currently, there are ~325,000 patients in China suffering from RP,
and the condition may be inherited in an autosomal-recessive,
autosomal-dominant, X-linked or mitochondrial pattern (4–6). To
date, >60 genes have been identified in non-syndromic RP,
including 41 genes associated with an autosomal-recessive form
(arRP), according to the RetNet database (https://sph.uth.edu/RetNet). These genes encode for
proteins involved in a variety of diverse functional pathways in
the neural retina, including photoreceptor development,
phototransduction, retinoid cycle, cilia, photoreceptor outer
segment development and intracellular protein transport (3,7);
dysregulation of these pathways generally results in RP (8). Typical patients with RP display
features of night blindness, retinal pigment epithelium atrophy,
visual field constriction, pale optic discs and bone spicule
pigmentation. Although tremendous progress has been made in the
study of RP-associated genes, the identified genes account for
merely 60% of all clinical cases of RP, while the pathogenic
variants remain elusive in the remaining cases (9). RP is highly heterogenous, making its
accurate diagnosis difficult, and diagnosis only depends on
clinical traits for certain cases. In recent years, tremendous
advances have been made in molecular biology to identify
disease-associated genes for Mendelian disorders. Furthermore,
numerous studies demonstrated that exome sequencing is a powerful,
efficient method for identifying pathogenic genes of monogenic
disorders, including RP (10–12).
In the present study, whole-exome sequencing (WES)
was used to identify the pathogenic variant in a non-consanguineous
Chinese family with arRP. A novel homozygous frameshift variant in
the cyclic nucleotide-gated channel subunit alpha 1 (CNGA1) gene
was identified in the patient with RP.
Materials and methods
Subjects
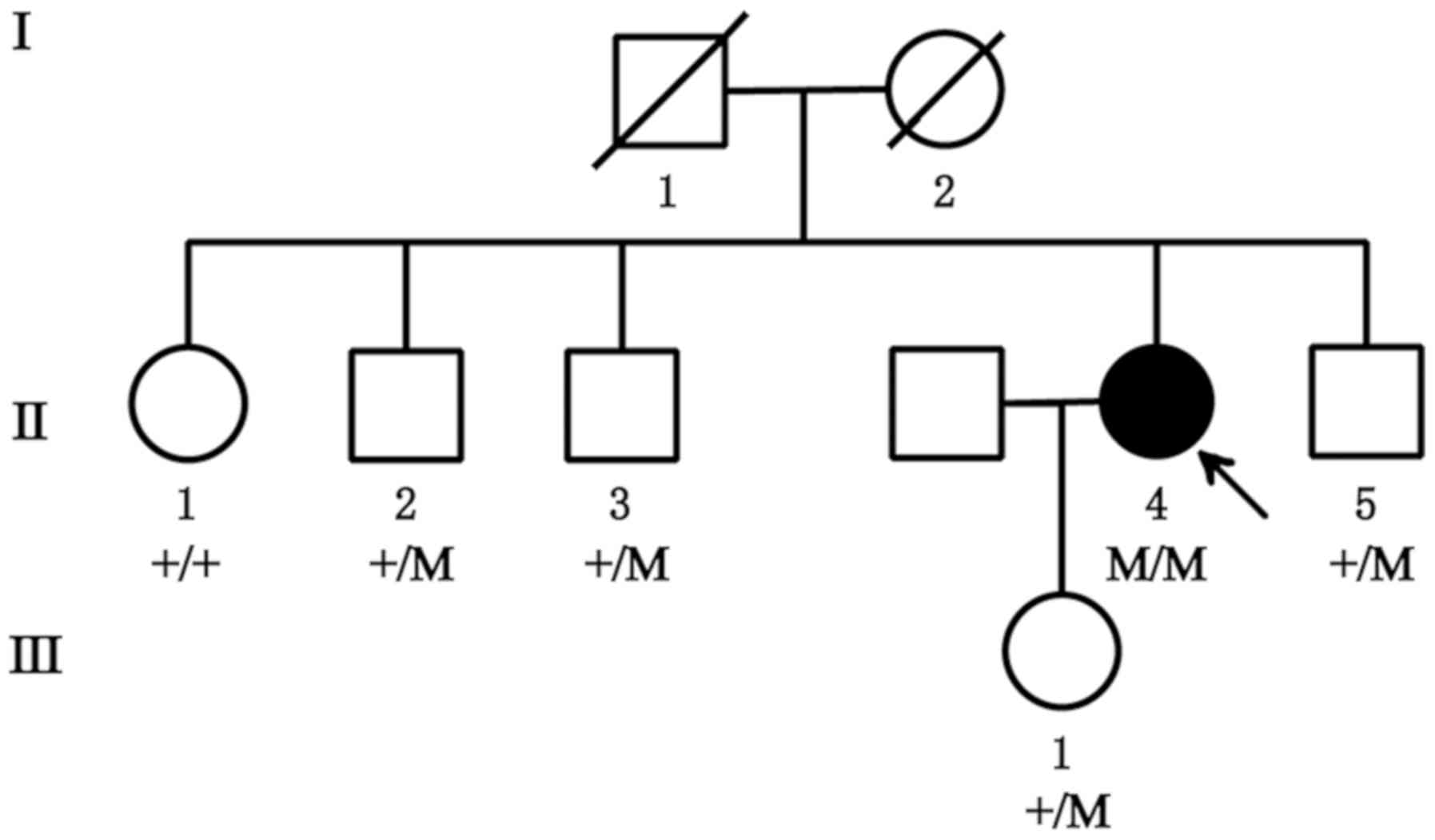
A Chinese family was recruited from the First
Hospital of Jilin University (Changchun, China), including a total
of seven members, with one affected patient and six unaffected
individuals (Fig. 1). The control
subjects were recruited from Sichuan Provincial People's Hospital
(Chengdu, China). All control subjects underwent ophthalmic
examination and exhibited no symptoms of RP. The recruitment of the
control subjects was previously described (13). The study was implemented in
conformity to the tenets of the Declaration of Helsinki and
approved by the Institutional Review Boards of Sichuan Provincial
People's Hospital (Chengdu, China) and the First Hospital of Jilin
University (Changchun, China). Written informed consent was
obtained from all participants or parents of children prior to
their inclusion in the present study.
Clinical diagnosis
Comprehensive ophthalmic examinations were performed
on the proband (age, 47 years), the proband's only daughter (age,
23 years), the proband's three brothers (age, 54, 51 and 44 years,
respectively), and the proband's only sister (age, 57 years) in the
present study. The examinations included visual acuity testing,
computerized visual field measurement, tests of dark adaptation and
color vision, optical coherence tomography (OCT) and fundus
imaging.
DNA isolation
Peripheral blood samples were collected in EDTA
tubes from the proband (II:4), the proband's daughter (III:1) and
the proband's brothers (II:2, II:3 and II:5). Genomic DNA was
extracted using the salt precipitation method, involving lysis,
precipitation, washing and resuspension (14). DNA samples were stored at −20°C
until use.
WES
The DNA of case no. II:4 (the proband) was analyzed
by WES at Novogene Biotechnology Inc., with a mean read depth of
target regions of 100X. The sequenced samples were prepared
according to the Illumina standard protocol (Illumina, Inc.).
First, genomic DNA was randomly fragmented to an average size of
180–280 bp by a Covaris S220 sonicator (Covaris, Inc.). The
remaining overhangs were converted into blunt ends via exonuclease
polymerase activities. Subsequently, DNA fragments were
end-repaired and phosphorylated, followed by A-tailing and ligation
at the 3′ends with paired-end adaptors (Illumina, Inc.). DNA
fragments with ligated adapter molecules on both ends were
selectively enriched by PCR. After PCR amplification, libraries
were hybridized with a liquid phase containing a biotin-labeled
probe and magnetic beads with streptomycin were used to capture the
exons of genes. Captured libraries were enriched by PCR to add
index tags to prepare for sequencing. Products were purified using
an AMPure XP system (Beckman Coulter, Inc.) and quantified using
the Agilent high-sensitivity DNA assay on the Agilent Bioanalyzer
2100 system (Agilent Technologies, Inc.). Finally, the DNA library
was sequenced on Illumina HiSeq4000 for paired-end 150 bp
reads.
Variant validation
Sanger sequencing was used to confirm the identified
mutation in CNGA1. All exons of CNGA1 were amplified from the
genomic DNAs from subjects II:2-5 and III:1 in the recruited family
by PCR using standard conditions (15). Amplified products were purified and
sequenced with forward and reverse primers (15) by using the BigDye®
Terminator v3.1 Cycle Sequencing kit (Applied Biosystems; Thermo
Fisher Scientific, Inc.) on a 3730 ABI DNA sequencer (Thermo Fisher
Scientific, Inc.).
Results
Clinical phenotype
A Chinese family with RP, without any history of
consanguineous marriage, was recruited for the present study. The
pedigree chart is provided in Fig.
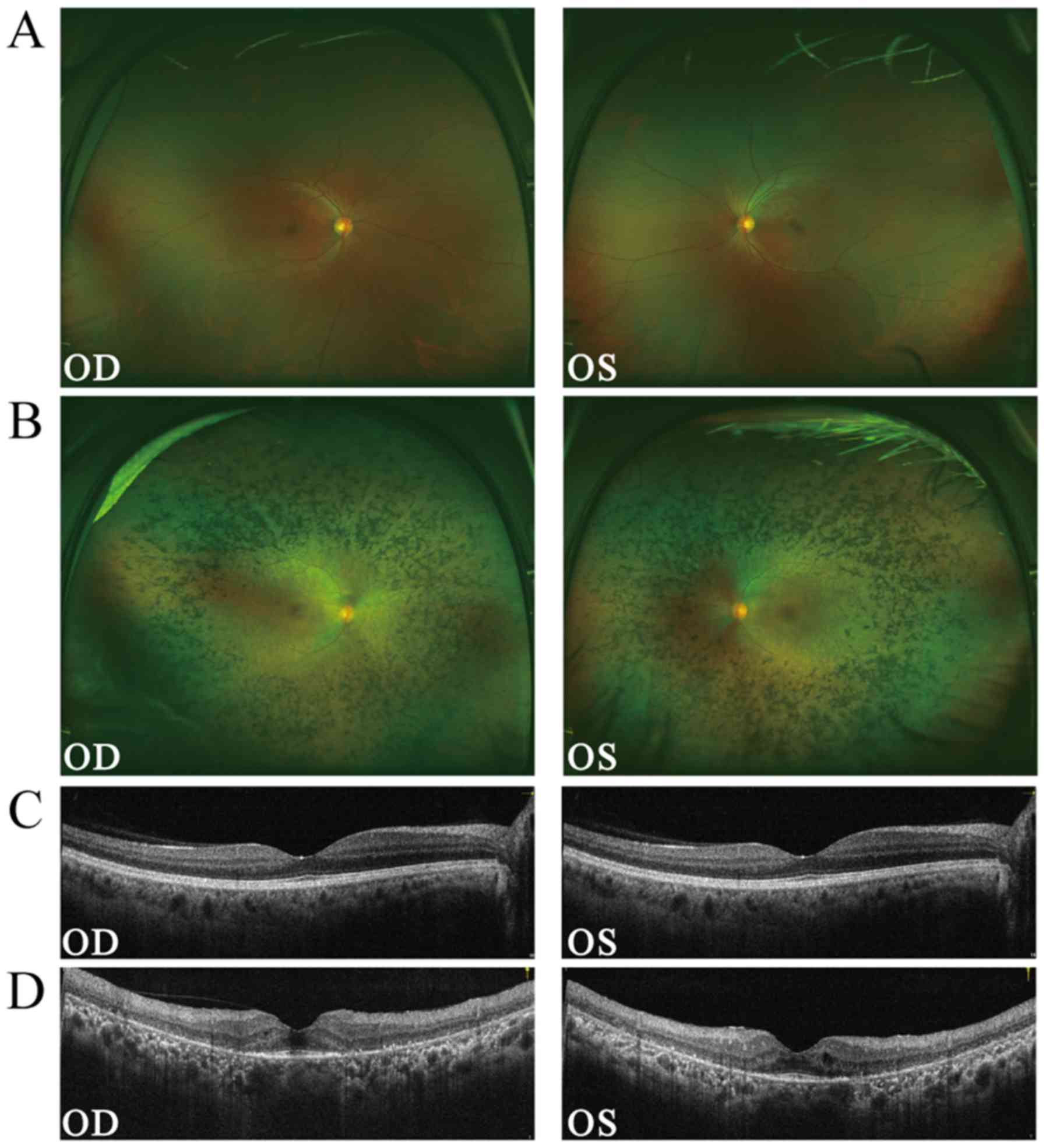
1. Examination of the fundus revealed a normal fundus in
subject no. III:1, the proband's daughter (Fig. 2A). By contrast, the proband (II:4)
exhibited several classic characteristics of RP, including
attenuated blood vessels, pale optic discs and paravascular bone
spicule pigmentation (Fig. 2B).
OCT revealed thinning of the outer nuclear layer and thinning of
the reflex of the retinal pigment epithelium-Bruch's membrane
complex (Fig. 2C). The visual
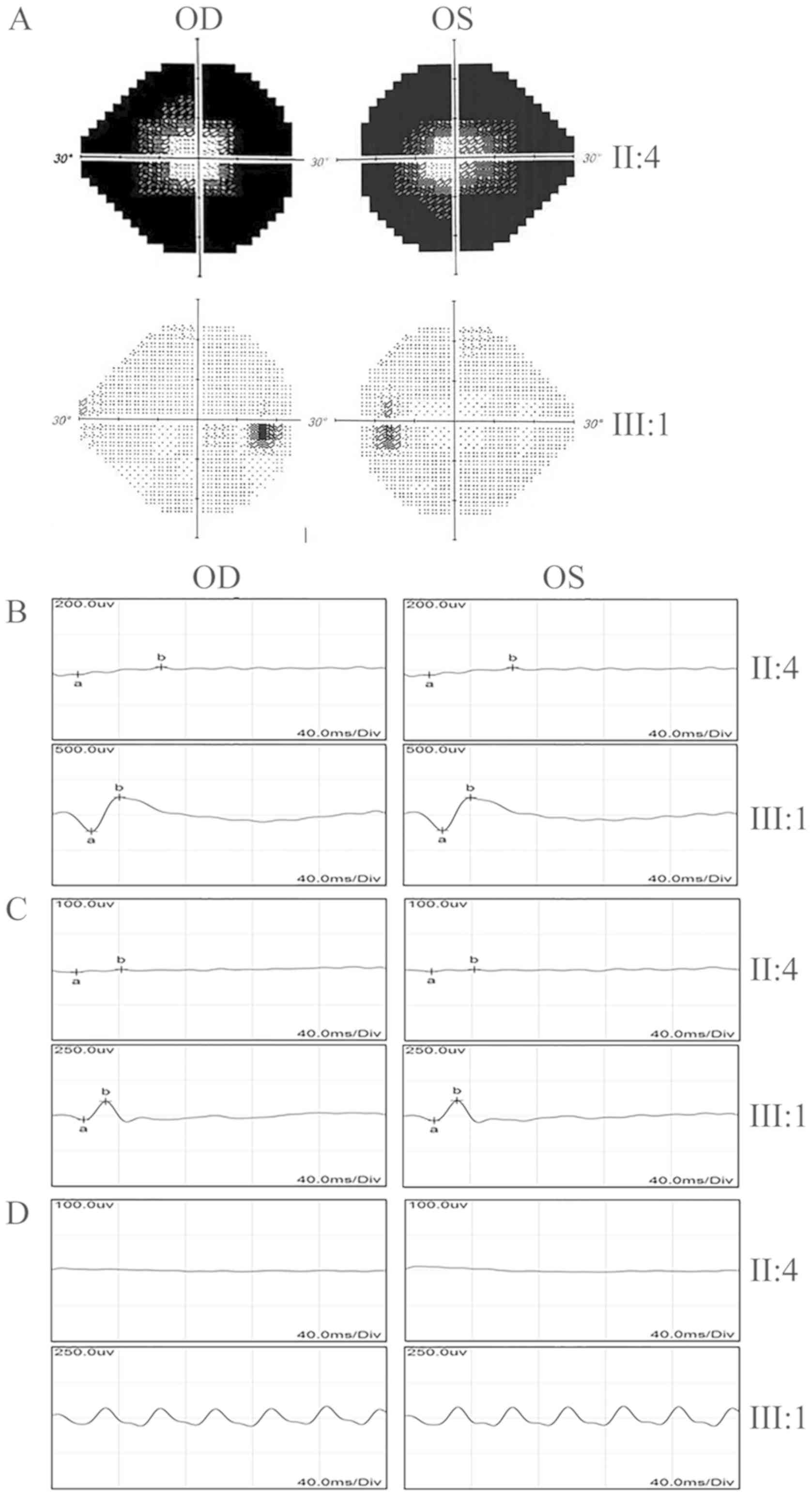
field test indicated a significant loss of peripheral vision in the
proband, while the vision was normal in the unaffected individuals
(II:2, 3, 5 and III:1; Fig. 3A).
Electroretinography (ERG) recordings were barely detectable in the
proband under either the scotopic or photopic conditions,
indicating an almost complete loss of the function of both rods and
cones (Fig. 3B-D). The proband's
daughter (III:1) had normal visual responses in the ERG tests. In
summary, while all of the healthy unaffected family members had
normal vision, the affected individual exhibited the typical
clinical symptoms of late-stage RP.
WES and data analysis
To identify the genetic variant causing the RP
condition in the family, WES was performed for the proband (subject
no. II:4). In total, 24,702 SNPs in coding regions or splicing
junctions (2,797 nonsynonymous SNPs, 3,349 synonymous SNPs and
18,556 other types of SNP) and 659 indels were identified. Since
neither the proband's parents nor her siblings were affected, the
pathogenic variant was more likely to be recessive. Thus, the
recessive genetic mode was applied to analyze those SNPs. In order
to define the pathogenic variant, the homozygous and compound
heterozygous variants were closely followed, particularly
frameshift insertions or deletions in the coding region, splicing
acceptor and donor site variants, as well as non-synonymous
variants that were more likely to be pathogenic. Of note, a
homozygous single nucleotide deletion variant in exon 6 of CNGA1
(c.265delC; Chr4:47951883-47951884) was identified in subject no.
II:4. This variant causes a frameshift of the open reading frame
and loss of function of the protein. CNGA1 is an essential protein
involved in the phototransduction cascade in rod photoreceptors.
Without CNGA1, the phototransduction cascade would fail in the rods
(16). Therefore, this variant was
a top candidate subjected to further validation.
Variant validation and co-segregation
analysis
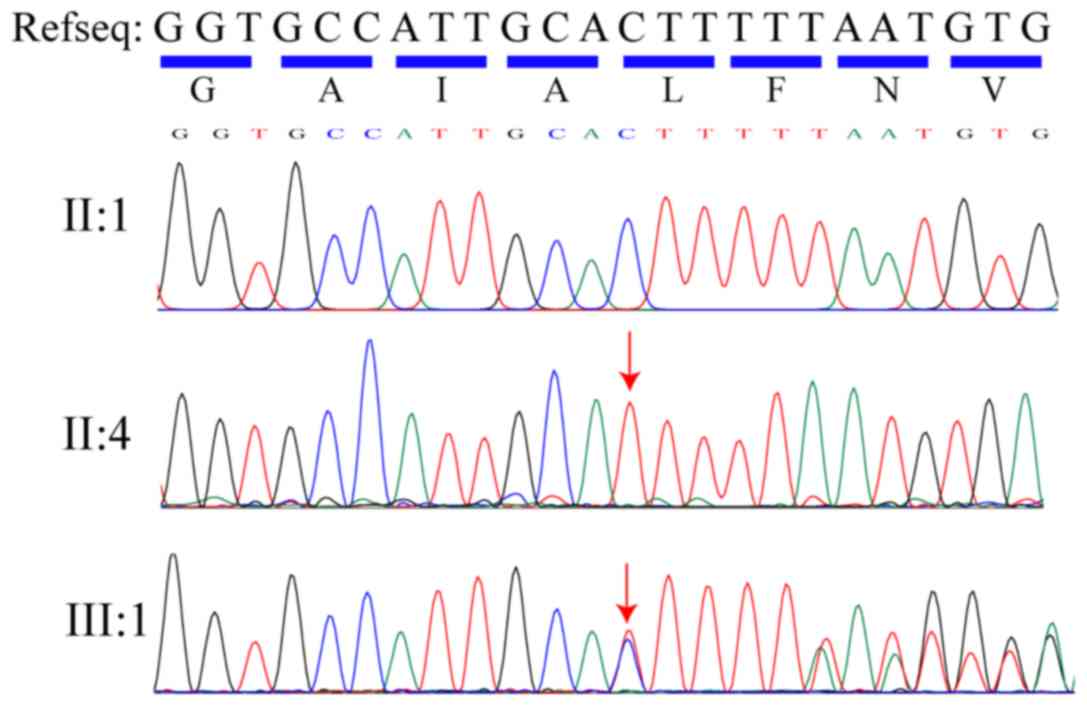
The homozygous variant, c.265delC (p.L89Ffs*3) in
CNGA1, was confirmed using direct Sanger sequencing in the affected
individual (II:4; Fig. 4). It was
also determined that, while the proband's daughter (III:1) and one
of the proband's brothers (II:2) were heterozygous carriers of
c.265delC (p.L89Ffs*3), the other two brothers of the proband (II:3
and II:5) had no variant at this position. The homozygous recessive
variant matched the genotype/phenotype in this family. Furthermore,
this variant was absent from 600 ethnicity-matched normal control
samples (median age, 61; age range, 40–78 years) that underwent
Sanger sequencing. Therefore, it was concluded that the homozygous
CNGA1 c.265delC variant (p.L89Ffs*3) is the variant responsible for
the RP condition in the proband.
Discussion
RP exhibits similar clinical symptoms between cases
and ultimately leads to severe blindness, but RP is highly
heterogenous from the genetic point of view. The exact clinical
symptoms for each patient with RP may vary from case to case.
Clinicians may face challenges to diagnose the condition solely
based on clinical appearance in certain cases (14). In the past decade, WES has proven
successful in efficiently identifying variants from inherited
diseases (10–12), markedly improving the accurate
diagnosis of RP.
In the present study, a pedigree with RP was
screened by using exome sequencing and a homozygous variant,
c.265delC (p.L89Ffs*3), was identified in the CNGA1 gene. CNGA1,
also known as RP49 or CNCG1, is a member of the subfamily of cyclic
nucleotide-gated cation channels. The CNGA1 gene, located at
chromosome 4p12, contains 11 exons. CNGA1 encodes the α-subunit of
the rod cGMP-gated channel (CNG) and is predominantly expressed in
rod photoreceptors. CNGA1 consists of six putative transmembrane
domains, a pore region and a cGMP-binding domain (17,18).
Rod CNG is a multi-subunit protein composed of CNGA1 and CNGB1 at a
ratio of 3:1 (19). CNG is a
voltage-independent and nonselective ion channel that is activated
by the direct binding of cGMP (20,21).
CNGs are involved in the final step of the phototransduction
cascade (22,23). In addition to its function as a
channel protein in the phototransduction cascade, CNGA1 is also
required for the formation of the structure of rod photoreceptor
outer segments (24).
Variants in the CNGA1 gene have been reported to
cause arRP in different ethnic populations (6,15,25,26).
The affected individuals with CNGA1 variants display classic
features of RP. In the present study, a novel homozygous variant,
c.265delC (p.L89Ffs*3) in the CNGA1 gene, was identified in a
Chinese family with arRP. PolyPhen2
(genetics.bwh.harvard.edu/pph2/) predicted that the variant may
damage the function of the protein. This frameshift variant causes
truncation of CNGA1 close to the N-terminus and may lead to
complete loss of function. This is consistent with no detectable
ERGs in the proband (II:4). Although CNGA1 is exclusively expressed
in rods, the affected patient in the present study also exhibited
impaired cone functions, as demonstrated by the photopic and
flicker ERG. The reason for this may be that rods account for ~95%
of all photoreceptors in the human retina, and in the late stage of
RP, substantial rod loss may undermine the physical support of
cones provided by rods in the retina. In the absence of this
physical support, cones may not function properly and can
degenerate progressively. The formation of outer segments may also
be affected in this patient. It may be possible to reveal the exact
phenotype arising from this variant by an animal model with the
CNGA1 frameshift variant (16). To
date, there is no treatment available for RP caused by CGNA1
mutations. Gene therapy, however, may be a promising treatment in
the future.
In conclusion, the present study revealed a novel
homozygous variant (p.L89Ffs*3) in the CNGA1 gene in a Chinese
family with arRP using WES. The present study demonstrated that WES
is a cost-effective method to screen disease-causing variants in
arRP. The present results expand the spectrum of known variants
associated with RP and may facilitate the molecular diagnosis of RP
prior to potential gene therapy for patients carrying this variant
in the future.
Acknowledgements
Not applicable.
Funding
This research project was supported by the National
Precision Medicine Project (grant no. 2016YFC0905200 to HZ), the
National Natural Science Foundation of China (grant nos. 81371030,
81570882 and 81770935 to HZ), and the Science and Technology of
Jilin Province (grant no. 20200201470JC to LW).
Availability of data and materials
The datasets used and analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LW, JH and HBZ conceived the study. TDZ, YQL, PZ and
LL performed the experiments. LW recruited the patients. HBZ and BG
analyzed the data. TDZ and HBZ wrote the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Institutional Review
Boards of Sichuan Provincial People's Hospital (Chengdu, China) and
the First Hospital of Jilin University (Changchun, China). Written
informed consent was obtained from all participants or parents of
children prior to their inclusion in the present study.
Patient consent for publication
Written informed consent for publication was
obtained from all family members participating in the present
study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Narayan DS, Wood JP, Chidlow G and Casson
RJ: A review of the mechanisms of cone degeneration in retinitis
pigmentosa. Acta Ophthalmol. 94:748–754. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zobor D and Zrenner E: Retinitis
pigmentosa-A review. Pathogenesis, guidelines for diagnostics and
perspectives. Ophthalmologe. 109:501–514; quiz 515. 2012.(In
German). View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nash BM, Wright DC, Grigg JR, Bennetts B
and Jamieson RV: Retinal dystrophies, genomic applications in
diagnosis and prospects for therapy. Transl Pediatr. 4:139–163.
2015.PubMed/NCBI
|
|
4
|
Bunker CH, Berson EL, Bromley WC, Hayes RP
and Roderick TH: Prevalence of retinitis pigmentosa in maine. Am J
Ophthalmol. 97:357–365. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grøndahl J: Estimation of prognosis and
prevalence of retinitis pigmentosa and usher syndrome in Norway.
Clin Genet. 31:255–264. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Paloma E, Martinez-Mir A, Garcia-Sandoval
B, Ayuso C, Vilageliu L, Gonzàlez-Duarte R and Balcells S: Novel
homozygous mutation in the alpha subunit of the rod cGMP gated
channel (CNGA1) in two Spanish sibs affected with autosomal
recessive retinitis pigmentosa. J Med Genet. 39:E662002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Daiger SP, Bowne SJ and Sullivan LS:
Perspective on genes and mutations causing retinitis pigmentosa.
Arch Ophthalmol. 125:151–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Berson EL: Retinitis pigmentosa. The
friedenwald lecture. Invest Ophthalmol Vis Sci. 34:1659–1676.
1993.PubMed/NCBI
|
|
9
|
Xu Y, Guan L, Shen T, Zhang J, Xiao X,
Jiang H, Li S, Yang J, Jia X, Yin Y, et al: Mutations of 60 known
causative genes in 157 families with retinitis pigmentosa based on
exome sequencing. Hum Genet. 133:1255–1271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ng SB, Buckingham KJ, Lee C, Bigham AW,
Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et
al: Exome sequencing identifies the cause of a mendelian disorder.
Nat Genet. 42:30–35. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao F, Wu J, Xue A, Su Y, Wang X, Lu X,
Zhou Z, Qu J and Zhou X: Exome sequencing reveals CCDC111 mutation
associated with high myopia. Hum Genet. 132:913–921. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hu YS, Song H, Li Y, Xiao ZY and Li T:
Whole-exome sequencing identifies novel mutations in genes
responsible for retinitis pigmentosa in 2 nonconsanguineous Chinese
families. Int J Ophthalmol. 12:915–923. 2019.PubMed/NCBI
|
|
13
|
Liu X, Wu Y, Miao Z, Zhang H, Gong B, Zhu
X, Huang L, Shi Y, Hao F, Ma S, et al: A novel deletion downstream
of the PAX6 gene identified in a Chinese family with congenital
aniridia. Ophthalmic Genet. 39:428–436. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang M, Gan D, Huang X and Xu G: Novel
compound heterozygous mutations in CNGA1in a Chinese family
affected with autosomal recessive retinitis pigmentosa by targeted
sequencing. BMC Ophthalmol. 16:1012016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Q, Zulfiqar F, Riazuddin SA, Xiao X,
Ahmad Z, Riazuddin S and Hejtmancik JF: Autosomal recessive
retinitis pigmentosa in a Pakistani family mapped to CNGA1 with
identification of a novel mutation. Mol Vis. 10:884–889.
2004.PubMed/NCBI
|
|
16
|
Hüttl S, Michalakis S, Seeliger M, Luo DG,
Acar N, Geiger H, Hudl K, Mader R, Haverkamp S, Moser M, et al:
Impaired channel targeting and retinal degeneration in mice lacking
the cyclic nucleotide-gated channel subunit CNGB1. J Neurosci.
25:130–138. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dhallan RS, Macke JP, Eddy RL, Shows TB,
Reed RR, Yau KW and Nathans J: Human rod photoreceptor cGMP-gated
channel: Amino acid sequence, gene structure, and functional
expression. J Neurosci. 12:3248–3256. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pittler SJ, Lee AK, Altherr MR, Howard TA,
Seldin MF, Hurwitz RL, Wasmuth JJ and Baehr W: Primary structure
and chromosomal localization of human and mouse rod photoreceptor
cGMP-gated cation channel. J Biol Chem. 267:6257–6262.
1992.PubMed/NCBI
|
|
19
|
Shuart NG, Haitin Y, Camp SS, Black KD and
Zagotta WN: Molecular mechanism for 3:1 subunit stoichiometry of
rod cyclic nucleotide-gated ion channels. Nat Commun. 2:4572011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Craven KB and Zagotta WN: CNG and HCN
channels: Two peas, one pod. Annu Rev Physiol. 68:375–401. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fesenko EE, Kolesnikov SS and Lyubarsky
AL: Induction by cyclic GMP of cationic conductance in plasma
membrane of retinal rod outer segment. Nature. 313:310–313. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yau KW: Phototransduction mechanism in
retinal rods and cones. The friedenwald lecture. Invest Ophthalmol
Vis Sci. 35:9–32. 1994.PubMed/NCBI
|
|
23
|
Dryja TP, Finn JT, Peng YW, McGee TL,
Berson EL and Yau KW: Mutations in the gene encoding the alpha
subunit of the rod cGMP-gated channel in autosomal recessive
retinitis pigmentosa. Proc Natl Acad Sci USA. 92:10177–10181. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tosi J, Davis RJ, Wang NK, Naumann M, Lin
CS and Tsang SH: shRNA knockdown of guanylate cyclase 2e or cyclic
nucleotide gated channel alpha 1 increases photoreceptor survival
in a cGMP phosphodiesterase mouse model of retinitis pigmentosa. J
Cell Mol Med. 15:1778–1787. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Iwanami M, Oshikawa M, Nishida T,
Nakadomari S and Kato S: High prevalence of mutations in the EYS
gene in Japanese patients with autosomal recessive retinitis
pigmentosa. Invest Ophthalmol Vis Sci. 53:1033–1040. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gonzalez-del Pozo M, Borrego S, Barragan
I, Pieras JI, Santoyo J, Matamala N, Naranjo B, Dopazo J and
Antiñolo G: Mutation screening of multiple genes in Spanish
patients with autosomal recessive retinitis pigmentosa by targeted
resequencing. PLoS One. 6:e278942011. View Article : Google Scholar : PubMed/NCBI
|