Introduction
Van der Woude Syndrome (VWS; OMIM. no. 119300,
http://www.omim.org/) is a prevalent form of
orofacial cleft syndrome, accounting for ~2% of all cleft lip (CL)
and cleft palate (CP) cases, and with an incidence of
1:35,000-1:100,000 worldwide (1,2).
Congenital lip pits, simultaneously occurring with CL and/or CP,
are the most common clinical feature of VWS, accounting for 80% of
patients (3).
Numerous studies have demonstrated that pathogenic
variants and polymorphisms in interferon regulatory factor 6
(IRF6; OMIM. no. 607199) contribute to the development of
VWS (4–10). Although the gene modifier effect
can explain the phenotypic variability of VWS, current
understanding of the modifier genes of IRF6 remains incomplete and
limited to a case-controlled genomic variant study (11). Additionally, the role of microRNA
variants in the coding and non-coding regions of the genome in CL
and CP pathogenesis has also been studied (12–15).
Advances in next generation sequencing have allowed
the analyses of several diseases from a multi-omics perspective,
particularly in oncology studies (16,17).
For instance, trio whole-exome sequencing (WES) is an established
method for accurate detection of variants in genomic coding regions
(18). Moreover, transcriptome
sequencing has greatly facilitated gene expression studies
(19). As a result, multi-omics
analysis is increasingly applied to fundamental research and
clinical diagnosis (20,21).
The aim of the present study was to initially
identify the genetic cause of VWS, and then to implement a
multi-omic analysis in order to identify possible associations
between different gene expression spectrums and phenotypic
diversity in a three-generation family with VWS.
Materials and methods
Subjects
In September 2018, a three-generation Chinese
family, including a 4-month-old boy with CP, was recruited by the
General Clinic of Beijing Stomatology Hospital, affiliated to
Capital Medical University (Beijing, China). Routine physical
examinations and a family history survey were conducted, with
subsequent collection and storage of peripheral blood samples from
eight family members, including the boy.
Chromosome karyotyping and chromosomal
microarray analysis (CMA)
Conventional karyotyping by G-banding, was performed
on a blood sample from the proband boy according to standard
operational procedures, in order to detect broad chromosomal
anomalies. In addition, total genomic DNA (1 µg) was extracted from
200 µl peripheral blood using the DNA Blood Midi/Mini kit (Qiagen
GmbH), according to the manufacturer's protocols. A CMA was carried
out with a CytoScan 750K SNP Array (Affymetrix; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol, in
order to determine genomic copy number variants (CNV) with clinical
significance based on the interpretation guideline (22). Data was collected and analyzed
using GeneChip Scanner 3000 (Affymetrix; Thermo Fisher Scientific,
Inc.) with Affymetrix GeneChip Convert Console (v.1.1) software
(Affymetrix; Thermo Fisher Scientific, Inc.).
WES and data analysis
DNA samples (1 µg) were extracted from blood samples
(200 µl) of all participants in the family using the DNA Blood
Midi/Mini kit (Qiagen GmbH), which then underwent quality control
using agarose gel electrophoresis and UV spectrophotometry. Trio
WES was carried out on the proband boy and his parents, as
previously described (18). DNA
fragments were hybridized and captured by xGen Exome Research Panel
(Integrated DNA Technologies, Inc.), according to the
manufacturer's protocol. The libraries were tested for enrichment
by quantitative PCR, and the size, distribution and concentration
were determined using an Agilent Bioanalyzer 2100 (Agilent
Technologies, Inc.). The NovaSeq 6000 platform (Illumina, Inc.),
along with 150 bp pair-end reads, was used for the genomic
sequencing of DNA with ~300 pM per sample using NovaSeq Reagent
kit.
Sequencing raw reads (quality level %Q30 > 89%)
were aligned to the human reference genome (accession no.
hg19/GRCh37; ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/)
using the Burrows-Wheeler Aligner tool and the PCR duplicates were
removed using Picard v1.57 (http://picard.sourceforge.net/). Variant calling was
performed with the Verita Trekker® Variants Detection
system (v2.0; Berry Genomics, Inc.) and Genome Analysis Toolkit
(https://software.broadinstitute.org/gatk/). Variants
were then annotated and interpreted using ANNOVAR (v2.0) and
Enliven® Variants Annotation Interpretation systems
(Berry Genomics, Inc.) (23),
based on common guidelines by American College of Medical Genetics
and Genomics (24). To validate
variants, Sanger sequencing was introduced as a confirmatory
method. Three-dimensional structure prediction was conducted with
Modeller V9.21 (https://salilab.org/modeller/). The comparative
modeling method with default parameters was used to construct the
model based on the wild-type structure (PDB ID, 3DSHA; http://www.rcsb.org/structure/3DSH).
Whole-transcriptome sequencing and
data analysis
RNA was then extracted from blood samples of five
affected individuals and a healthy external control using Tempus™
Blood RNA Tube and Tempus™ Spin RNA Isolation kit (Thermo Fisher
Scientific, Inc.). RNA purity was checked by
NanoPhotometer® spectrophotometer (Implen GmbH). In
addition, RNA integrity assessed using an RNA Nano 6000 assay kit
on a Bioanalyzer 2100 system (Agilent Technologies, Inc.).
Sequencing libraries with sample indexing were
generated using the NEBNext® Ultra™ RNA Library Prep kit
for Illumina (New England Biolabs, Inc.), according to the
manufacturer's instructions. Briefly, mRNA was purified and a cDNA
library was synthesized by reverse transcription, which was then
fragmented into 250~300 bp. After PCR amplification, clustering was
performed on the cBot Cluster Generation system, using a TruSeq PE
Cluster kit v3-cBot-HS (cat. no. 401-3001; Illumina, Inc.).
Finally, the library preparations, ~300 pM per sample, were
sequenced on a HiSeq instrument (Illumina, Inc.), and 150 bp
paired-end reads were generated.
Raw reads were firstly processed to produce clean
reads via the removal of reads containing adapters, reads
containing ploy-N and low quality reads from raw data. At the same
time, Q20, Q30 and GC content in the clean data were calculated.
Then, data were mapped to the reference genome (accession no.
hg19/GRCh37; ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/),
quantification of gene expression levels was conducted using
featureCounts v1.5.0-p3 (25).
Differential expression analysis between two groups of patients,
Group 1 and −2, was performed using the DESeq2 R package v1.16.1
(26) and genes with an adjusted
P<0.05 were considered differentially expressed (with the
threshold: |log2(fold-change)|>0.5). In addition,
Gene Ontology (GO) enrichment analysis of differentially expressed
genes was performed by the clusterProfiler R package (27), in which gene length bias was
corrected. GO terms with a corrected P-value <0.05 were
considered significantly enriched by differentially expressed genes
(28). The heatmap was generated
using the pheatmap package (29).
Results
Clinical data
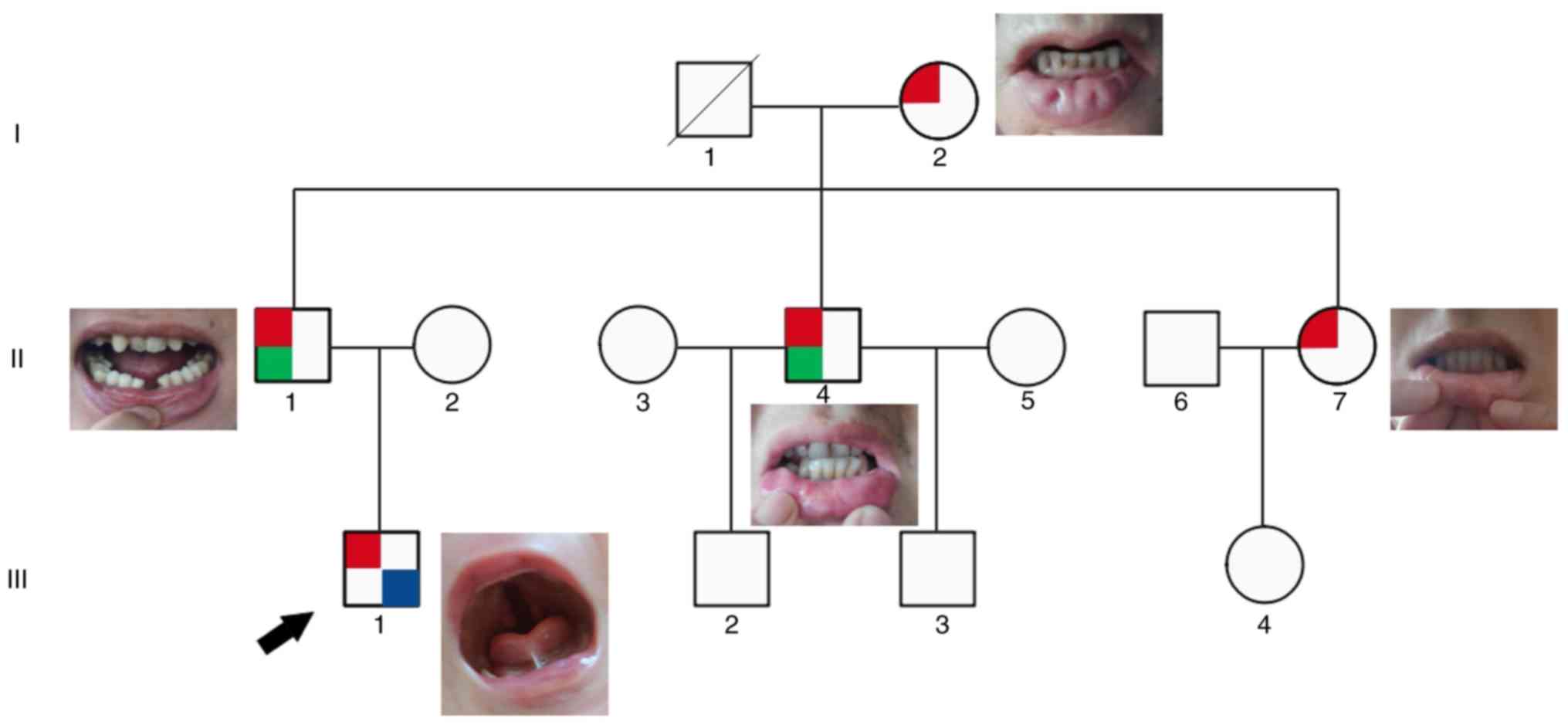
In this family, the affected patients included: i)
III-1, a 4-month-old proband with CP and bilateral lower lip pits;
ii) II-1, 38 years old with bilateral lower lip pits with gland
(removed) and congenital hypodontia; iii) II-4, 37 years old with
bilateral lower lip pits with gland (removed) and hypodontia; iv)
II-7, 32 years old with bilateral lower lip pits; and v) I-2, 60
years old with bilateral lower lip pits. There are notable
phenotypic heterogeneity among them, as demonstrated in Fig. 1 and Table I. The proband was the only patient
with CP, whereas the four adult patients also had differences in
phenotypic severity, mainly in the presence of hypodontia.
 | Table I.Clinical features of individuals from
three generations of a family with Van der Woude syndrome. |
Table I.
Clinical features of individuals from
three generations of a family with Van der Woude syndrome.
| Individual | Age | Clinical
features | Phenotype |
|---|
| I-1 | 32 years | Premature death
from myocardial infarction | / |
| I-2 | 60 years | Bilateral lower lip
pits | 1 |
| II-1 | 38 years | Bilateral lower lip
pits with gland, removed by surgery at age 5; congenitally missing
4 teeth | 1,2 |
| II-4 | 37 years | Bilateral lower lip
pits with gland, removed by surgery at age 4; teeth not
aligned | 1,2 |
| II-7 | 32 years | Bilateral lower lip
pits | 1 |
| III-1 | 4 months | Bilateral lower lip
pits; cleft palate | 1,3 |
| III-3 | 4.5 years | Two buccal fistulas
on neck, with surgical treatment | / |
| III-4 | 7 years | Currently in tooth
replacement period; no other indications; tooth development to be
determined | / |
Karyotyping and CMA
The karyotype of the boy was (46, XY), with no CNV
with clinical significance detected by CMA (data not shown).
WES
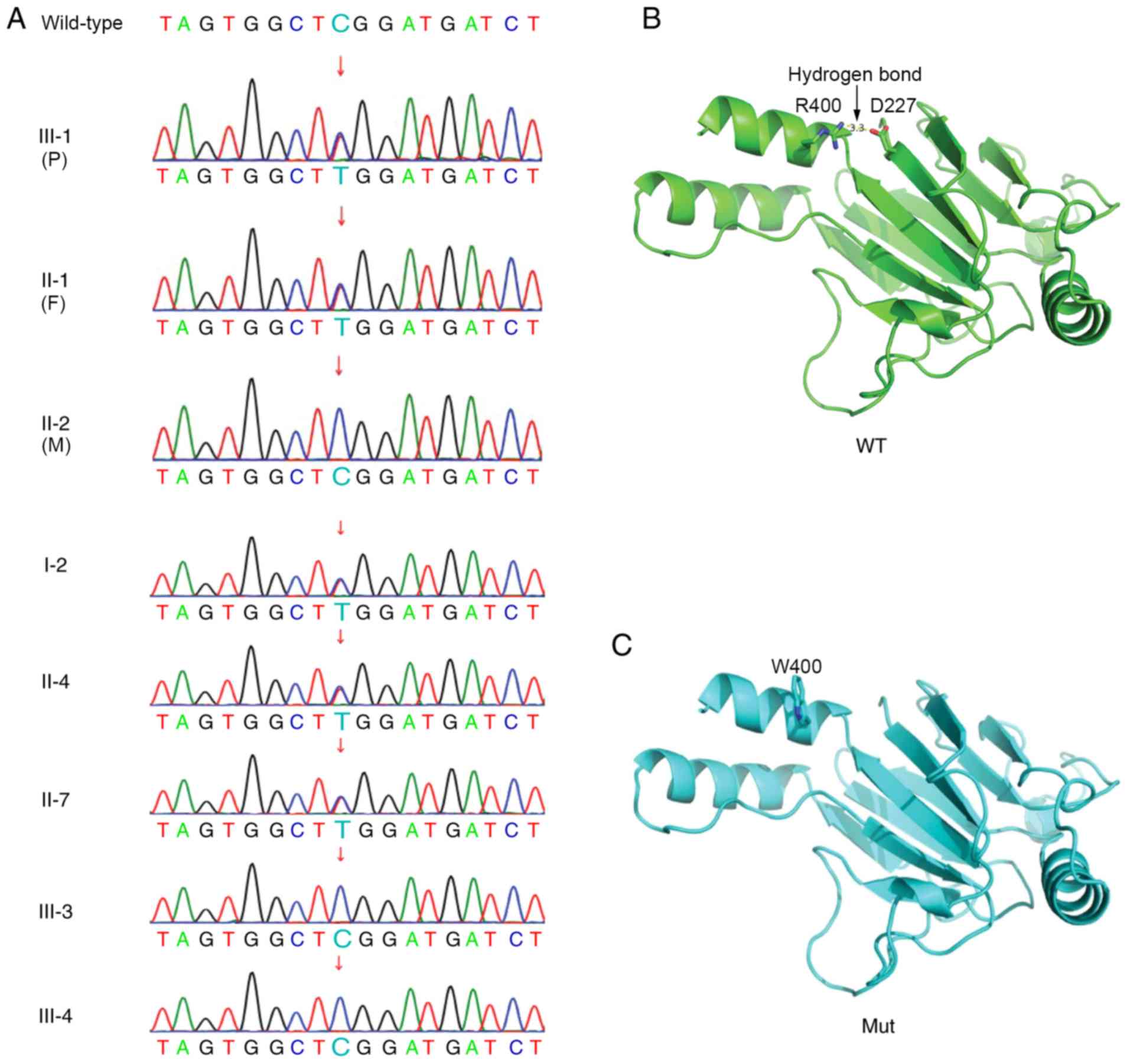
Trio WES demonstrated that the proband boy carried a
paternally inherited heterozygous 1198 C>T variant (translating
to R400W at the amino acid level) in exon 9 of the IRF6 gene
(Fig. 2A). Sanger sequencing also
indicated all five affected individuals in this family (I-2, II-1,
II-4, II-7 and III-1) shared the same variant. However, the
remaining three unaffected individuals carried the wild-type IRF6
gene (Fig. 2A). Therefore, genetic
examination demonstrated that variants and phenotypes in this
family were co-segregated.
Structural prediction
The R400W variant turned a positively charged amino
acid (R) into a polar amino acid with a benzene ring (W).
Structural prediction suggested that, compared with the wild-type
protein, the W400 residue in the mutant could not form hydrogen
bonds and mediate electrostatic interactions with D227, thereby
compromising overall structural stability (Fig. 2B and C).
Whole-transcriptome sequencing
The affected patients were divided into two groups
according to phenotypic differences, mainly reflected in lip
fistula tube formation and hypodontia. Group 1 included II-1 and
II-4, whereas Group 2 consisted of I-2 and II-7. The four-month-old
proband was excluded from the comparison due to the large age gap
that could affect the results. To determine whether specific
molecular pathways associated with IRF6 could account for the
phenotypic differences between the two pairs, gene expression
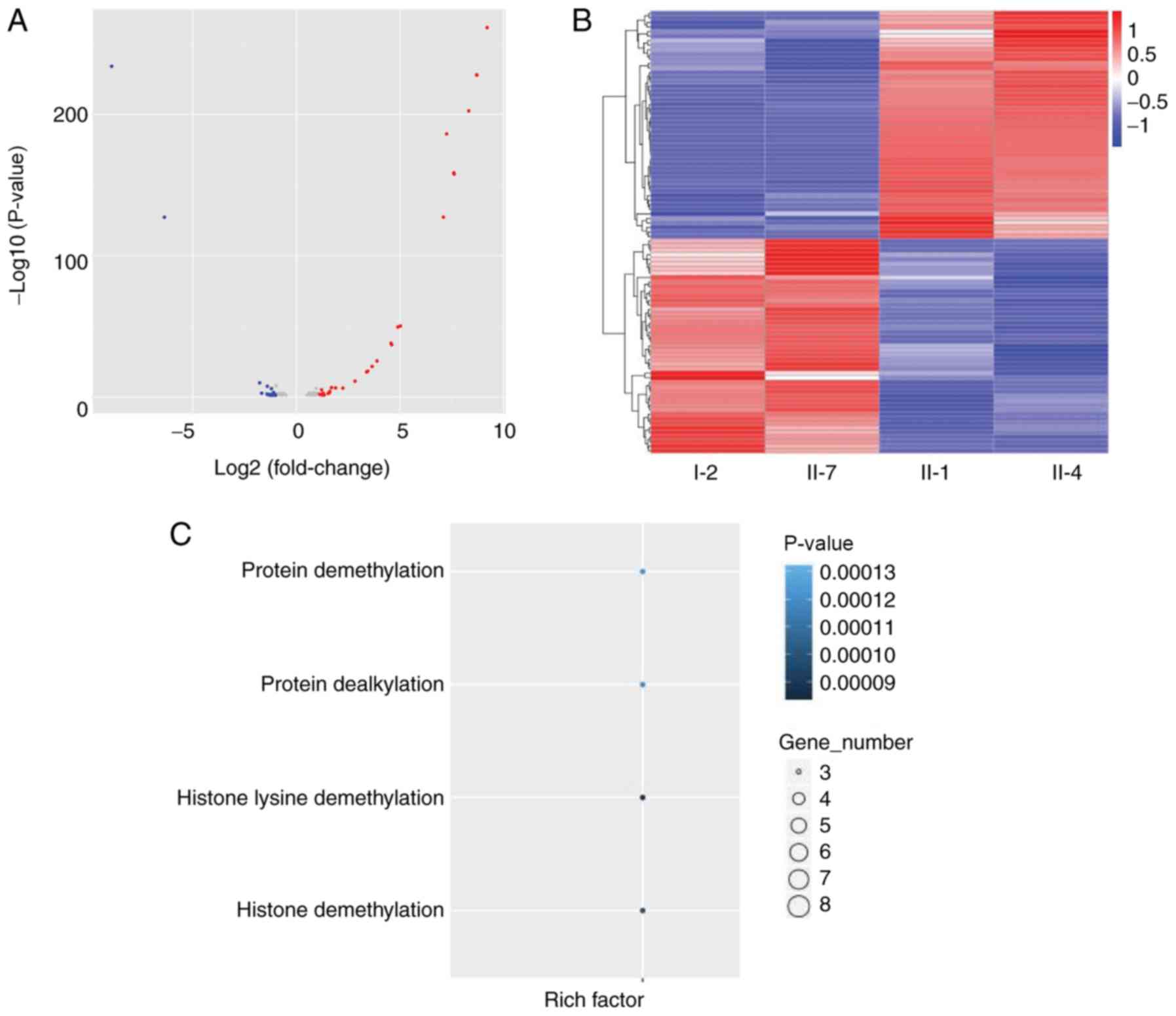
analysis was initially carried out. A volcano plot used to
visualize the different expressing genes between the two groups. As
presented in Fig. 3A, the red dots
represent the upregulated genes (Group 1 vs. 2), whereas the blue
dots represent the downregulated genes (Group 1 vs. 2).
In addition, the distribution of some significantly
upregulated and downregulated expression genes is also presented in
a heatmap (Fig. 3B). The
horizontal coordinate was the sample, and the vertical coordinate
was the differential gene; the left side clusters the gene
according to the expression similarity degree, gene expression
gradually upregulates from blue to red. The top 10 differentially
expressed genes, other than those related to sex determination, are
summarized in Table II. The
results of GO enrichment analysis are displayed in Fig. 3C, which demonstrated that the most
significantly expressed pathways between the two groups were
associated with ‘protein demethylation’, ‘protein dealkylation’,
‘histone lysine demethylation’ and ‘histone demethylation’.
| Table II.Top 10 genes (excluding
gender-related genes) expressing significant differences between
Group 1 (II-1 and II-4) and Group 2 (I-2 and II-7). |
Table II.
Top 10 genes (excluding
gender-related genes) expressing significant differences between
Group 1 (II-1 and II-4) and Group 2 (I-2 and II-7).
| Gene | Group 1, FKPM | Group 2, FPKM | Log2
(fold-change) | P-value | P-adj |
|---|
| ADARB2 | 0.02 | 4.53 | −6.37 |
2.73×10−13 |
4.03×10−12 |
| TMEM176A | 1.67 | 6.92 | −1.77 |
1.09×10−13 |
8.12×10−11 |
| PAM | 5.59 | 11.61 | −0.97 |
7.59×10−12 |
5.31×10−9 |
| PRSS23 | 1.26 | 2.63 | −0.98 |
1.98×10−11 |
1.34×10−8 |
| TMEM176B | 8.97 | 26.83 | −1.41 |
3.40×10−11 |
2.19×10−8 |
| PLA2G7 | 3.13 | 0.79 | 1.69 |
2.98×10−10 |
1.84×10−7 |
| MYBPH | 1.59 | 0.32 | 1.89 |
4.74×10−10 |
2.82×10−7 |
| C20orf27 | 21.33 | 10.95 | 0.95 |
1.55×10−9 |
8.55×10−7 |
| LGALS9B | 1.13 | 2.91 | −1.21 |
2.34×10−9 |
1.24×10−6 |
| MYO7A | 0.64 | 0.26 | 1.22 |
1.29×10−8 |
6.61×10−6 |
Discussion
VWS is an autosomal dominant disorder, with patients
usually presenting with CL and CP. Unlike non-syndromic CL and CP,
individuals with VWS exhibit bilateral and paramedian lower-lip
fistulae, or occasionally, small mounds with a sinus tract deriving
from a mucous gland of the lip (30). Moreover, hypodontia affecting
maxillary incisors, canines and premolars also occurs in patients
with VWS (15.5–86%), together with other dental anomalies that may
cause malocclusions (31).
IRF6 gene mutations are commonly found in individuals with
VWS (32).
The present study reported the case of a family with
VWS spanning three generations. Five individuals (I-2, II-1, II-4,
II-7 and III-1) were affected, with bilateral lower lip pits as the
common symptom, and presented intrafamilial phenotypic variability,
mainly in whether they displayed CP and hypodontia. WES suggested
that all affected subjects carried a reported heterozygous variant
c.1198C>T (R400W) of the IRF6 gene, whereas the
unaffected did not (5). Protein
structure analysis was carried out using prediction software, which
suggested that this mutation might interrupt the formation of
hydrogen bonds and electrostatic interactions between R400 and
D227, thereby compromising overall structural stability. A previous
study also described a recurrent c.1198C>T (R400W) mutation in
IRF6 in Chinese families with VWS (7). According to the American College of
Medical Genetics and Genomics criteria for variant interpretation
(24), this variant has been
determined as pathogenic (with evidence levels of PS1+PS2+PS4).
IRF6 contributes to the regulation of craniofacial
development and epidermal proliferation (33). Moreover, numerous studies have
indicated that variants in IRF6 that interrupt orofacial
development are the main cause of VWS (34,35).
De Lima et al (32) carried
out a sequencing analysis on 307 families with VWS and observed
IRF6 exon variants in 68% of these, with ~80% of IRF6 variants
located in exons 3, 4, 7 and 9. Peyrard-Janvid et al
(36) suggested that variants in
IRF6 could account for ~70% of familial cases of VWS, with a
further 17% of patients resulting from grainy head like
transcription factor 3 (GRHL3) variants.
IRF6 belongs to the IRF family of transcription
factors and contains a highly conserved helix-turn-helix
DNA-binding domain and a less conserved protein-binding domain.
Most IRF6 variants are missense and result in protein truncation
(37). The IRF family is composed
of nine members, most of which are involved in mediating interferon
responses following viral infection, as well as in innate immune
responses (38). The IRF6 gene is
2,171 bp in length and includes 10 exons, of which exons 1, 2 and
10 are non-coding regions (39).
The coding regions have a total length of 1,404 bp and encode 467
amino acids (37). Specifically,
exons 3 and 4 encode the DNA-binding domain, with exons 7 and 8
encoding the protein-binding domain of IRF6 (40).
Phenotypic variability in VWS families with IRF6
variant occurs frequently (11).
For instance, a previous study identified the pathogenic IRF6
variant 265A>G (K89E) in three affected members. However, the
newborn proband was diagnosed with popliteal pterygium syndrome
(PPS), whereas the mother presented with classic VWS and the
maternal grandfather had VWS with minor signs of PPS (30).
In the present study, the five patients with VWS
displayed varying phenotypic severity. A gene expression analysis
was therefore carried out on the four adult affected patients, who
all lived in similar circumstances. Differentially expressed genes
were identified between the two groups. Among these, myosin-binding
protein H (OMIM. no. 160795) is expressed in a pattern specific to
skeletal muscle (41). Myosin VII
A (OMIM. no. 276903) is an unconventional myosin that is
responsible for the formation and maintenance of epithelial cells
in several human and animal organs (42). Adenosine deaminase RNA specific
(ADAR) B2 (OMIM. no. 602065), which had the most significant
expression difference between Group 1 and 2, is a member of the
double-stranded RNA adenosine deaminase family of RNA-editing
enzymes and can inhibit in vitro RNA editing by other ADAR
family members (43). GO analysis
suggested that these differentially expressed genes were associated
with pathways related to protein and histone modifications. Whether
these genes play a role in VWS phenotypic variability and
interactions with IRF6 will require further study. Animal models
may need to be developed and other ‘omic’ methods may have to be
employed.
Furthermore, several previously reported genes
related to bone formation, including Wnt family, fibroblast growth
factor family, the Sonic hedgehog pathway, β-catenin, GRHL3, AP-2
complex subunit α, receptor interacting serine/threonine kinase 4,
and TGF-β activated kinase 1 (33,36,44–46)
were examined. There was no significant difference in expression in
these genes between the two groups (Table SI), along with the results shown
in Fig. 3, suggesting that VWS
phenotypic variability may predominantly be associated with
epigenetic levels and protein posttranslational modifications.
However, this also requires further in-depth research, including
investigation into epigenetic and protein modification (47), to confirm.
In conclusion, using WES, a heterozygous 1198 C>T
IRF6 variant was identified in five affected members of a Chinese
family with VWS. Protein structure prediction suggested overall
structure stability was compromised in the R400W mutant protein.
Gene expression and GO enrichment analysis also provided insight
into a possible mechanism, such as epigenetic modification, to
explain phenotypic variability in VWS. However, small sample size
was a major limitation of this study. Therefore, a larger study
sample is necessary, and further functional experiments are needed
to validate the present findings.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by The National Key
Research and Development Program of China (grant no.
2017YFC1001700) and The Maternal and Child Development Special
Program of Haidian Maternal and Child Healthcare Hospital (grant
no. 201805).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the authors on reasonable request.
Authors' contributions
KY and DZ recruited subjects and designed the study.
KY and XD wrote the manuscript, XD also analyzed the sequencing
data. JZ conducted chromosomal karyotyping and chromosomal
microarray analysis. JW and YT carried out whole-exome sequencing.
YY and LL conducted the whole transcriptome sequencing and data
analysis. All authors read and approved the final manuscript.
Ethics approval and consent for
participation
Ethical approval was obtained from The Medical
Ethics Committee of The School of Stomatology, Capital Medical
University. All participants, or their guardians, signed written
informed consent, including permission to publish anonymized test
results.
Patient consent for publication
Consent was obtained from participants for the
publication of this report and any accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sudhakara Reddy R, Ramesh T, Vijayalaxmi
N, Lavanya Reddy R, Swapna LA and Rajesh Singh T: Van der woude
syndrome-a syndromic form of orofacial clefting. J Clin Exp Dent.
4:2925–e128. 2012.
|
|
2
|
Angiero F, Farronato D, Ferrante F, Paglia
M, Crippa R, Rufino L, Trevisiol A, Mazzola RF and Blasi S:
Clinical, histomorphological and therapeutic features of the van
der woude syndrome: Literature review and presentation of an
unusual case. Eur J Paediatr Dent. 19:70–73. 2018.PubMed/NCBI
|
|
3
|
Ural A, Bilgen F, Çakmakli S and
Bekerecioğlu M: Van der woude syndrome with a novel mutation in the
IRF6 gene. J Craniofac Surg. 30:e465–e467. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kwa MQ, Huynh J, Reynolds EC, Hamilton JA
and Scholz GM: Disease-associated mutations in IRF6 and RIPK4
dysregulate their signalling functions. Cell Signal. 27:1509–1516.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang X, Liu J, Zhang H, Xiao M, Li J, Yang
C, Lin X, Wu Z, Hu L and Kong X: Novel mutations in the IRF6 gene
for van der woude syndrome. Hum Genet. 113:382–386. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ghassibe M, Bayet B, Revencu N,
Verellen-Dumoulin C, Gillerot Y, Vanwijck R and Vikkula M:
Interferon regulatory factor-6: A gene predisposing to isolated
cleft lip with or without cleft palate in the Belgian population.
Eur J Hum Genet. 13:1239–1242. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ye XQ, Jin HX, Shi LS, Fan MW, Song GT,
Fan HL and Bian Z: Identification of novel mutations of IRF6 gene
in Chinese families with van der woude syndrome. Int J Mol Med.
16:851–856. 2005.PubMed/NCBI
|
|
8
|
Du X, Tang W, Tian W, Li S, Li, X Liu L,
Zheng X, Chen X, Lin Y and Tang Y: Novel IRF6 mutations in Chinese
patients with van der woude syndrome. J Dent Res. 85:937–940. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khandelwal KD, Ishorst N, Zhou H, Ludwig
KU, Venselaar H, Gilissen C, Thonissen M, van Rooij IA, Dreesen K,
Steehouwer M, et al: Novel IRF6 mutations detected in orofacial
cleft patients by targeted massively parallel sequencing. J Dent
Res. 96:179–185. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao H, Zhang M, Zhong W, Zhang J, Huang
W, Zhang Y, Li W, Jia P, Zhang T, Liu Z, et al: A novel IRF6
mutation causing non-syndromic cleft lip with or without cleft
palate in a pedigree. Mutagenesis. 33:195–202. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Leslie EJ, Mancuso JL, Schutte BC, Cooper
ME, Durda KM, L'Heureux J, Zucchero TM, Marazita ML and Murray JC:
Search for genetic modifiers of IRF6 and genotype-phenotype
correlations in van der woude and popliteal pterygium syndromes. Am
J Med Genet A. 161:2535–2544. 2013.
|
|
12
|
Kumari P, Singh SK and Raman R: A novel
non-coding RNA within an intron of CDH2 and association of its SNP
with non-syndromic cleft lip and palate. Gene. 658:123–128. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gajera M, Desai N, Suzuki A, Li A, Zhang
M, Jun G, Jia P, Zhao Z and Iwata J: MicroRNA-655-3p and
microRNA-497-5p inhibit cell proliferation in cultured human lip
cells through the regulation of genes related to human cleft lip.
BMC Med Genomics. 12:702019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Suzuki A, Abdallah N, Gajera M, Jun G, Jia
P, Zhao Z and Iwata J: Genes and microRNAs associated with mouse
cleft palate: A systematic review and bioinformatics analysis. Mech
Dev. 150:21–27. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thieme F and Ludwig KU: The role of
noncoding genetic variation in isolated orofacial clefts. J Dent
Res. 96:1238–1247. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu GM, Ji X, Lu TC, Duan LW, Jia WY, Liu
Y, Sun ML and Luo YG: Comprehensive multi-omics analysis identified
core molecular processes in esophageal cancer and revealed GNGT2 as
a potential prognostic marker. World J Gastroenterol. 25:6890–6901.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kiebish MA, Cullen J, Mishra P, Ali A,
Milliman E, Rodrigues LO, Chen EY, Tolstikov V, Zhang L,
Panagopoulos K, et al: Multi-Omic serum biomarkers for prognosis of
disease progression in prostate cancer. J Transl Med. 18:102020.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang K, Shen M, Yan Y, Tan Y, Zhang J, Wu
J, Yang G, Li S, Wang J, Ren Z, et al: Genetic analysis in fetal
skeletal dysplasias by trio whole-exome sequencing. BioMed Res Int.
2019:24925902019.PubMed/NCBI
|
|
19
|
Jiang Z, Zhou X, Li R, Michal JJ, Zhang S,
Dodson MV, Zhang Z and Harland RM: Whole transcriptome analysis
with sequencing: Methods, challenges and potential solutions. Cell
Mol Life Sci. 72:3425–3439. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Argelaguet R, Velten B, Arnol D, Dietrich
S, Zenz T, Marioni JC, Buettner F, Huber W and Stegle O:
Multi-Omics factor analysis-a framework for unsupervised
integration of multi-omics data sets. Mol Syst Biol. 14:e81242018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hasin Y, Seldin M and Lusis A: Multi-Omics
approaches to disease. Genome Biol. 18:832017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Riggs ER, Andersen EF, Cherry AM, Kantarci
S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, et
al: Technical standards for the interpretation and reporting of
constitutional copy-number variants: A joint consensus
recommendation of the American college of medical genetics and
genomics (ACMG) and the clinical genome resource (ClinGen). Genet
Med. 22:245–257. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from next-generation
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zytnicki M: Mmquant: How to count
multi-mapping reads? BMC Bioinformatics. 18:4112017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu G, Wang LG, Han Y and He QY:
ClusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
The Gene Ontology Consortium, . The Gene
Ontology Resource: 20 years and still GOing strong. Nucleic Acids
Res. 47:D330–D338. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang X, Yao X, Qin C, Luo P and Zhang J:
Investigation of the molecular mechanisms underlying metastasis in
prostate cancer by gene expression profiling. Exp Ther Med.
12:925–932. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Busche A, Hehr U, Sieg P and
Gillessen-Kaesbach G: Van der woude and popliteal pterygium
syndromes: Broad intrafamilial variability in a three generation
family with mutation in IRF6. Am J Med Genet A. 170:2404–2407.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peralta-Mamani M, Terrero-Perez A, Dalben
G, Rubira CMF, Honorio HM and Rubira-Bullen IF: Treatment of lower
lip pits in van der woude syndrome: A systematic review. Int J Oral
Maxillofac Surg. 47:421–427. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
de Lima RL, Hoper SA, Ghassibe M, Cooper
ME, Rorick NK, Kondo S, Katz L, Marazita ML, Compton J, Bale S, et
al: Prevalence and nonrandom distribution of exonic mutations in
interferon regulatory factor 6 in 307 families with van der woude
syndrome and 37 families with popliteal pterygium syndrome. Genet
Med. 11:241–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Oberbeck N, Pham VC, Webster JD, Reja R,
Huang CS, Zhang Y, Roose-Girma M, Warming S, Li Q, Birnberg A, et
al: The RIPK4-IRF6 signalling axis safeguards epidermal
differentiation and barrier function. Nature. 574:249–253. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ingraham CR, Kinoshita A, Kondo S, Yang B,
Sajan S, Trout KJ, Malik MI, Dunnwald M, Goudy SL, Lovett M, et al:
Abnormal skin, limb and craniofacial morphogenesis in mice
deficient for interferon regulatory factor 6 (Irf6). Nat Genet.
38:1335–1340. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Biggs LC, Naridze RL, DeMali KA, Lusche
DF, Kuhl S, Soll DR, Schutte BC and Dunnwald M: Interferon
regulatory factor 6 regulates keratinocyte migration. J Cell Sci.
127:2840–2848. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Peyrard-Janvid M, Leslie EJ, Kousa YA,
Smith TL, Dunnwald M, Magnusson M, Lentz BA, Unneberg P, Fransson
I, Koillinen HK, et al: Dominant mutations in GRHL3 cause van der
woude syndrome and disrupt oral periderm development. Am J Hum
Genet. 94:23–32. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kondo S, Schutte BC, Richardson RJ, Bjork
BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S,
Sander A, et al: Mutations in IRF6 cause van der woude and
popliteal pterygium syndromes. Nat Genet. 32:285–289. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hixon K, Rhea L, Standley J, Canady FJ,
Canady JW and Dunnwald M: Interferon regulatory factor 6 controls
proliferation of keratinocytes from children with van der woude
syndrome. Cleft Palate Craniofac J. 54:281–286. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li S, Zhang X, Chen D, Zhao W, Zhang X,
Jiao J, Guo L, Yin L, Song X, Liang C and Sun C: Association
between genotype and phenotype of virulence gene in van der woude
syndrome families. Mol Med Rep. 17:1241–1246. 2018.PubMed/NCBI
|
|
40
|
Malik S, Wilcox ER and Naz S: Novel lip
pit phenotypes and mutations of IRF6 in van der woude syndrome
patients from Pakistan. Clin Genet. 85:487–491. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vaughan KT, Weber FE, Ried T, Ward DC,
Reinach FC and Fischiman DA: Human myosin-binding protein H
(MyBP-H): Complete primary sequence, genomic orgnization, and
chromosomal localization. Genomics. 16:34–40. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bahloul A, Michel V, Hardelin JP, Nouaille
S, Hoos S, Houdusse A, England P and Petit C: Cadherin-23, myosin
VIIa and harmonin, encoded by usher syndrome type I genes, form a
ternary complex and interact with membrane phospholipids. Hum Mol
Genet. 19:3557–3565. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen CX, Cho DS, Wang Q, Lai F, Carter KC
and Nishikura K: A third member of the RNA-specific adenosine
deaminase gene family, ADAR3, contains both single- and
double-stranded RNA binding domains. RNA. 6:755–767. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ke CY, Mei HH, Wong FH and Lo LJ: IRF6 and
TAK1 coordinately promote the activation of HIPK2 to stimulate
apoptosis during palate fusion. Sci Signal. 12:eaav76662019.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Maili L, Letra A, Silva R, Buchanan EP,
Mulliken JB, Greives MR, Teichgraeber JF, Blackwell SJ, Ummer R,
Weber R, et al: PBX-WNT-P63-IRF6 pathway in nonsyndromic cleft lip
and palate. Birth Defects Res. 112:234–244. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kousa YA, Fuller E and Schutte BC: IRF6
and AP2A interaction regulates epidermal development. J Invest
Dermatol. 138:2578–2588. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sun Y, Weng Y, Zhang C, Liu Y, Kang C, Liu
Z, Jing B, Zhang Q and Wang Z: Glycosylation of dentin matrix
protein 1 is critical for osteogenesis. Sci Rep. 5:175182015.
View Article : Google Scholar : PubMed/NCBI
|