Introduction
Nonalcoholic steatohepatitis (NASH) is the
inflammatory subtype nonalcoholic fatty liver disease (NAFLD). It
is a representative phenotype of metabolic syndrome (MetS) in the
liver and is involved in disease progression leading to cirrhosis
and need for liver transplantation (1–4).
Emerging evidences claim that the degree of hepatic fibrosis is
considered the most crucial predictor of overall mortality in
patients with NASH (5,6). This indicates that the ultimate
medical goal in the treatment of NASH would be to clinically
prevent the development of liver fibrosis. Thus, a variety of
pharmacological strategies have been proposed, but no
evidence-based pharmacotherapies have been established for
MetS-related liver fibrosis including NASH.
Plasminogen activator inhibitor-1 (PAI-1), a member
of the serine protease inhibitor (serpin) gene family, is the major
physiologic regulator of the plasmin-based pericellular proteolytic
cascade (7,8). PAI-1 is synthesized by numerous types
of cells including cardiac myocytes, lipocytes, vascular
endothelial cells, macrophages, and fibroblasts. It also acts as a
potent inhibitor of fibrinolysis by regulating urokinase
plasminogen activator (uPA)/tissue plasminogen activator
(tPA)/plasmin/matrix metalloproteinase (MMP) proteolytic activity,
and fibrin levels. It is therefore extensively involved in
fibrogenesis in multiple organs such as skin, lung, heart, kidney,
and liver (9–16). Indeed, fibrotic livers show a
significantly higher level of PAI-1 (13), while PAI-1 deficiency ameliorates
bile duct ligation-induced cholestatic liver fibrosis with
promotion of collagen degradation (14). A recent clinical report has
demonstrated that higher plasma levels of PAI-1 were strongly
associated with NASH severity in obese patients (15). Additionally, siRNA-mediated
inhibition of PAI-1 was shown to exert a protective effect on
murine liver fibrosis and to alleviate the fibrotic properties in
hepatic stellate cells (HSCs), a major component of the fibrotic
process in the liver (16). This
evidence, therefore, further supports the concept that PAI-1 is
possible to be suitable as a therapeutic target for the liver
fibrosis.
TM5275{5-chloro-2-[({2-[4-(diphenylmethyl)piperazin-1-yl]
−2-oxoethoxy}acetyl)amino]benzoate} is an orally bioavailable small
molecule PAI-1 inhibitor (17,18).
It exerts powerful antithrombotic activity in both rodents and
nonhuman primates (cynomolgus monkey) (17,18).
Of great importance, TM5275 does not interfere with other
serpin/serine protease systems including 1-antitrypsin/trypsin and
α2-antiplasmin/plasmin and causes no obvious toxicity in the liver,
kidney, hematopoietic system, central nervous system, or
cardiovascular system of rodents and primates (17–19).
Hence, it has no detrimental effects on hemostatic function, which
are the most common adverse effects of anticoagulation agents; this
suggests that TM5275 specifically acts on PAI-1 with limited
toxicity (17–19). Actually, TM5441, a derivative of
TM5275, has been reported to show a suppressive effect in murine
fatty liver models (20,21). However, the direct effect of TM5275
on liverfibrosis development under the condition of MetS in
conjunction with the proliferation and activation of HSCs has not
been investigated.
The present study aimed to explore the effect of the
PAI-1 inhibitor TM5275 in improving the pathology of MetS-based
fibrosis and to determine the therapeutic mechanisms in HSC biology
using two rat liver fibrosis models under the condition of MetS:
Choline-deficient, L-amino acid-defined diet (CDAA)-fed rats or
porcine serum (PS)-induced fibrotic Otsuka Long-Evans Tokushima
Fatty (OLETF) diabetic rats.
Materials and methods
Animals and reagents
Six-week-old male Fischer-344 rats, ten-week-old
male OLETF and Long-Evans Tokushima Otsuka (LETO), littermate
controls for OLETF, were purchased from Japan SLC, Inc. The PAI-1
inhibitor TM5275 was provided by Toshio Miyata (Tohoku University
Graduate School of Medicine). Recombinant rat PAI-1 was purchased
from PeproTech. Conventional chemical agents were obtained from
Nacalai Tesque. The rat HSC cell line HSC-T6 was purchased from the
Japanese Cancer Research Resources Bank.
Animal treatment
Two different in vivo models of liver
fibrosis were established in rats: CDAA-fed rats or PS-mediated
fibrotic OLETF rats. In the CDAA-fed model, the Fisher-344 rats
received a CDAA diet (CLEA Japan) for 12 weeks, and the treatment
group (n=6) was orally administered 50 mg/kg/day of TM5275 in the
drinking water. A positive control group (n=6) was administered the
same amount of vehicle during the CDAA feeding period, and the
negative control group (n=6) consisted of the rats fed a
choline-supplemented amino acid (CSAA) diet (CLEA Japan) and
treated with vehicle. In the PS-mediated fibrotic OLETF rats, the
OLETF rats were intraperitoneally administered 1 ml/kg PS (Rockland
Immunochemicals, Inc.) twice a week for 6 weeks. Treatment groups
(n=6 each) received administration of 50 or 100 mg/kg/day of TM5275
in the drinking water. A positive control group (n=6) was
administered the same amount of vehicle during the PS injection. As
a negative control group (n=6), LETO rats received a PS injection
and were treated with vehicle in the same manner as the positive
control group. All rats were housed in stainless steel mesh cages
under the following controlled conditions: Temperature: 23±3°C;
relative humidity: 50±20%; 10–15 air changes/h; and 12-h day/night
cycle. The rats had ad libitum access to tap water throughout the
study period. At the end of the experiment, all rats underwent the
following procedures: Euthanasia by intraperitoneal injection of
pentobarbital sodium (200 mg/kg), opening of the abdominal cavity,
blood collection via puncture of the aorta and harvesting of liver
for histological and molecular evaluation. Rats were subsequently
decapitated to assure for death.
Serum markers, such as alanine aminotransferase
(ALT), were assessed by routine laboratory methods. All animal
procedures were performed according to the criteria outlined in the
Guide for the Care and Use of Laboratory Animals prepared by the
National Academy of Sciences. All experiments were approved by the
Animal Care and Use Committee of Nara Medical University (Protocol
no. 10034).
Estimation of glycemic status
At the experiment's conclusion, insulin sensitivity
and insulin resistance were evaluated according to the quantitative
insulin sensitivity check index (QUICKI) and the homeostasis model
assessment of insulin resistance (HOMA-IR), respectively, as
previously described (22).
Immunohistochemical staining and
semi-quantification
Liver specimens were fixed in 10% formalin and
embedded in paraffin. Sections of 5-µm thickness were stained with
hematoxylin and eosin (H&E) and Sirius-Red. Histological scores
for steatosis, lobular inflammation and hepatocyte ballooning were
calculated according to the NAFLD Activity Score (23); steatosis (0; <5%, 1; 5–33%, 2;
>33–66%, 3; >66%), lobular inflammation (0; no foci, 1; <2
foci/200-fold, 2; 2–4 foci/200-fold, 3; >4 foci/200-fold),
hepatocyte ballooning (0; none, 1; few balloon cells, 2; many
cells/prominent ballooning). These scoring were performed for 5
fields per each section. A primary antibody against α-SMA
(ab124964; 1:1,000 dilution) (Abcam) was applied for
immunostaining. Specifically, staining was performed according to
the supplier's recommendations. The quantitative analyses for liver
fibrosis and α-SMA-positive area were performed for 5 fields per
each section in high-power fields at 400-fold magnification by
applying NIH ImageJ software.
Measurement of protein levels of
hepatic collagen content and TGF-β1
After equalizing the protein concentration from
frozen liver samples to 200 mg, hepatic collagen content and TGF-β1
protein were measured using Sircol Soluble Collagen Assays
(Biocolor Ltd.) and TGF-β1 ELISA kits (Bender MedSystems GmbH)
according to the manufacturer's instructions.
Cell culture and WST-1 assay
Rat HSCs (HSC-T6) (cat no. SCC069) were purchased
from Merck KGaA. Cells were cultured in Dulbecco's Modified Eagle's
Medium (Nacalai) supplemented with 10% fetal bovine serum and 2 mM
l-glutamine in a 95% air/5% CO2 humidified atmosphere at
37°C. Mycoplasma testing was performed using MycoProbe®
Mycoplasma Detection kit (R&D Systems, Inc.) according to the
manufacturer's protocol. For the assay, HSC-T6 cells
(1×104) were seeded in a 96-well plate. Subsequently,
the cells were treated with different concentrations of recombinant
rat PAI-1 or 10 ng/ml of rat TGF-β1 (Abcam) and/or different doses
of TM5275 for 24 h. Premix WST-1 Cell Proliferation Assay System
(Takara Bio Inc.) was applied for cell proliferation analysis.
RNA extraction and real-time
polymerase chain reaction
Total RNA was isolated from the liver tissues and
HSC-T6 cells using a RNeasy Mini kit (Qiagen). HSC-T6 cells were
incubated in the presence or absence of rat TGF-β1 (10 ng/ml)
and/or TM5275 (100 µM) for 12 h. The mRNA levels of Tgfb1,
Col1a1, and Serpine1 were measured by quantitative
polymerase chain reaction (qPCR) using an Applied Biosystems
StepOnePlus™ Real-Time PCR® system (Applied Biosystems).
Primer sequences were as follows: Tgfb1, forward
5′-CGGCAGCTGTACATTGACTT-3′ and reverse 5′-AGCGCACGATCATGTTGGAC-3′;
Col1a1, forward 5′-AGCTCCTGGGCCTATCTGATGA-3′ and reverse
5′-AATGGTGCTCTGAAACCCTGATG-3′; and Serpine1, forward
5′-AGGGGCAGCAGATAGACAGA-3′ and reverse 5′-CACAGGGAGACCCAGGTAAA-3′.
Relative gene expression levels were determined using
glyceraldehyde-3-phosphate dehydrogenase (Gapdh): Forward
5′-CGACCACTTTGTCAAGCTCA-3′ and reverse 5′-AGGGGAGATTCAGTGTGGTG-3′;
as the internal control. The predicted cycle threshold (CT) values
were exported for analysis. All reactions were performed using a
1:10 diluted cDNA while mRNA expression levels were estimated using
the 2ΔΔCT method by normalization to Gapdh and
showed relative values to each control group.
Protein extraction and western
blotting
Proteins were extracted from 106 cultured
HSC-T6 cells. HSC-T6 cells were incubated in the presence or
absence of rat TGF-β1 (10 ng/ml) and/or TM5275 (100 µM) for 12 h.
For this purpose, T-PER Tissue Protein Extraction Reagent as lysis
buffer supplemented with proteinase and phosphatase inhibitors
(Thermo Fisher Scientific Inc.) was used. The protein concentration
was measured by protein assay (BioRad), and all samples were
normalized to 50 µg. Cellular proteins were separated by SDS-PAGE
(Thermo Fisher Scientific Inc.) and transferred to an Invitrolon
PVDF membrane (Thermo Fisher Scientific Inc.). And then membranes
were blocked with 5% bovine serum albumin in Tris-buffered saline
with Tween-20 for 1 h. The AKT (#9272; 1:1,000 dilution),
phosphorylated AKT (Ser473) (p-AKT) (#9271; 1:1,000 dilution),
extracellular signal-regulated kinase (ERK1/2) (#9102; 1:1,000
dilution), phosphorylated ERK1/2 (Thr202/Tyr204) (p-ERK1/2) (#4370;
1:1,000 dilution), or SMAD2/3 (#3102; 1:1,000 dilution),
phosphorylated SMAD2 (Ser465/Ser467)/3 (Ser423/425) (p-SMAD2/3)
(#8828; 1:1,000 dilution), GAPDH (#5174; 1:1,000 dilution) and
β-actin (Actin) (#4967; 1:10,000 dilution) (Cell Signaling
Technology) were used as primary antibodies Amersham ECL IgG,
HRP-linked F(ab)2 fragment (GE Healthcare Life Sciences; 1:5,000
dilution) was applied as a secondary antibody. The bands were
visualized using Clarity Western ECL Substrate (BioRad).
Statistical analysis
The data were subjected to a one-way analysis of
variance followed by Bonferroni's multiple-comparison test, where
appropriate. Bartlett's test was used to determine the homology of
variance. All tests were two-tailed, and P<0.05 was considered
to indicate a statistically significant difference.
Results
TM5275, a PAI-1 inhibitor, attenuates
the progression of liver fibrosis in CDAA-fed rats
Initially, we assessed the effect of a PAI-1
inhibitor in the in vivo development of liver fibrosis in
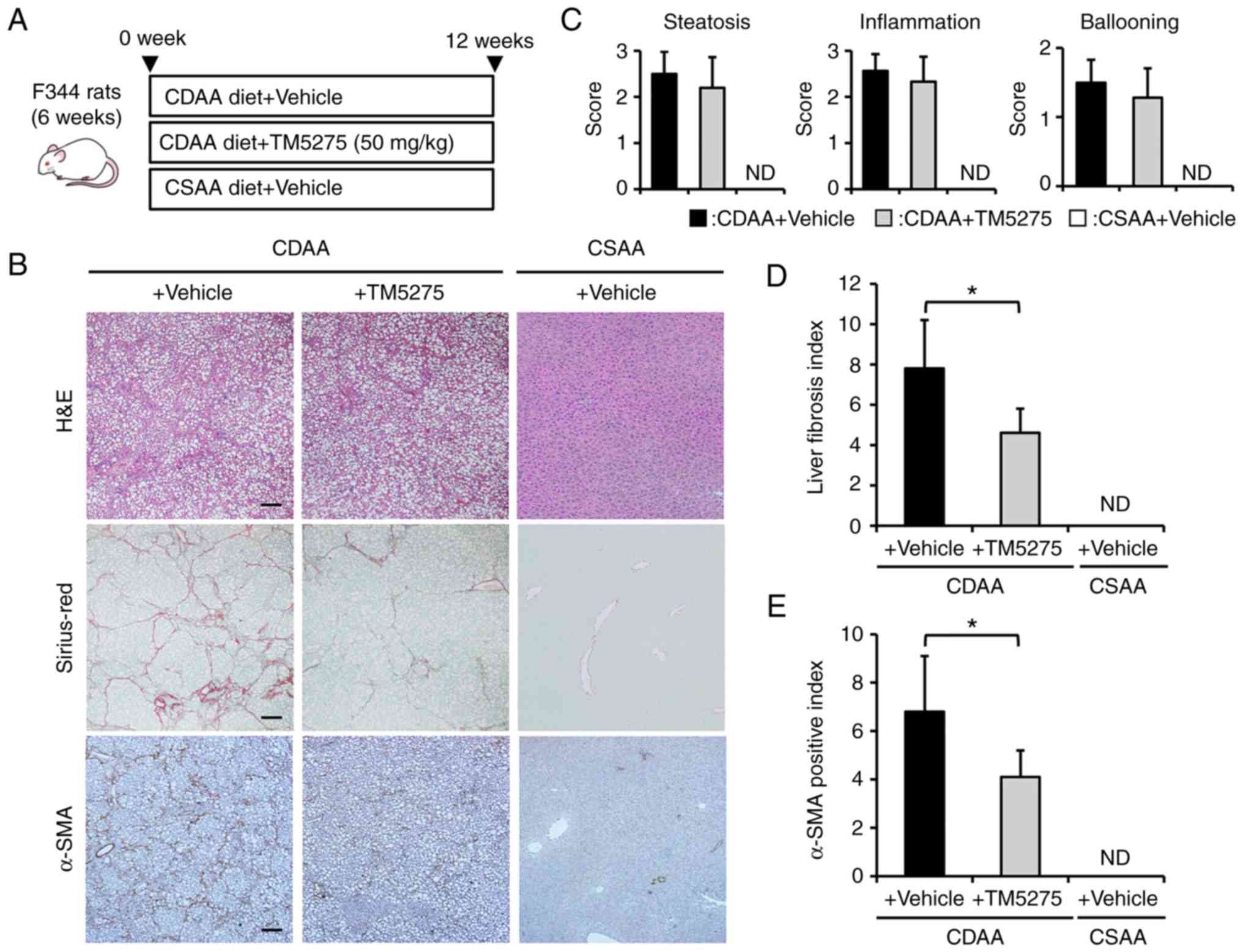
CDAA-fed rats (Fig. 1A). As shown
in Fig. 1B, 12 weeks of CDAA
feeding induced prominent hepatic steatosis and fibrosis with
collagen fiber deposition, which were histologically indicated by
H&E and Sirius-Red staining, respectively, as principal
phenotypic changes of NASH. Moreover, immunohistochemistry for
α-SMA revealed that CDAA-fed mice showed an increase in
immunopositive activated HSCs compared with CSAA-fed negative
control rats. Continuous oral administration of TM5275, a
pharmacological PAI-1 inhibitor, did not affect the histological
changes in CDAA-fed rats according to the NAFLD activity score,
which includes steatosis, lobular inflammation, and hepatocyte
ballooning (Fig. 1B and C). By
contrast, treatment with TM5275 significantly attenuated liver
fibrosis development in CDAA-fed rats (Fig. 1B). Computer-assisted
semiquantitative analysis demonstrated a decrease to one-half the
fibrotic area of the liver in CDAA-fed rats treated with TM5275
compared with vehicle-treated rats (Fig. 1D). The CDAA-fed rats who were
administered TM5275 also showed a marked decrease in
α-SMA-immunopositive areas compared with those treated with vehicle
in accordance with attenuated liver fibrosis (Fig. 1E). This suppressed fibrogenesis
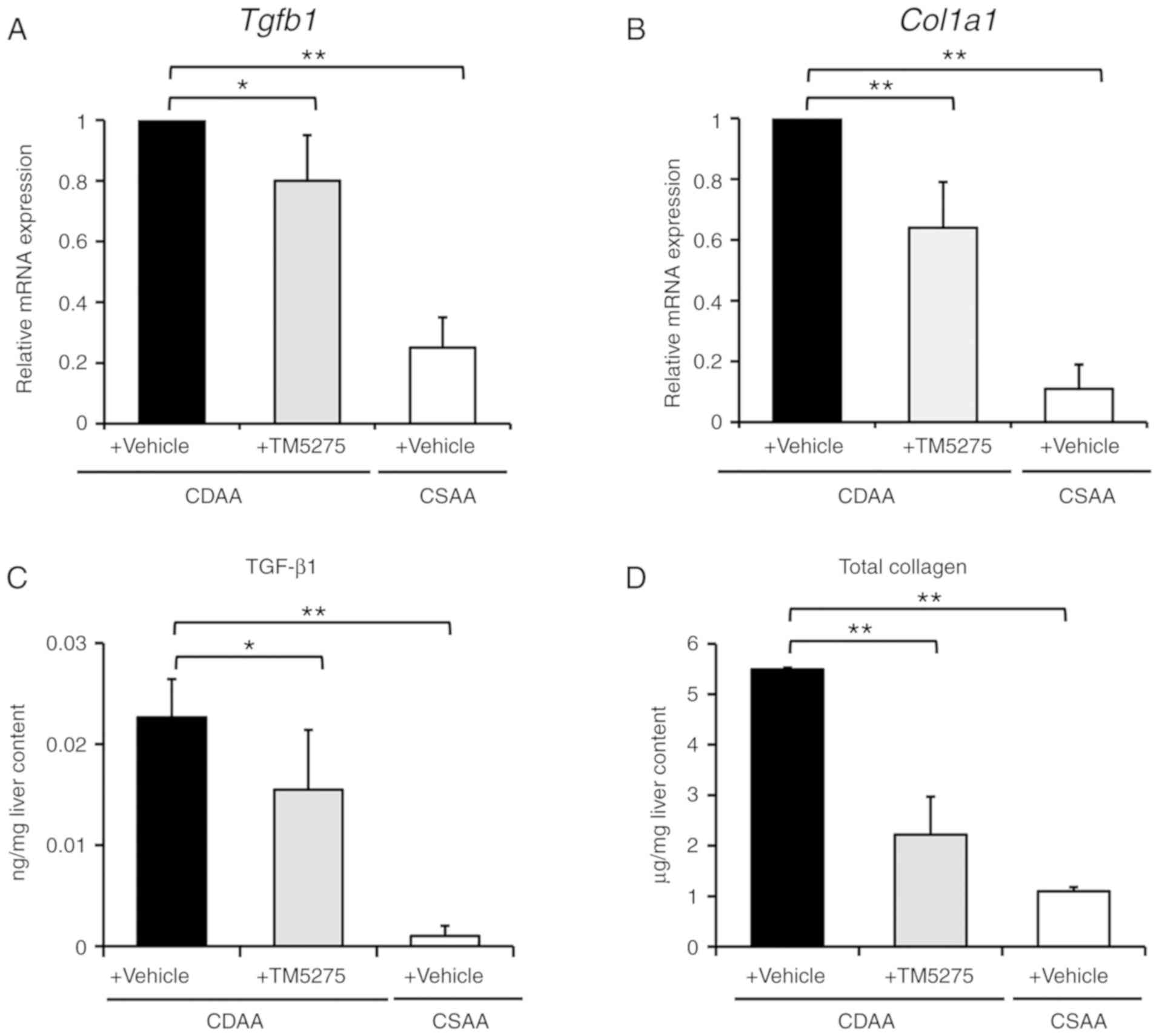
coincided with decreases in the hepatic expression of profibrogenic
genes, including Tgfb1 and Col1a1 (Fig. 2A and B), and reduced hepatic levels
of TGF-β1 and total collagen (Fig. 2C
and D). These findings suggest that the PAI-1 inhibitor TM5275
can potentially inhibit liver fibrosis development without
affecting hepatic steatosis and inflammation, especially in
CDAA-induced steatohepatitis.
TM5275 exerts an inhibitory effect on
porcine serum-induced liver fibrosis in diabetic OLETF rats
To evaluate whether the PAI-1 inhibitor TM5275
exerts antifibrotic effects independently of its antisteatotic and
anti-inflammatory properties, we next examined the effect of TM5275
on porcine serum (PS)-induced liver fibrosis development in OLETF
genetically engineered diabetic rats (Fig. 3A). We have reported that this model
could induce progression of liver fibrosis with limited hepatic
steatosis and inflammation (24).
As shown in Fig. 3B, OLETF rats
showed impaired insulin sensitivity and insulin resistance, which
were calculated according to QUICKI and HOMA-IR, respectively, and
these impairments remained unchanged by 12 weeks of treatment with
TM5275. Moreover, OLETF rats showed a mild elevation in serum
alanine aminotransferase (ALT) as a result of hepatic steatosis,
and neither low-nor high-dose treatments with TM5275 significantly
altered the increased serum ALT level in OLETF rats. As previously
reported, PS administration induced a profound progression of liver
fibrosis in diabetic OLETF rats (Fig.
3C). Despite the above insufficiencies in terms of liver enzyme
levels and glucose tolerance, it was noteworthy that TM5275 could
remarkably suppress PS-mediated liver fibrosis development in OLETF
rats in a dose-dependent manner (Fig.
3C and D). As in the CDAA-fed rats, α-SMA-immunopositive areas
were decreased along with attenuation of liver fibrosis in
PS-mediated rats who were administered TM5275 (Fig. 3C and D). These data support the
hypothesis that a PAI-1 inhibitor attenuates liver fibrosis through
direct action upon activated HSCs.
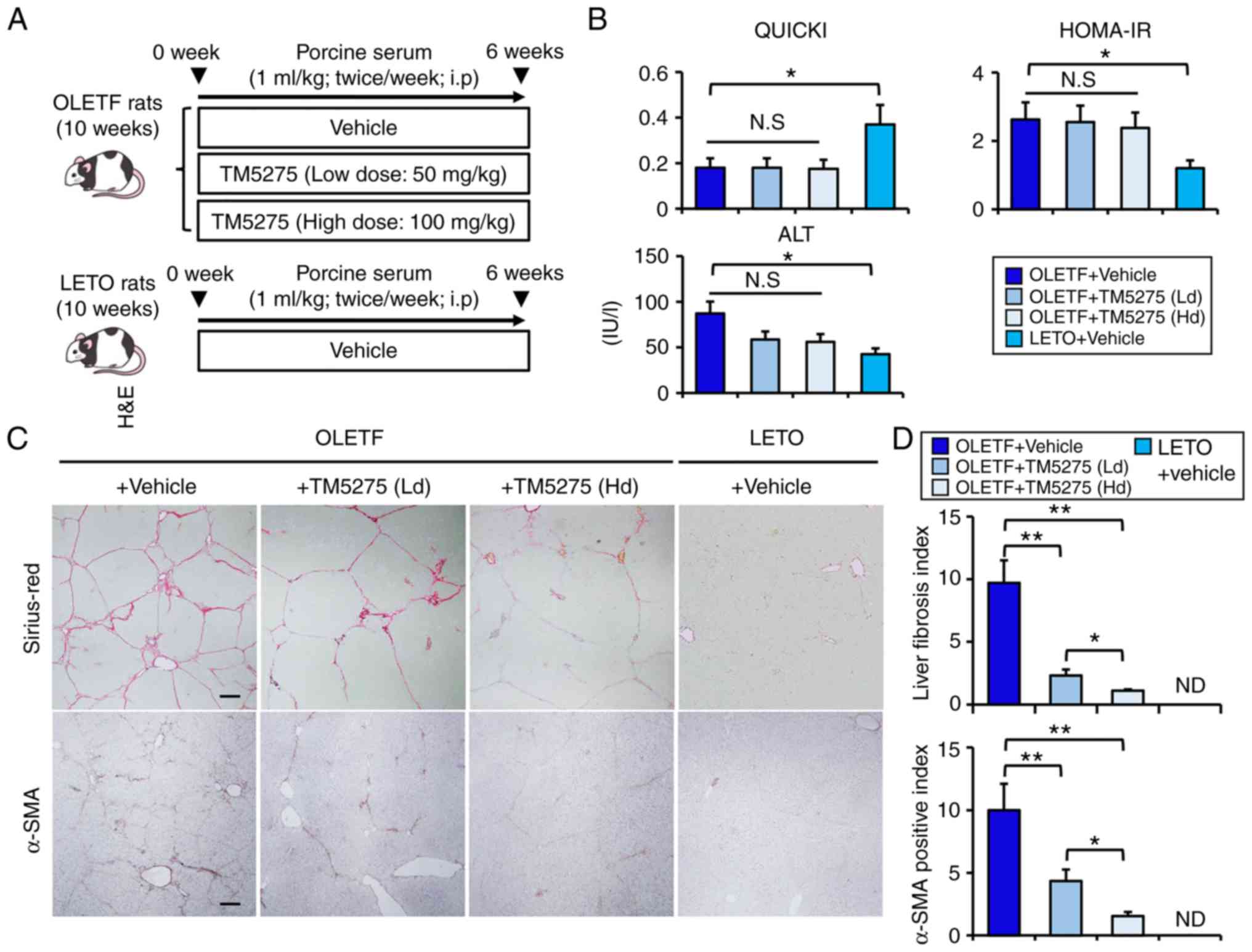 | Figure 3.Effects of TM5275 on porcine
serum-induced liver fibrosis in diabetic OLETF rats. (A) Schematic
of porcine serum-induced liver fibrosis in diabetic rat models. (B)
The values of QUICKI, HOMA-IR and serum levels of ALT in
experimental rats. (C) Representative images of liver sections
stained with Sirius-Red and α-SMA. Scale bar, 50 µm. (D)
Semi-quantitation of Sirius Red- and α-SMA-positive area by
National Institutes of Health ImageJ software (version 1.52). All
quantitative analyses were performed for 5 fields per each section
in high-power fields at ×400 magnification. Data are presented as
the mean ± SD (n=6). *P<0.05 and **P<0.01 as indicated.
QUICKI, Quantitative Insulin Sensitivity Check Index; HOMA-IR,
homeostasis model assessment-insulin resistance; ALT, alanine
aminotransferase; SMA, smooth muscle actin; Ld, low dose; Hd, high
dose; N.S., not significant; ND, not detected; OLETF, Otsuka
Long-Evans Tokushima Fatty; LETO, Long-Evans Tokushima Otsuka; wk,
week. |
TM5275 suppresses proliferative and
fibrogenic activities in rat activated hepatic stellate cells
Next, we examined the direct effect of TM5275 on
HSC-T6 cells, a rat HSC line, in vitro to elucidate the
molecular mechanism underlying the antifibrogenic activity of this
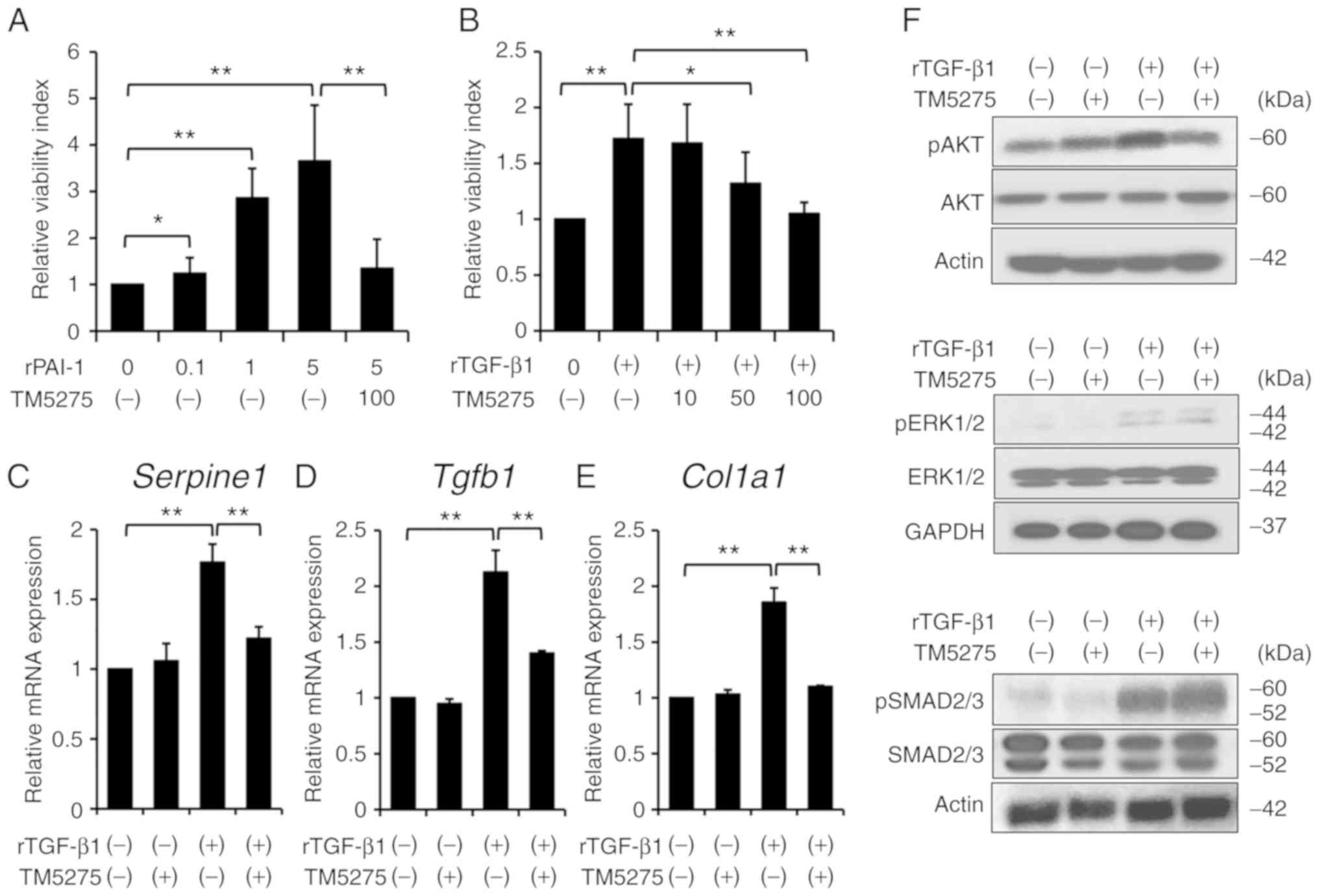
compound. A cell proliferation assay revealed that recombinant
PAI-1 stimulated the proliferation of HSC-T6 cells in a
dose-dependent manner, which was efficiently suppressed by
treatment with TM5275 (Fig. 4A).
Interestingly, TM5275 also suppressed recombinant TGF-β1
(rTGF-β1)-mediated HSC-T6 proliferation in a dose-dependent manner
(Fig. 4B). Therefore, we next
assessed the interaction between TGF-β1 and PAI-1 in HSC-T6 cells
and found that an rTGF-β1-mediated stimulus upregulated mRNA
expression of Serpine1, the gene that encodes PAI-1
(Fig. 4C). Moreover, TM5275
significantly inhibited rTGF-β1-stimulated upregulation of
Serpine1 mRNA expression (Fig.
4C). These results indicate that TM5275 has potential to
suppress HSC-T6 cell proliferation by inhibiting the upregulation
of PAI-1 stimulated by TGF-β1. In addition to its effect on cell
proliferation, TM5275 markedly repressed the rTGF-β1-induced
increases in the mRNA levels of the fibrosis-related genes
Tgfb1 and Col1a1 (Fig.
4D and E). Analysis of intracellular signaling revealed that
rTGF-β1 increased the protein expression of pAKT, pERK1/2, and
pSMAD2/3 in HSC-T6 cells, whereas treatment with TM5275 could
suppress rTGF-β-stimulated phosphorylation of AKT but not that of
ERK1/2 and SMAD2/3 (Fig. 4F).
Discussion
Currently, a variety of medical approaches targeting
MetS-related liver fibrosis are in the pipeline. Increasing
evidence for the ability to address the reversibility of liver
fibrosis has led to intense interest in comprehending the
regulation of collagen degradation and resolution. It has been
recognized that liver fibrosis is characterized by the
transdifferentiation of HSCs into activated myofibroblasts,
proliferative arrest of hepatocytes, and the accumulation of
extracellular matrix (ECM) (25).
Thus, a reversion of these sequences has been emphasized as major
treatment strategies for liver fibrosis. In the present study, we
demonstrated that TM5275, a novel PAI-1 inhibitor, could attenuate
liver fibrosis in two mechanistically different models of liver
fibrosis, CDAA-fed NASH rats and PS-mediated diabetic rats. The
CDAA-fed rats characteristically display a steatohepatitis-based
liver fibrosis while they don't show a typical diabetic status
including insulin resistance and hyperglycemia (26). On the other hand, PS-mediated
diabetic rats develop liver fibrosis with insulin resistance and
diabetes, but they show a limited steatosis and inflammation
(24). In our results, TM5275
attenuated liver fibrosis development with decreased expressions of
α-SMA in both models without affecting steatosis and inflammation
in CDAA-fed rats as well as insulin resistance in PS-mediated
diabetic rats. These suggest the possibility that TM5275 directly
affect liver fibrosis development via HSC activation under the MetS
condition.
Several studies have suggested that PAI-1 could
potentially affect both collagen production and degradation
(27–29). It has also been reported that HSCs
are a key source of PAI-1 and that there is an imbalance between
PAs and PAI-1 at different stages of liver fibrosis development; at
an early stage, uPA and expression of its receptor are
predominantly elevated, while during the cirrhotic stage, increased
expression of PAI-1 hampers uPA activity, which results in the
inhibition of plasmin generation and attenuation of excess ECM
degradation (30–32). Moreover, PAI-1 has also been shown
to regulate the proliferation, adhesion, and migration of various
types of cells, which stimulates the migration of leukocytes and
collagen-producing cells into damaged tissue (27–29).
This indicates that PAI-1 itself might also promote ECM synthesis.
Actually, our data from in vitro experiments demonstrated
that stimulation of recombinant PAI-1 induced the proliferation of
HSC-T6 cells in a dose-dependent manner, which was abrogated by
treatment with TM5275. These results support the idea that PAI-1
directly augmented the proliferative activity of HSC.
Furthermore, PAI-1 gene expression is regulated by
various biological factors including TGF-β, IL-1β, epidermal growth
factor, insulin-like growth factor 1, platelet-derived growth
factor, and basic fibroblast growth factor (33–37).
Of the numerous key regulatory factors, TGF-β1 is a principle
mediator of HSC activation (25).
Previous basic studies of the genetic overexpression of TGF-β have
revealed a vital contribution of TGF-β1 to HSC activation and
fibrogenesis (38,39). The TGF-β1 signaling pathway is
mediated by the SMAD family, but other effectors such as
phosphatidylinositol-3 kinase (PI3K), mitogen-activated protein
kinase (MAPK), and nuclear factor κB (NF-κB), which also have key
roles in cell proliferation, apoptosis, differentiation and ECM
synthesis may be involved, and can independently regulate SMAD
expression (40). Importantly,
upon its activation and trans-differentiation, TGF-β1
transcriptionally controls the gene expression of PAI-1 as well as
that of α1 and α2 type 1 procollagen, and tissue inhibitor of
metalloproteinase-1 and −2 (25).
It has been confirmed that TGF-β1 induces PAI-1 synthesis, and a
TGF-β1 response element has been found in the promoter of the PAI-1
gene; in addition, multiple lines of evidence have determined that
PAI-1 expression might be regulated in connection with several
signaling pathways. For instance, continuous stimulation of TGF-β
prominently induced not only phosphorylation of Smad2/3 but also
upregulation of PAI-1 (41,42).
Moreover, TGF-β also stimulates the activation of c-Src kinase,
which triggers to phosphorylation of caveolin-1, an upstream
repressor of EGFR and the RhoA/ROCK pathway and leads to upregulate
PAI-1 expression (43).
Additionally, in another study, TGF-β administration induced
progressive fibrosing steatohepatitis in mice, and interestingly,
this process was reversed by a PPARα agonist, which functioned via
AMPK-mediated induction of SHP gene expression with a marked
decrease in the mRNA and protein expression of PAI-1 and other
fibrotic markers (44). In line
with these findings, the results of our in vitro experiments
showed that stimulation with recombinant TGF-β1 significantly
increased Serpine1 gene expression in HSC-T6 cells, which
was abrogated by treatment with TM5275. Consistently, with
decreased Serpine1 expression, TM5275 significantly
suppressed TGF-β1-stimulated HSCs proliferation. Interestingly, we
discovered that TM5275 inhibited the phosphorylation of AKT
stimulated by TGF-β1, but it did not alter the phosphorylation of
ERK1/2 and SMAD2/3. We speculate the possibility that TM5275 may
directly PAI-1/PI3K/AKT pathway in HSC-T6 cells because of its
suppressive effect PAI-1-stimulated HSC-T6 cell viability. The
definitive molecular mechanism responsible for these heterogenous
effects of TM5275 on TGF-β1 signaling in HSCs has been uncertain
until now, as PAI-1 seemed to be multifunctional and have multiple
interactions.
Several limitations are apparent in the present
study. First, the functional role of PAI-1 in liver fibrosis
development is still controversial, although PAI-1 has been found
to be involved in the process of fibrotic progression in various
tissues. For example, a recent report showed that either genetic
deletion or pharmacologic inhibition of PAI-1 ameliorated
methionine- and choline-deficient diet-induced hepatic steatosis
but did not affect hepatic inflammation or fibrosis (21). These conflicting effects of PAI-1
inhibition on steatohepatitis suggest that the impact of PAI-1
regulation on tissue fibrosis, especially in the liver, is highly
dependent on the tissue type and experimental model. Second, this
study elucidated the preventive effects of a PAI-1 inhibitor on the
development of liver fibrosis, while the pharmacological properties
of fibrinolysis and liver regeneration in an established model of
liver fibrosis are still obscure. Future studies should address
whether TM5275 could induce fibrinolysis and efficient liver
regeneration in other models of cirrhosis.
Taken together, our data show that TM5275, a novel
PAI-1 inhibitor, appears to have a protective effect on liver
fibrosis development in rat fibrosis models under the condition of
MetS. Notably, this agent has the potential to directly suppress
HSC proliferation and profibrogenic activity by blockade of
TGF-β/PAI-1 binding. These findings suggest a possibility that
TM5275 confers a clinical benefit for liver fibrosis with MetS, but
there is a major issue to be addressed for future clinical
application. Representatively, this agent has a risk to foster the
bleeding tendency especially in cirrhotic patients because of its
antithrombotic action. Further studies are warranted to determine
the influences of PAI-1 inhibitor on other organs in chronic liver
diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by Japan Society for
the Promotion of Science KAKENHI (grant no. JP2646101).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Author's contributions
RN, KK and HY conceived and designed the current
study, and outlined the proof. RN, KK and MK designed the model and
the computational framework, and analyzed the data. RN, TN, KM, HK,
MK, HT, YA, AD, KA and NN contributed to animal breeding, sample
collection and performed the experiments. RN and KK validated the
results. TM provided the TM5275 compound and interpreted the data.
RN and KK wrote the manuscript in consultation with TM and HY. HY
supervised the current study. RN acquired the funding. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All animal procedures were performed according to
the criteria outlined in the Guide for the Care and Use of
Laboratory Animals prepared by the National Academy of Sciences.
All experiments were approved by the Animal Care and Use Committee
of Nara Medical University (protocol no. 10034).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Farrell GC and Larter CZ: Nonalcoholic
fatty liver disease: From steatosis to cirrhosis. Hepatology.
43:2948–S112. 2006. View Article : Google Scholar
|
|
2
|
Angulo P: Nonalcoholic fatty liver
disease. N Engl J Med. 346:1221–1231. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sanyal AJ, Neuschwander-Tetri BA and
Tonascia L: End points must be clinically meaningful for drug
development in nonalcoholic fatty liver disease. Gastroenterology.
150:11–13. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Younossi ZM, Koenig AB, Abdelatif D, Fazel
Y, Henry L and Wymer M: Global epidemiology of nonalcoholic fatty
liver disease-Meta-analytic assessment of prevalence, incidence,
and outcomes. Hepatology. 64:73–84. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dulai PS, Singh S, Patel J, Soni M, Prokop
LJ, Younossi Z, Sebastiani G, Ekstedt M, Hagstrom H, Nasr P, et al:
Increased risk of mortality by fibrosis stage in nonalcoholic fatty
liver disease: Systematic review and meta-analysis. Hepatology.
65:1557–1565. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Angulo P, Kleiner DE, Dam-Larsen S, Adams
LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC,
Lafferty HD, Stahler A, et al: Liver fibrosis, but No other
histologic features, is associated with long-term outcomes of
patients with nonalcoholic fatty liver disease. Gastroenterology.
149:389–397 e10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Erickson LA, Ginsberg MH and Loskutoff DJ:
Detection and partial characterization of an inhibitor of
plasminogen activator in human platelets. J Clin Invest.
74:1465–1472. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Providence KM and Higgins PJ: PAI-1
expression is required for epithelial cell migration in two
distinct phases of in vitro wound repair. J Cell Physiol.
200:297–308. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Toriseva M and Kähäri VM: Proteinases in
cutaneous wound healing. Cell Mol Life Sci. 66:203–224. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lazar MH, Christensen PJ, Du M, Yu B,
Subbotina NM, Hanson KE, Hansen JM, White ES, Simon RH and Sisson
TH: Plasminogen activator inhibitor-1 impairs alveolar epithelial
repair by binding to vitronectin. Am J Respir Cell Mol Biol.
31:672–678. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takeshita K, Hayashi M, Iino S, Kondo T,
Inden Y, Iwase M, Kojima T, Hirai M, Ito M, Loskutoff DJ, et al:
Increased expression of plasminogen activator inhibitor-1 in
cardiomyocytes contributes to cardiac fibrosis after myocardial
infarction. Am J Pathol. 164:449–456. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma LJ and Fogo AB: PAI-1 and kidney
fibrosis. Front Biosci (Landmark Ed). 14:2028–2041. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Clouthier DE, Comerford SA and Hammer RE:
Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like
syndrome in PEPCK-TGF-beta1 transgenic mice. J Clin Invest.
100:2697–2713. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang H, Zhang Y and Heuckeroth RO: PAI-1
deficiency reduces liver fibrosis after bile duct ligation in mice
through activation of tPA. FEBS Lett. 581:3098–3104. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ajmera V, Perito ER, Bass NM, Terrault NA,
Yates KP, Gill R, Loomba R, Diehl AM and Aouizerat BE; NASH
Clinical Research Network, : Novel plasma biomarkers associated
with liver disease severity in adults with nonalcoholic fatty liver
disease. Hepatology. 65:65–77. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu PF, Chen H, Zhong W, Lin Y, Zhang X,
Chen YX and Xie WF: Adenovirus-mediated transfer of siRNA against
PAI-1 mRNA ameliorates hepatic fibrosis in rats. J Hepatol.
51:102–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Izuhara Y, Yamaoka N, Kodama H, Dan T,
Takizawa S, Hirayama N, Meguro K, van Ypersele de Strihou C and
Miyata T: A novel inhibitor of plasminogen activator inhibitor-1
provides antithrombotic benefits devoid of bleeding effect in
nonhuman primates. J Cereb Blood Flow Metab. 30:904–912. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tashiro Y, Nishida C, Sato-Kusubata K,
Ohki-Koizumi M, Ishihara M, Sato A, Gritli I, Komiyama H, Sato Y,
Dan T, et al: Inhibition of PAI-1 induces neutrophil-driven
neoangiogenesis and promotes tissue regeneration via production of
angiocrine factors in mice. Blood. 119:6382–6393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Imai J, Yahata T, Ichikawa H, Ibrahim AA,
Yazawa M, Sumiyoshi H, Inagaki Y, Matsushima M, Suzuki T, Mine T,
Ando K, et al: Inhibition of plasminogen activator inhibitor-1
attenuates against intestinal fibrosis in mice. Intest Res.
18:219–228. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee SM, Dorotea D, Jung I, Nakabayashi T,
Miyata T and Ha H: TM5441, a plasminogen activator inhibitor-1
inhibitor, protects against high fat diet-induced non-alcoholic
fatty liver disease. Oncotarget. 8:89746–89760. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Henkel AS, Khan SS, Olivares S, Miyata T
and Vaughan DE: Inhibition of plasminogen activator inhibitor 1
attenuates hepatic steatosis but does not prevent progressive
nonalcoholic steatohepatitis in mice. Hepatol Commun. 2:1479–1492.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Katz A, Nambi SS, Mather K, Baron AD,
Follmann DA, Sullivan G and Quon MJ: Quantitative insulin
sensitivity check index: A simple, accurate method for assessing
insulin sensitivity in humans. J Clin Endocrinol Metab.
85:2402–2410. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kleiner DE, Brunt EM, Van Natta M, Behling
C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS,
Unalp-Arida A, et al: Design and validation of a histological
scoring system for nonalcoholic fatty liver disease. Hepatology.
41:1313–1321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kaji K, Yoshiji H, Kitade M, Ikenaka Y,
Noguchi R, Yoshii J, Yanase K, Namisaki T, Yamazaki M, Moriya K, et
al: Impact of insulin resistance on the progression of chronic
liver diseases. Int J Mol Med. 22:801–808. 2008.PubMed/NCBI
|
|
25
|
Tsuchida T and Friedman SL: Mechanisms of
hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol.
14:397–411. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ozutsumi T, Namisaki T, Shimozato N, Kaji
K, Tsuji Y, Kaya D, Fujinaga Y, Furukawa M, Nakanishi K, Sato S, et
al: Combined treatment with sodium-glucose cotransporter-2
inhibitor (Canagliflozin) and dipeptidyl peptidase-4 inhibitor
(teneligliptin) alleviates NASH progression in A non-diabetic rat
model of steatohepatitis. Int J Mol Sci. 21:21642020. View Article : Google Scholar
|
|
27
|
Gleizes PE, Munger JS, Nunes I, Harpel JG,
Mazzieri R, Noguera I and Rifkin DB: TGF-beta latency: Biological
significance and mechanisms of activation. Stem Cells. 15:190–197.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mulligan-Kehoe MJ, Schwartz GN and
Zacharski LR: The functions of plasminogen activator inhibitor-1:
Do we have all the pieces of PAI? Thromb Res. 117:483–486. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bajou K, Noël A, Gerard RD, Masson V,
Brunner N, Holst-Hansen C, Skobe M, Fusenig NE, Carmeliet P, Collen
D and Foidart JM: Absence of host plasminogen activator inhibitor 1
prevents cancer invasion and vascularization. Nat Med. 4:923–928.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang LP, Takahara T, Yata Y, Furui K, Jin
B, Kawada N and Watanabe A: Increased expression of plasminogen
activator and plasminogen activator inhibitor during liver
fibrogenesis of rats: Role of stellate cells. J Hepatol.
31:703–711. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Seki T, Imai H, Uno S, Ariga T and
Gelehrter TD: Production of tissue-type plasminogen activator
(t-PA) and type-1 plasminogen activator inhibitor (PAI-1) in mildly
cirrhotic rat liver. Thromb Haemost. 75:801–807. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Leyland H, Gentry J, Arthur MJ and Benyon
RC: The plasminogen-activating system in hepatic stellate cells.
Hepatology. 24:1172–1178. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dennler S, Itoh S, Vivien D, ten Dijke P,
Huet S and Gauthier JM: Direct binding of Smad3 and Smad4 to
critical TGF beta-inducible elements in the promoter of human
plasminogen activator inhibitor-type 1 gene. EMBO J. 17:3091–3100.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Okada H, Woodcock-Mitchell J, Mitchell J,
Sakamoto T, Marutsuka K, Sobel BE and Fujii S: Induction of
plasminogen activator inhibitor type 1 and type 1 collagen
expression in rat cardiac microvascular endothelial cells by
interleukin-1 and its dependence on oxygen-centered free radicals.
Circulation. 97:2175–2182. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Crandall DL, Groeling TM, Busler DE and
Antrilli TM: Release of PAI-1 by human preadipocytes and adipocytes
independent of insulin and IGF-1. Biochem Biophys Res Commun.
279:984–988. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wilkins-Port CE, Ye Q, Mazurkiewicz JE and
Higgins PJ: TGF-beta1 + EGF-initiated invasive potential in
transformed human keratinocytes is coupled to a
plasmin/MMP-10/MMP-1-dependent collagen remodeling axis: Role for
PAI-1. Cancer Res. 69:4081–4091. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Su EJ, Fredriksson L, Schielke GP,
Eriksson U and Lawrence DA: Tissue plasminogen activator-mediated
PDGF signaling and neurovascular coupling in stroke. J Thromb
Haemost. 7 (Suppl 1):S155–S158. 2009. View Article : Google Scholar
|
|
38
|
Hellerbrand C, Stefanovic B, Giordano F,
Burchardt ER and Brenner DA: The role of TGFbeta1 in initiating
hepatic stellate cell activation in vivo. J Hepatol. 30:77–87.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kanzler S, Lohse AW, Keil A, Henninger J,
Dienes HP, Schirmacher P, Rose-John S, zum Büschenfelde KH and
Blessing M: TGF-beta1 in liver fibrosis: An inducible transgenic
mouse model to study liver fibrogenesis. Am J Physiol.
276:G1059–G1068. 1999.PubMed/NCBI
|
|
40
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Motojima M, Kakuchi J and Yoshioka T:
Association of TGF-beta signaling in angiotensin II-induced PAI-1
mRNA upregulation in mesangial cells: Role of PKC. Biochim Biophys
Acta. 1449:217–226. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dong C, Zhu S, Alvarez RJ and
Goldschmidt-Clermont PJ: Angiotensin II induces PAI-1 expression
through MAP kinase-dependent, but TGF beta and PI3
kinase-independent pathway. J Heart Lung Transplant. 20:226–227.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Samarakoon R, Higgins SP, Higgins CE and
Higgins PJ: TGF-beta1-induced plasminogen activator inhibitor-1
expression in vascular smooth muscle cells requires pp60(c-src)/
EGFR(Y845) and Rho/ROCK signaling. J Mol Cell Cardiol. 44:527–538.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chanda D, Lee CH, Kim YH, Noh JR, Kim DK,
Park JH, Hwang JH, Lee MR, Jeong KH, Lee IK, et al: Fenofibrate
differentially regulates plasminogen activator inhibitor-1 gene
expression via adenosine monophosphate-activated protein
kinase-dependent induction of orphan nuclear receptor small
heterodimer partner. Hepatology. 50:880–892. 2009. View Article : Google Scholar : PubMed/NCBI
|