Introduction
Diabetes mellitus (DM) is a chronic endocrine and
metabolic disease that affects human health and is a heavy burden
on individuals, their families and society (1). An epidemiological survey conducted in
China estimated that the overall prevalence of type I and type II
diabetes in Chinese adults in 2010 was 11.6% (1).
The destruction of islet β cells plays an important
role in the occurrence and development of DM. The inflammation
theory of islet β-cell destruction has drawn considerable attention
(2–5). As important regulators in the process
of inflammation, inflammatory cytokines directly or indirectly
damage islet β cells in different ways. The destruction of
pancreatic islet β cells in type 1 DM (T1DM) is associated with
inflammatory cytokines, including interleukin-1β (IL-1β),
interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) (2). The potential mechanisms of islet
β-cell damage in type 2 DM (T2DM) include the induction of
inflammatory cytokines, oxidative stress induced by high glucose
and lipids, and amyloid deposition in islets (3). Although some progress has been made
in the study of the destruction of pancreatic islet β cells induced
by inflammatory cytokines in recent years (4,5), the
molecular mechanisms by which these factors cause damage remain
unclear.
It has been reported that the mitogen-activated
protein kinase (MAPK) pathway may be involved in the inflammatory
cytokine-induced destruction of pancreatic islet β cells (1,6), but
the specific roles of different MAPK signaling pathways, namely the
extracellular signal-regulated kinase 1/2 (ERK1/2), p38 and c-jun
N-terminal kinase (JNK) pathways, remain unclear, and their mutual
regulation requires further clarification. Previous studies have
focused on only one or two pathways (7–13)
and the role of the MAPK signaling system is not yet fully
understood.
In our previous studies, it was found that IL-1β
inhibited the secretion of insulin under glucose stimulation in
βTC-6 cells, and the mechanism of insulin secretion was associated
with the inhibition of ERK1/2 (14,15).
However, in those studies, only the effect of IL-1β on the ERK1/2
pathway was examined and the roles of JNK and p38 signaling
pathways in the insulin secretory function of pancreatic β cells
remain unclear. In addition, the response of βTC-6 cells to glucose
stimulation is relatively weak.
Our previous studies showed that the optimal
concentration of glucose for the stimulation of βTC-6 cells was
1.38 mmol/l, but the peak value of insulin secretion after
stimulation was only 26% higher than the base value (14,15).
Min6 pancreatic β cells are more sensitive to glucose than βTC-6
cells in the study of insulin secretion (16). Therefore, the present study aimed
to further investigate the role of the three MAPK signaling
pathways in the IL-1β-induced inhibition of insulin-secretion
response in Min6 cells under glucose stimulation.
Materials and methods
Cell culture and treatment
Min6 cells (The Cell Bank of Type Culture Collection
of the Chinese Academy of Sciences) were cultured in Dulbecco's
modified Eagle's medium (DMEM, Hyclone; GE Healthcare life
Sciences) containing 25 mmol/l glucose supplemented with 15% fetal
bovine serum (M), 10 U/l penicillin and 10 U/l streptomycin (both
Shanghai Jingtian Biotechnology Co., Ltd.). Cells were cultured at
37°C in a 5% CO2 incubator, and the culture medium was
changed every 2 days. Cells were passaged at a 1:3 ratio for 4–7
days.
The survival rate of the Min6 cells in the cell
culture was measured using an MTT assay and the survival rate was
found to be >90%. MTT solution (Beyotime Institute of
Biotechnology, China) was added to the cells, which were then
incubated in the dark at 37°C. After 4 h, the liquid was absorbed
and discarded, the formazan crystals were dissolved by the addition
of 150 µl DMSO (Beyotime Institute of Biotechnology) and the
absorbance was measured at 490 nm.
Cells were digested by 0.25% trypsin (Hyclone;
Cytiva) for 30–60 sec at 37°C, which was seeded into 12-well plates
at 1×105 cells/well. After 72 h, the cells were adherent
to the plate walls and were used for glucose-stimulated insulin
secretion (GSIS) assays.
GSIS with and without IL-1β
Min6 cells were washed twice with phosphate-buffered
saline (PBS) and the medium was changed to glucose-free Krebs
buffered HEPES (KRBH; NaCl 119 mmol/l, KCl 4.74 mmol/l,
NaHCO3 25 mmol/l, MgSO4 1.19 mmol/l,
CaCl2 2.54 mmol/l, HEPES 10 mmol/l, pH 7.4). The cells
were then cultured for 60 min at 37°C in KRBH with glucose
concentrations of 0.0, 5.5, 11.1 and 22.2 mmol/l. The conditioned
culture medium was collected and used for the measurement of
insulin concentration via enzyme-linked immunosorbent assay
(ELISA).
To evaluate the effect of IL-1β on GSIS, Min6 cells
were incubated in DMEM at 37°C for 72 h and then IL-1β (Peprotech,
Inc.) at concentrations of 0.00, 0.25 and 2.50 ng/ml was added to
the culture medium. After 24 h at 37°C, the culture medium was
removed, and the cells were washed twice with PBS. KRBH buffer
containing 0.1% bovine serum albumin (Sigma-Aldrich; Merck KGaA)
without glucose was then added and the cells were incubated for 60
min at 37°C, prior to culture in KRBH medium containing 22.2 mmol/l
glucose for 60 min at 37°C. The procedures for the untreated group
(without glucose or IL-1β treatment) were the same as for the
intervention group. The conditioned culture medium was collected
and its insulin concentration was measured by ELISA.
Western blotting
Cells were collected, lysed with RIPA buffer
(ProteinTech Group, Inc.) containing phosphatase inhibitor and
protease inhibitors, and then centrifuged at 1,049 × g for 10 min
at 4°C. The cell lysate was collected and an equivalent volume (50
µl) of SDS loading buffer (Beyotime Institute of Biotechnology) was
added. BCA protein assay was used to determine the protein
concentration. The mixture was then heated in a water bath for 5
min at 95°C, and then refrigerated with ice immediately afterwards.
Using 10% polyacrylamide gel as a separating gel and 4%
polyacrylamide gel as stacking gel, the samples (30 µg protein per
lane) were electrophoresed for 1 h at 110 V. Transfer buffer was
used to balance the gel and nitrocellulose membrane for 10 min,
after which electrophoretic protein transfer was conducted for 1.5
h at 200 mV and the membrane was blocked in 5% skimmed milk at room
temperature for 1 h. The following primary antibodies were then
added: Anti-Erk1 (pT202/pY204) + Erk2 (pT185/pY187; 1:10.000, cat.
no. ab50011, Abcam), anti-p38 (phosphorylated (p) T180 + Y182;
1:1,000, cat. no. ab195049, Abcam), anti-JNK1 + JNK2 + JNK3 (p-T183
+ T183 + T221; 1:1,000, cat. no. ab124956, Abcam), anti-ERK1 + ERK2
(1:1,000, ab17942, Abcam), anti-p38 (1:1,000, cat. no. ab31828,
Abcam), anti-JNK1 + JNK2 + JNK3 (1:2000, cat. no. ab208035, Abcam),
or β-actin antibody (1:1,000, cat. no. ab8226, Abcam), and samples
were incubated overnight at 4°C. Secondary antibodies (anti-rabbit
IgG, HRP-linked antibody; cat. no. ab7074, Abcam) were then added
at a dilution of 1:2,000 and samples were incubated at room
temperature for 1 h. Finally, visualization was achieved using an
enhanced chemiluminescence analysis system (Merck KGaA) and blots
were quantified by densitometry using ImageJ v1.8.0 software
(National Institutes of Health). Each experiment was performed in
triplicate.
ELISA
An ELISA kit (cat. no. ml001983-1, Mlbio) was used
to detect the concentration of insulin in the conditioned culture
medium, and each sample was assessed in triplicate. The mean
optical density (OD) value for each sample was used to calculate
the insulin concentration. The OD values were determined using a
microplate reader (Sigma960; Metertech Inc.). All experiments were
performed strictly in accordance with the manufacturer's protocol.
The standard curves were constructed using CurveExpert v1.3
software (Hyams Development). The correlation coefficients were
≥0.999.
Reverse transcription-quantitative
PCR
Total RNA was extracted from Min6 cells using
Eastep® Super Total RNA reagent (Promega Corporation)
according to the manufacturer's protocols. The purity of the total
RNA was examined and RNA quantification performed by detecting the
absorbance at 260 and 280 nm using a spectrophotometer
(Biophotometer; Eppendorf). The cDNA was synthesized by reverse
transcription using the GoScript™ Reverse Transcription System
(Promega Corporation) and corresponding genes were amplified by
employing SYBR-Green Μaster Μix (cat. no. KK4601, KAPA;
Sigma-Aldrich; Merck KGaA). The thermocycling program was: 95°C for
5 min, followed by 35 cycles of denaturization at 95°C for 30 sec,
subsequent annealing at 50°C for 1 min and extension at 72°C for 1
min, followed by a final extension at 72°C for 10 min. The PCR
primers were synthesized by Takara Bio, Inc. and their sequences
were as follows: Insulin 1 forward, 5′-CGTTGAAATGCCACTGAAGCTACT-3′
and reverse, 5′-TTGCTGTGACTCCCCTGCT-3′; GAPDH forward,
5′-TTTGTCAAGCAGCACCTTTGT-3′ and reverse,
5′-CTCCACCCAGCTCCAGTTGT-3′. The mRNA level of insulin 1 was
normalized to that of GAPDH and was calculated using the
2−ΔΔCq method (17).
The experiments were performed in triplicate.
Cytotoxicity assays
Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc.) assay was used to measure cell viability.
Following treatment, 10 µl CCK-8 reagent was added to the cells in
each well and incubated for 3 h at 37°C. The absorbance was then
measured using a microplate reader at 450 nm. The experiment was
performed in triplicate.
Measurement of reactive oxygen species
(ROS)
Intracellular ROS production was measured using a
DCFH-DA probe (Beyotime Institute of Biotechnology). Following
treatment, the medium was removed from the cells, which were then
incubated with 10 µM DCFH-DA for 30 min at 37°C. The cell
fluorescence was detected by flow cytometry (FlowMax v2.8.2,
Sysmex).
Statistical analysis
Technical triplicates were performed for each
experiment, with a minimum of three biological replicates for each
study. Data are expressed as the mean ± standard deviation. One-way
ANOVA was used to analyze differences among groups followed by
Bonferroni's post hoc test to analyze differences between two
groups. Statistical analysis was performed using GraphPad Prism 6
software (GraphPad Software, Inc.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Effect of glucose stimulation on
insulin secretion in Min6 cells
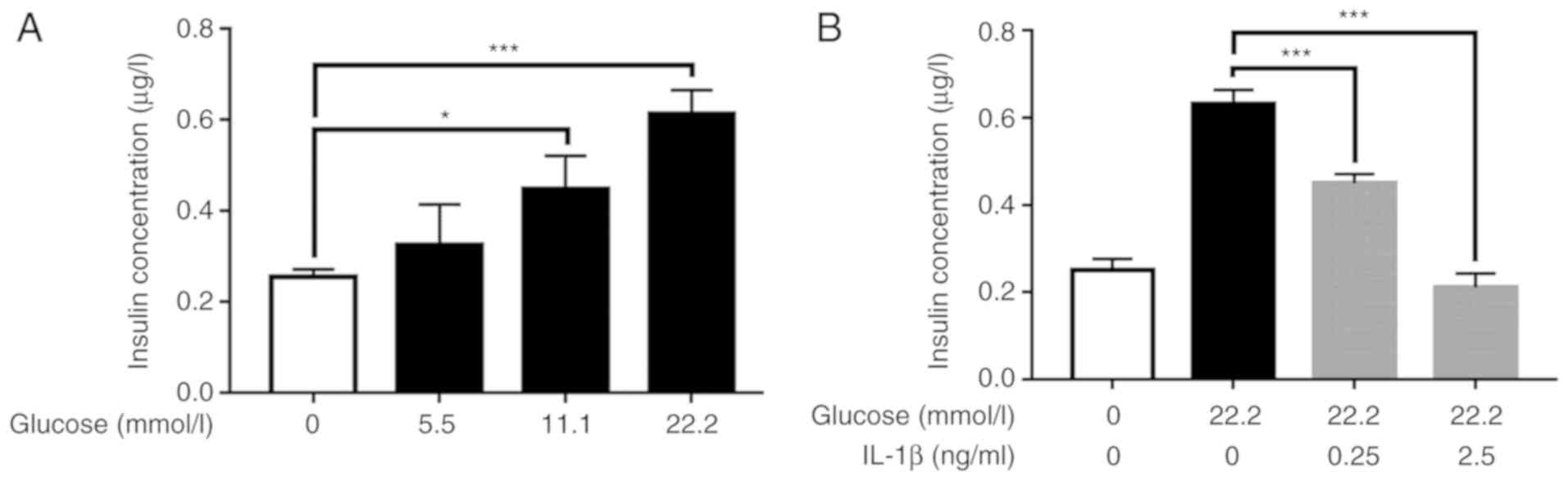
The insulin level in the conditioned culture medium
was 0.25±0.02 µg/l without glucose stimulation, and 0.33±0.09,
0.45±0.07 and 0.61±0.05 µg/l with 5.5, 11.1 and 22.2 mmol/l glucose
stimulation, respectively. There was a significant difference in
insulin level among the groups (F=18.38, P=0.0006). The insulin
level in the conditioned culture medium was highest following
stimulation with 22.2 mmol/l glucose. The insulin level in the 22.2
mmol/l glucose stimulation group was 241% of that in the
glucose-free group (0.61±0.05 vs. 0.25±0.02 µg/l, P=0.0003)
(Fig. 1A).
Effect of glucose stimulation on
ERK1/2, JNK and p38 phosphorylation in Min6 cells
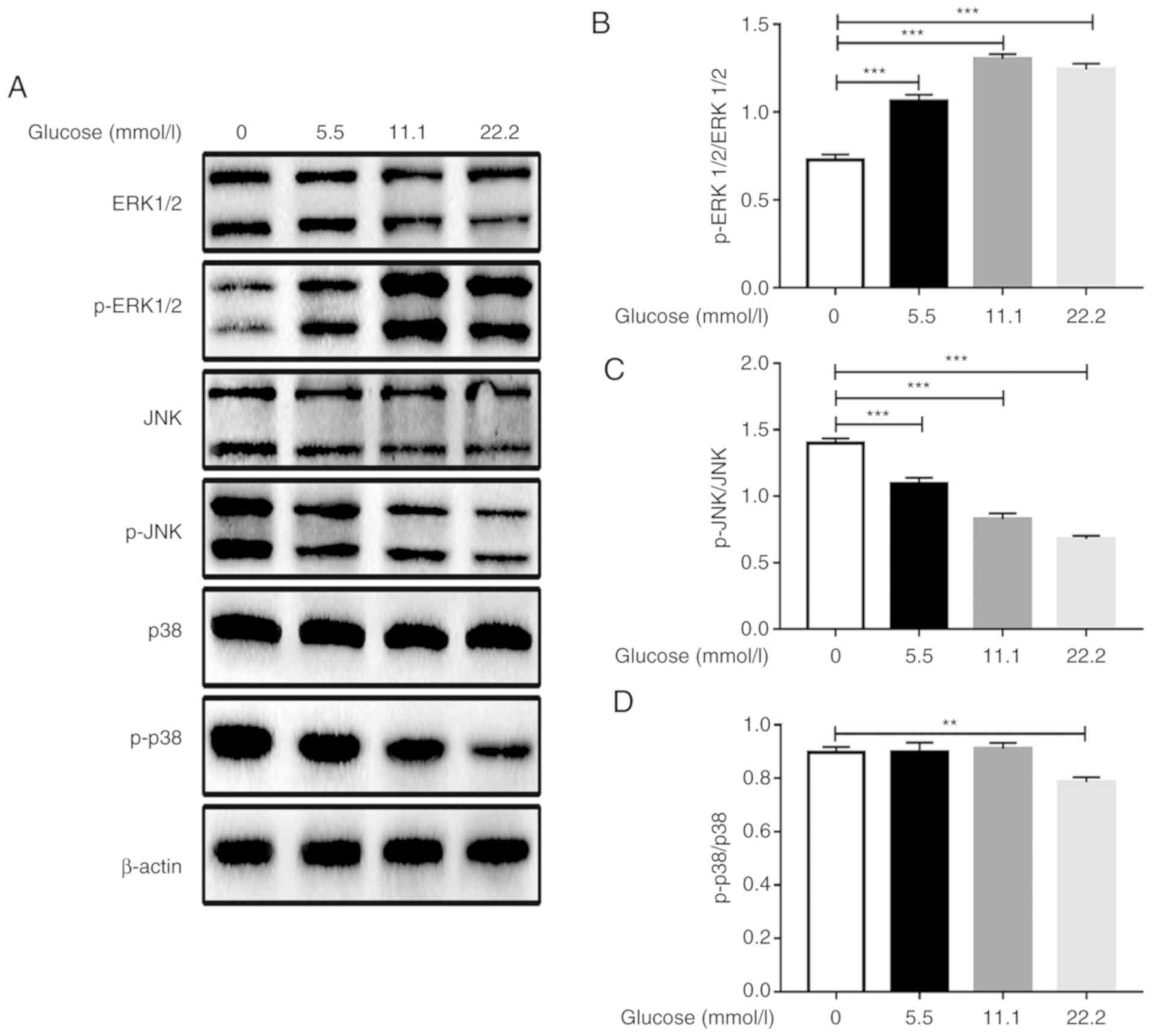
Following stimulation with glucose, the level of
ERK1/2 phosphorylation was increased compared with that in the
glucose-free group and appeared to peak in the 11.1 mmol/l glucose
stimulation group. However, glucose stimulation inhibited JNK and
p38 phosphorylation in Min6 cells. As the glucose concentration
increased the phosphorylation levels of JNK decreased in a
concentration-dependent manner. By contrast, the level of p38
phosphorylation was only reduced in the 22.2 mmol/l glucose
stimulation group (Fig. 2).
Effect of IL-1β on GSIS in Min6
cells
When no IL-1β pretreatment was performed, the
insulin level in the conditioned culture medium was 0.25±0.03 µg/l
without glucose stimulation and 0.63±0.03 µg/l under 22.2 mmol/l
glucose stimulation. The insulin level was reduced to 0.45±0.02
µg/l when 0.25 ng/ml IL-1β was added prior to GSIS and 0.21±0.03
µg/l when 2.5 ng/ml IL-1β was added prior to GSIS. There was a
significant difference in insulin level among these groups
(F=142.1, P<0.001). The 2.5 ng/ml IL-1β group exhibited the
highest inhibitory effect on the GSIS of Min6 cells, with a
reduction in the insulin level of 66% compared with the glucose
stimulation only group (0.21±0.03 vs. 0.63±0.03 µg/l, P=0.0001);
the insulin level of Min6 cells in the 0.25 ng/ml IL-1β group was
decreased by 28% compared with that in the glucose stimulation only
group (0.45±0.02 vs. 0.63±0.03 µg/l, P=0.0001) (Fig. 1B).
Effect of IL-1β on ERK1/2, JNK, p38
phosphorylation induced by glucose stimulation in Min6 cells
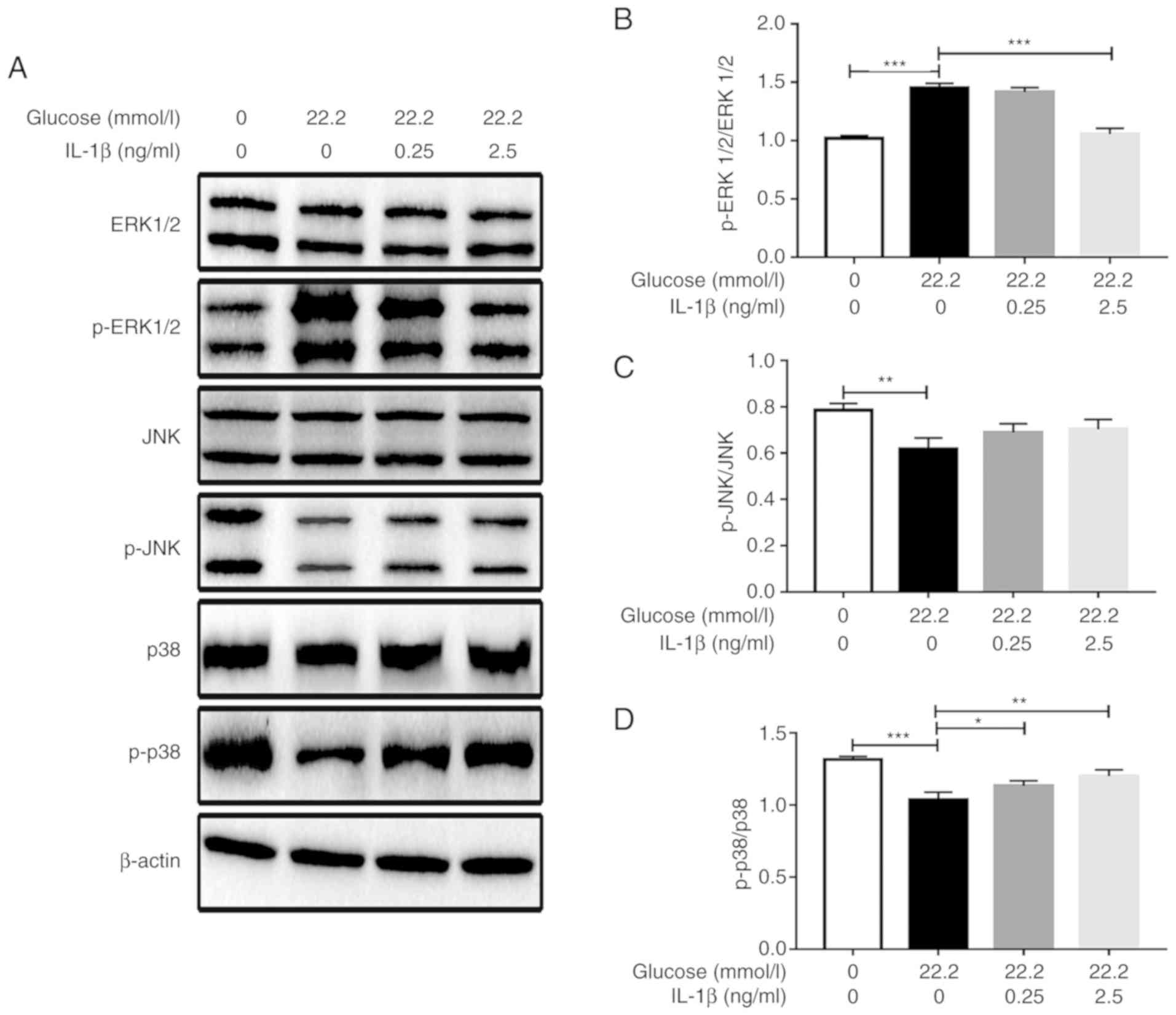
As presented in Fig.
3, 22.2 mmol/l glucose stimulated ERK1/2 phosphorylation in
Min6 cells and IL-1β inhibited the glucose-induced phosphorylation.
Pretreatment with 2.5 ng/ml IL-1β significantly reduced the level
of ERK1/2 phosphorylation compared with that in the cells only
stimulated with glucose. However, 22.2 mmol/l glucose inhibited p38
phosphorylation in Min6 cells, and IL-1β attenuated the
glucose-induced inhibition of p38 phosphorylation. The p38
phosphorylation levels of the 0.25 and 2.5 ng/ml IL-1β pretreatment
groups were increased compared with those in the cells only
stimulated with glucose, and the highest p38 phosphorylation level
was observed in the 2.5 ng/ml IL-1β group. However, IL-1β exhibited
no effect on the JNK signaling pathway as no significant changes in
JNK phosphorylation levels were induced by IL-1β following glucose
stimulation (Fig. 3).
Level of intracellular oxidative
stress
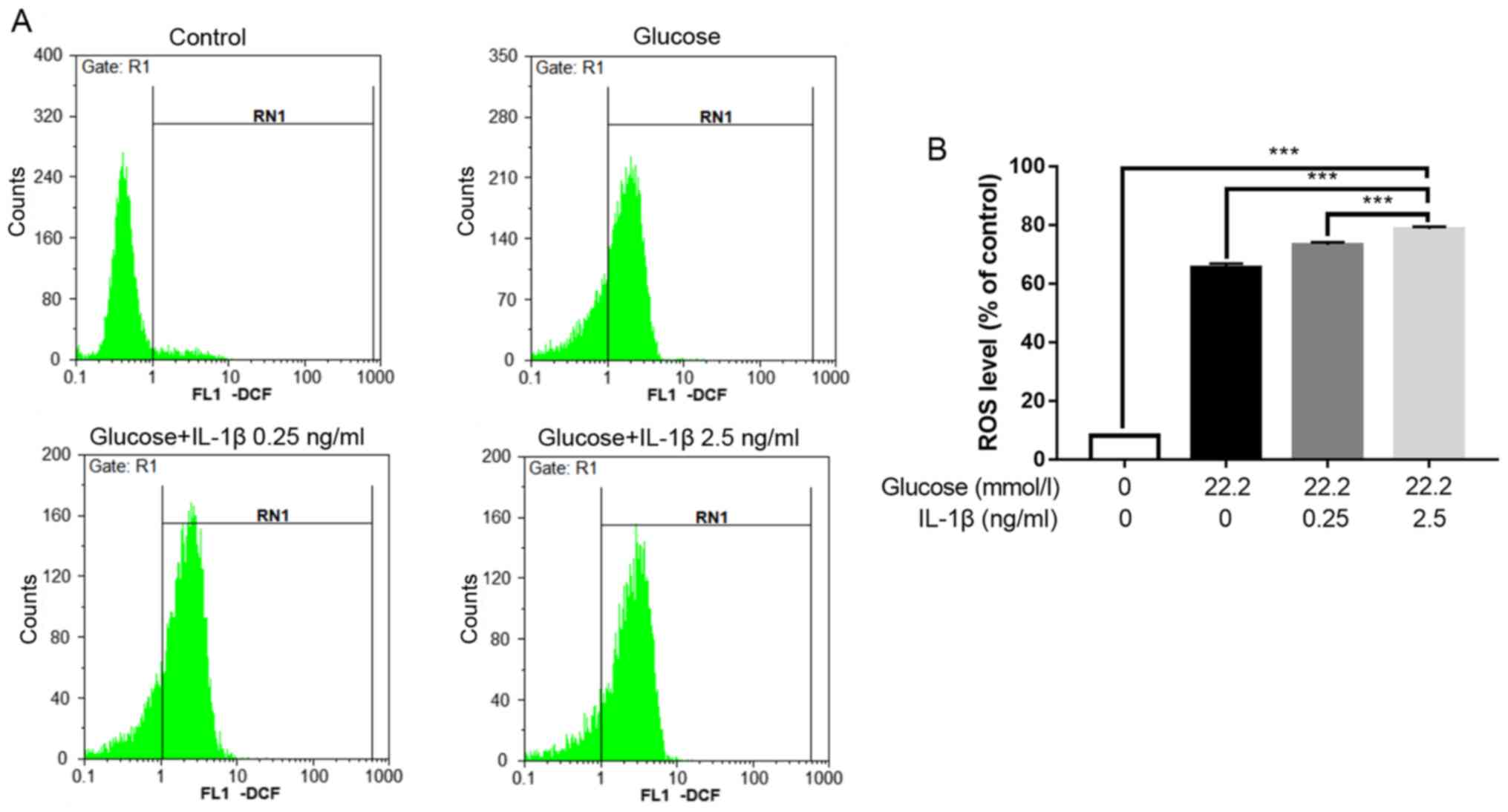
The levels of ROS were measured to evaluate the
oxidative stress of the cells. The hyperglycemic condition (22.2
mmol/l glucose) caused intracellular oxidative stress to the Min6
cells. When hyperglycemia was combined with IL-1β pretreatment, the
level of intracellular oxidative stress was elevated further and
increased with IL-1β concentration (Fig. 4).
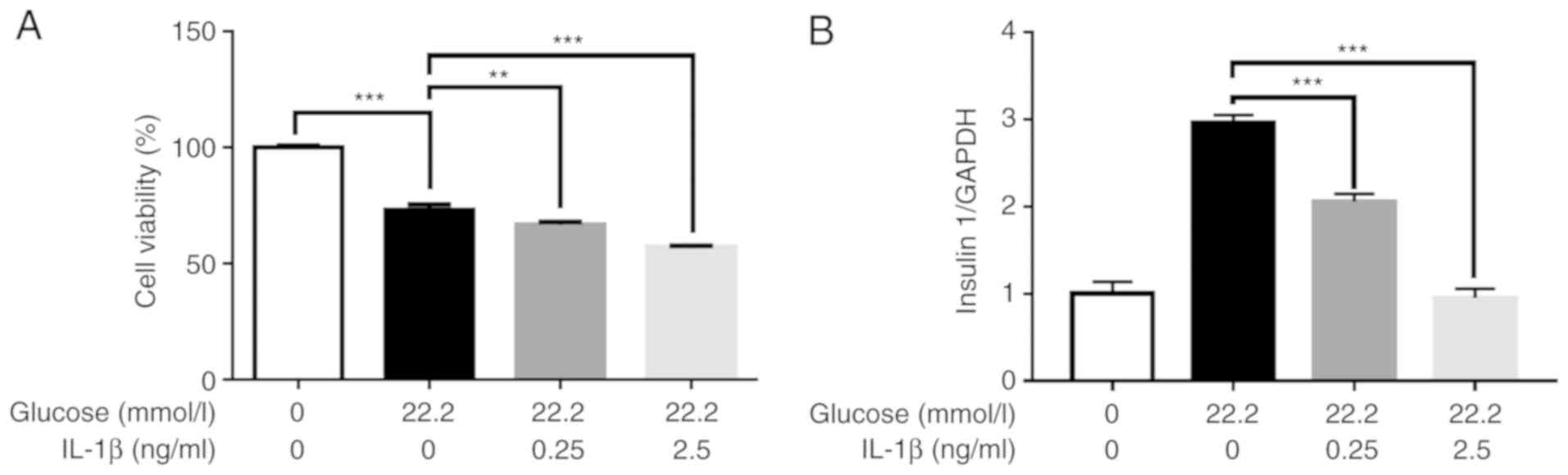
Effect of high glucose and IL-1β on
cell viability and insulin gene expression
The CCK8 assay was used to evaluate cell viability.
The results revealed that the viability of Min6 cells was decreased
following treatment with 22.2 mmol/l glucose alone or in
combination with different concentrations of IL-1β (Fig. 5A).
In order to determine whether the changes in the
levels of insulin secretion were caused by pretreatment with IL-1β
and not due to changes of cell viability, insulin mRNA levels in
the Min6 cells were further examined. The results revealed that the
changes in insulin mRNA levels were consistent with those of
insulin concentration (Fig. 5B).
These findings indicate that the decrease of insulin secretion was
caused by IL-1β.
Discussion
The present study aimed to investigate the roles of
different MAPK signal transduction pathways in the IL-1β-induced
inhibition of GSIS in Min6 mouse pancreatic cells. The results
revealed that insulin secretion was stimulated by various
concentrations of glucose in Min6 cells. Glucose stimulation
activated ERK1/2 phosphorylation and inhibited JNK and p38
phosphorylation in a concentration-dependent manner. The
inflammatory cytokine IL-1β inhibited GSIS and the GSIS-induced
activation of ERK1/2 phosphorylation but attenuated the
GSIS-induced inhibition of p38 phosphorylation. However, JNK
phosphorylation was neither activated nor inhibited by IL-1β.
MAPK signal transduction pathways comprise
serine/threonine protein kinases that exist in the majority of
cells and transduce extracellular stimuli to cells and their nuclei
(18). Numerous kinds of
extracellular stresses, including ultraviolet radiation, heat
shock, proinflammatory factors, specific antigens and other
stressors activate MAPK pathways and cause cell proliferation,
differentiation, transformation and apoptosis (19). MAPK signal transduction pathways
are highly conserved in cells, with prokaryotic cells and mammalian
cells having multiple parallel MAPK signaling pathways (20). At present, three MAPK signaling
pathways, namely the ERK1/2, JNK and p38 pathways, have been
clearly studied (20), but their
specific roles remain unclear.
Recent studies have shown that MAPK signaling
pathways may play an important role in the pathogenesis of
diabetes, especially in insulin secretion. For example, Liu et
al (21) revealed that
paeoniflorin (PF), a natural glycoside, attenuated the inhibitory
effect of streptozotocin (STZ) on the insulin secretion ability of
INS-1 cells. Furthermore, PF inhibited the STZ-induced
phosphorylation of p38 and JNK in INS-1 cells (21). Wei et al (22) reported that the single nucleotide
polymorphism rs2076878 of p38 was associated with insulin secretion
in the Chinese Han population, and revealed that the plasma insulin
levels of db/db mice were increased following administration of the
p38 MAPK inhibitor SB203580 for 9 weeks. A study of α-mangostin
revealed that it stimulated insulin secretion in INS-1 cells by
increasing phosphorylation in the phospho-phosphatidylinositol-3
kinase and ERK signaling cascades (23). In another study, secreted
frizzled-related protein-5 (Sfrp5) dose-dependently increased
glucose-stimulated insulin secretion but not basal insulin
secretion in INS-1E cells. In addition, Sfrp5 decreased JNK
signaling activity in INS-1E cells, suggesting that decreased JNK
activity may associated with the increased insulin secretion
induced by Sfrp5 (24). Youl et
al (25) demonstrated that an
MEK inhibitor completely abolished glucose-induced ERK1/2
phosphorylation and significantly decreased glucose-induced insulin
secretion in INS-1 pancreatic β-cells. The aforementioned studies
indicate that ERK1/2 phosphorylation promotes insulin secretion
while the phosphorylation of JNK and p38 inhibits insulin
secretion, and the results of the present study are in agreement
with the previous findings.
The inflammation theory of islet β cell destruction
has been widely researched, and inflammatory factors are known to
play an important role in dysfunctional insulin secretion and the
destruction of islet β cells (2,26).
Inflammatory cytokines, including IL-1β, TNF-α and IFN-γ, have been
shown to contribute to long term functional suppression and β-cell
apoptosis in T1DM and T2DM (27–29).
Notably, IL-1β contributes to β-cell failure and decreases insulin
secretion (30,31). β cells appear to be sensitive to
short pulses of cytokine exposure, as the incubation of rat islets
with IL-1β for 1 h resulted in the nitric oxide-dependent
inhibition of insulin secretion 18 h after cytokine removal
(32). IL-1β also inhibited GSIS
in Cohen diabetic rat islets through nitric oxide-induced
mitochondrial cytochrome c oxidase inhibition (33). Furthermore, when elevated serum
levels of IL-1β in diabetic rats were decreased by nitrite
administration, significantly increased insulin secretion was
observed (34). In a clinical
trial, a trend towards improved insulin secretion was observed in
patients treated with the anti-IL-1β antibody canakinumab,
supporting the hypothesis that insulin secretion is improved by
blocking IL-1β (35). Weaver et
al (36) revealed that GSIS
was attenuated by the inflammatory cytokines TNF-α, IL-1β and
IFN-γ, but protected by the NADPH-1 oxidase-1 inhibitor ML171 in
isolated mouse islets and murine β cell lines. Our previous study
found that IL-1β and/or IFN-γ inhibited insulin secretion by βTC-6
cells in a glucose stimulation test with a synergistic effect, and
the inhibitory effect of IL-1β on GSIS was dose-dependent (15). The present study revealed similar
results in Min6 cells.
It has been reported that insulin secretion in
vivo is associated with intracellular calcium (Ca2+) (37). In pancreatic β cells,
proinflammatory cytokines affect insulin secretion by regulating
Ca2+; they induce changes in intracellular
Ca2+ levels by depleting Ca2+ stores in the
endoplasmic reticulum (ER) and increasing extracellular
Ca2+ influx (38). In
mouse islets, exposure to TNF-α, IL-1β and IFN-γ has been shown to
disrupt the regulation of intracellular Ca2+ (39). Cytokine signaling has also been
demonstrated to disrupt β-cell glucose-stimulated Ca2+
influx and Ca2+ ER handling, leading to diminished
insulin secretion in response to glucose stimulation (40). Furthermore, the analysis of islets
from normal mice that underwent overnight exposure to IL-1β and
IL-6 via a cytokine-pump revealed deficiencies in Ca2+
handling and insulin secretion that were similar to observations
with islets exposed to cytokines in vitro (41).
Kim et al (7) demonstrated that TNF-α reduced
glucose-stimulated Ca2+ influx in INS-1 cells and
decreased GSIS, potentially by the activation of JNK and p38 MAPK
signaling. Similar findings have been reported for IL-1β, with
Ca2+ being indicated to participate in the
IL-1β-mediated activation of the JNK signaling pathway in
insulin-secreting cells (8). In
other studies, treatment with IL-1β increased the phosphorylation
of JNK in islets and Min-6 β cells (9), and elevated IL-1β induced apoptosis
through JNK1/2 activation-induced cellular Ca2+ movement
in human primary β-cells (10).
Furthermore, in a study of primary rat β cells and Min6 cells,
IL-1β promoted ER Ca2+ release by activating JNK and the
decreased activation of JNK provided protection against
IL-1β-mediated apoptosis via ER stress (11). Comparable results were not observed
in the present study when the JNK signaling pathway was assessed.
The potential reasons may be that human islet cells or higher IL-1β
concentrations were used in the other studies. However, the present
study suggested that IL-1β has the potential to activate the
glucose-stimulated JNK signaling pathway, although no significant
activation was detected.
However, the effects of IL-1β on ERK1/2 are
inconsistent. High glucose and IL-1β can lead to the apoptosis of
islet β cells and impairment of GSIS secretion, which are
associated with Ca2+ influx and activation of the ERK
signaling pathway (12). Burke
et al (13) revealed that
JNK and p38 were rapidly phosphorylated 15 min following the
exposure of pancreatic β-cells to IL-1β. By contrast, ERK was not
activated within 60 min. The present study revealed that IL-1β
inhibited the glucose-induced activation of ERK1/2 phosphorylation.
The reasons for these inconsistencies may be due to the different
concentrations and action times of IL-1β in the various studies.
The present study revealed that the phosphorylation of p38 was
activated by IL-1β in a concentration-dependent manner.
The present study has certain limitations. The
results only indicate that the mechanism by which inflammatory
cytokines impair insulin secretion in pancreatic β cells is
associated with ERK1/2 and p38 pathways. However, the changes of
upstream kinases such as MEK, Raf and Ras and their downstream
transcription factors remain to be elucidated. Furthermore, a cell
line rather than primary cells was used. Future studies will aim to
confirm the findings of the current study in rat primary cells.
In summary, the present study indicates that MAPK
signal transduction pathways participate in IL-1β-induced GSIS
inhibition in Min6 cells, with the ERK1/2 and p38 signaling
pathways appearing to have different effects. Activation of the
three MAPK pathways following glucose stimulation differs in Min6
cells and the effects of IL-1β on the three MAPK pathways also
differ, suggesting that these MAPK pathways play different roles in
the secretion of insulin by islet β cells, and that mutual
regulatory mechanisms may exist among them. The results are
valuable for elucidating the mechanism of islet β-cell destruction
and may aid the investigation of new intervention targets for the
protection of islet β-cells function in patients with DM.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 81560135).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HS designed and analyzed the experiments. YO read
the literature, analyzed data and wrote the manuscript. JS
performed the experiments. BN performed the supplementary
experiments (RT-qPCR, ROS detection and CCK-8 assays) and revised
the manuscript. ZZ searched the literature and performed
statistical analysis All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MAPK
|
mitogen-activated protein kinase
|
|
GSIS
|
glucose-stimulated insulin secretion:
IL-1β interleukin-1β
|
|
ERK1/2
|
extracellular signal-regulated kinase
1/2
|
|
JNK
|
c-jun N-terminal kinase
|
|
DM
|
diabetes mellitus
|
|
T1DM
|
type 1 DM
|
|
T2DM
|
type 2 DM
|
|
IFN-γ
|
interferon-γ
|
|
TNF-α
|
tumor necrosis factor-α
|
|
PBS
|
phosphate-buffered saline
|
|
KRBH
|
Krebs buffered HEPES
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
PF
|
paeoniflorin
|
|
Sfrp5
|
secreted frizzled-related
protein-5
|
|
ER
|
endoplasmic reticulum
|
|
ROS
|
reactive oxygen species
|
|
CCK-8
|
Cell Counting Kit-8
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
References
|
1
|
Xu Y, Wang L, He J, Bi Y, Li M, Wang T,
Wang L, Jiang Y, Dai M, Lu J, et al 2010 China Noncommunicable
Disease Surveillance Group, : Prevalence and control of diabetes in
Chinese adults. JAMA. 310:2973–959. 2013. View Article : Google Scholar
|
|
2
|
Kim KA and Lee MS: Recent progress in
research on β-cell apoptosis by cytokines. Front Biosci.
14:657–664. 2009. View
Article : Google Scholar
|
|
3
|
Ehses JA, Ellingsgaard H, Böni-Schnetzler
M and Donath MY: Pancreatic islet inflammation in type 2 diabetes:
From alpha and β cell compensation to dysfunction. Arch Physiol
Biochem. 115:240–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lambelet M, Terra LF, Fukaya M, Meyerovich
K, Labriola L, Cardozo AK and Allagnat F: Dysfunctional autophagy
following exposure to pro-inflammatory cytokines contributes to
pancreatic β-cell apoptosis. Cell Death Dis. 9:962018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Berchtold LA, Prause M, Størling J and
Mandrup-Poulsen T: Cytokines and Pancreatic β-Cell Apoptosis. Adv
Clin Chem. 75:99–158. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ammendrup A, Maillard A, Nielsen K,
Aabenhus Andersen N, Serup P, Dragsbaek Madsen O, Mandrup-Poulsen T
and Bonny C: The c-Jun amino-terminal kinase pathway is
preferentially activated by interleukin-1 and controls apoptosis in
differentiating pancreatic β-cells. Diabetes. 49:1468–1476. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim HE, Choi SE, Lee SJ, Lee JH, Lee YJ,
Kang SS, Chun J and Kang Y: Tumour necrosis factor-alpha-induced
glucose-stimulated insulin secretion inhibition in INS-1 cells is
ascribed to a reduction of the glucose-stimulated Ca2+
influx. J Endocrinol. 198:549–560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Størling J, Zaitsev SV, Kapelioukh IL,
Karlsen AE, Billestrup N, Berggren PO and Mandrup-Poulsen T:
Calcium has a permissive role in interleukin-1β-induced c-jun
N-terminal kinase activation in insulin-secreting cells.
Endocrinology. 146:3026–3036. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Edén D, Siegbahn A and Mokhtari D: Tissue
factor/factor VIIa signalling promotes cytokine-induced β cell
death and impairs glucose-stimulated insulin secretion from human
pancreatic islets. Diabetologia. 58:2563–2572. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Verma G, Bhatia H and Datta M: JNK1/2
regulates ER-mitochondrial Ca2+ cross-talk during
IL-1β-mediated cell death in RINm5F and human primary β-cells. Mol
Biol Cell. 24:2058–2071. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Q, Zhang H, Zhao B and Fei H: IL-1β
caused pancreatic β-cells apoptosis is mediated in part by
endoplasmic reticulum stress via the induction of endoplasmic
reticulum Ca2+ release through the c-Jun N-terminal
kinase pathway. Mol Cell Biochem. 324:183–190. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fei H, Zhao B, Zhao S and Wang Q:
Requirements of calcium fluxes and ERK kinase activation for
glucose- and interleukin-1β-induced β-cell apoptosis. Mol Cell
Biochem. 315:75–84. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Burke SJ, Goff MR, Updegraff BL, Lu D,
Brown PL, Minkin SC Jr, Biggerstaff JP, Zhao L, Karlstad MD and
Collier JJ: Regulation of the CCL2 gene in pancreatic β-cells by
IL-1β and glucocorticoids: Role of MKP-1. PLoS One. 7:e469862012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Niu B, Liu L, Su H, Xia X, He Q, Feng Y,
Xue Y and Yan X: Role of extracellular signal regulated kinase 1/2
signal transduction pathway in insulin secretion by β TC6 cells.
Mol Med Rep. 13:4451–4454. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Niu B, Su H, Xia XS, He Q, Xue YM and Yan
XM: The role of interleukin-1β and extracellular signal-regulated
kinase 1/2 in glucose-stimulated insulin secretion. Kaohsiung J Med
Sci. 33:224–228. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nie Y, Li J, Jin Y, Nyomba BLG, Cattini PA
and Vakili H: Negative effects of cyclic palmitate treatment on
glucose responsiveness and insulin production in mouse insulinoma
Min6 cells Are Reversible. DNA Cell Biol. 38:395–403. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔC(T)) Method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Anbazhagan K, Rabbind Singh A, Isabelle P,
Stella I, Céline AD, Bissac E, Bertrand B, Rémy N, Naomi T, Vincent
F, et al: Human pre-B cell receptor signal transduction: Evidence
for distinct roles of PI3kinase and MAP-kinase signalling pathways.
Immun Inflamm Dis. 1:26–36. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kyriakis JM and Avruch J: Mammalian MAPK
signal transduction pathways activated by stress and inflammation:
A 10-year update. Physiol Rev. 92:689–737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Singh R and Jwa NS: The rice MAPKK-MAPK
interactome: The biological significance of MAPK components in
hormone signal transduction. Plant Cell Rep. 32:923–931. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu Y, Han J, Zhou Z and Li D:
Paeoniflorin protects pancreatic β cells from STZ-induced damage
through inhibition of the p38 MAPK and JNK signaling pathways. Eur
J Pharmacol. 853:18–24. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wei X, Gu N, Feng N, Guo X and Ma X:
Inhibition of p38 mitogen-activated protein kinase exerts a
hypoglycemic effect by improving β cell function via inhibition of
β cell apoptosis in db/db mice. J Enzyme Inhib Med Chem.
33:1494–1500. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee D, Kim YM, Jung K, Chin YW and Kang
KS: Alpha-mangostin improves insulin secretion and protects INS-1
cells from streptozotocin-induced damage. Int J Mol Sci.
19:192018.
|
|
24
|
Carstensen-Kirberg M, Röhrig K, Niersmann
C, Ouwens DM, Belgardt BF, Roden M and Herder C: Sfrp5 increases
glucose-stimulated insulin secretion in the rat pancreatic β cell
line INS-1E. PLoS One. 14:e02136502019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Youl E, Bardy G, Magous R, Cros G, Sejalon
F, Virsolvy A, Richard S, Quignard JF, Gross R, Petit P, et al:
Quercetin potentiates insulin secretion and protects INS-1
pancreatic β-cells against oxidative damage via the ERK1/2 pathway.
Br J Pharmacol. 161:799–814. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pham MN, Hawa MI, Pfleger C, Roden M,
Schernthaner G, Pozzilli P, Buzzetti R, Scherbaum WA, Seissler J,
Kolb H, et al Action LADA Study Group, : Pro- and anti-inflammatory
cytokines in latent autoimmune diabetes in adults, type 1 and type
2 diabetes patients: Action LADA 4. Diabetologia. 54:1630–1638.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eizirik DL, Colli ML and Ortis F: The role
of inflammation in insulitis and β-cell loss in type 1 diabetes.
Nat Rev Endocrinol. 5:219–226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Alexandraki K, Piperi C, Kalofoutis C,
Singh J, Alaveras A and Kalofoutis A: Inflammatory process in type
2 diabetes: The role of cytokines. Ann N Y Acad Sci. 1084:89–117.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Padgett LE, Broniowska KA, Hansen PA,
Corbett JA and Tse HM: The role of reactive oxygen species and
proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y
Acad Sci. 1281:16–35. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Burke SJ, Stadler K, Lu D, Gleason E, Han
A, Donohoe DR, Rogers RC, Hermann GE, Karlstad MD and Collier JJ:
IL-1β reciprocally regulates chemokine and insulin secretion in
pancreatic β-cells via NF-κB. Am J Physiol Endocrinol Metab.
309:E715–E726. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao G, Dharmadhikari G, Maedler K and
Meyer-Hermann M: Possible role of interleukin-1β in type 2 diabetes
onset and implications for anti-inflammatory therapy strategies.
PLOS Comput Biol. 10:e10037982014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Corbett JA, Sweetland MA, Lancaster JR Jr
and McDaniel ML: A 1-hour pulse with IL-1β induces formation of
nitric oxide and inhibits insulin secretion by rat islets of
Langerhans: Evidence for a tyrosine kinase signaling mechanism.
FASEB J. 7:369–374. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Weksler-Zangen S, Aharon-Hananel G,
Mantzur C, Aouizerat T, Gurgul-Convey E, Raz I and Saada A: IL-1β
hampers glucose-stimulated insulin secretion in Cohen diabetic rat
islets through mitochondrial cytochrome c oxidase inhibition by
nitric oxide. Am J Physiol Endocrinol Metab. 306:E648–E657. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gheibi S, Bakhtiarzadeh F, Jeddi S,
Farrokhfall K, Zardooz H and Ghasemi A: Nitrite increases
glucose-stimulated insulin secretion and islet insulin content in
obese type 2 diabetic male rats. Nitric Oxide. 64:39–51. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rissanen A, Howard CP, Botha J and Thuren
T; Global Investigators, : Effect of anti-IL-1β antibody
(canakinumab) on insulin secretion rates in impaired glucose
tolerance or type 2 diabetes: Results of a randomized,
placebo-controlled trial. Diabetes Obes Metab. 14:1088–1096. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Weaver JR, Grzesik W and Taylor-Fishwick
DA: Inhibition of NADPH oxidase-1 preserves β cell function.
Diabetologia. 58:113–121. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sabatini PV, Speckmann T and Lynn FC:
Friend and foe: Β-cell Ca2+ signaling and the
development of diabetes. Mol Metab. 21:1–12. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ramadan JW, Steiner SR, O'Neill CM and
Nunemaker CS: The central role of calcium in the effects of
cytokines on β-cell function: Implications for type 1 and type 2
diabetes. Cell Calcium. 50:481–490. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dula SB, Jecmenica M, Wu R, Jahanshahi P,
Verrilli GM, Carter JD, Brayman KL and Nunemaker CS: Evidence that
low-grade systemic inflammation can induce islet dysfunction as
measured by impaired calcium handling. Cell Calcium. 48:133–142.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dickerson MT, Bogart AM, Altman MK, Milian
SC, Jordan KL, Dadi PK and Jacobson DA: Cytokine-mediated changes
in K+ channel activity promotes an adaptive Ca2+
response that sustains β-cell insulin secretion during
inflammation. Sci Rep. 8:11582018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
O'Neill CM, Lu C, Corbin KL, Sharma PR,
Dula SB, Carter JD, Ramadan JW, Xin W, Lee JK and Nunemaker CS:
Circulating levels of IL-1B+IL-6 cause ER stress and dysfunction in
islets from prediabetic male mice. Endocrinology. 154:3077–3088.
2013. View Article : Google Scholar : PubMed/NCBI
|