Introduction
Diabetic nephropathy (DN) is the most common cause
of end-stage renal disease worldwide (1). One of the main mechanisms of DN is
renin-angiotensin system (RAS) activation, and inhibition of the
RAS can slow the development of diabetic kidney injury (2). Angiotensin-converting enzyme 2 (ACE2)
is a component of the RAS that has counter-regulatory functions to
the classic ACE/angiotensin II (Ang II)/Ang II receptor type 1
(AT1R) axis (3). In human renal
tissues, ACE2 is highly expressed in renal tubular epithelial
cells, and low tubular ACE2 expression has been revealed to be
associated with the progression of renal fibrosis in DN (4). However, the mechanisms by which ACE2
protects against high-glucose-induced renal fibrosis remain
unclear. Moreover, circulating ACE2 has not been well studied in
patients with DN.
Tubulointerstitial fibrosis is essential in the
development of end-stage renal disease in DN. The renal tubular
epithelial to mesenchymal cell transition (EMT) plays a key role in
this process. It has been demonstrated that the transforming growth
factor-β (TGF-β)/SMAD family member (Smad) signaling pathway is a
potent initiator and regulator of EMT (5). Previous studies have revealed that
TGF-β-induced EMT is primarily activated by Smad3 phosphorylation
and inhibited by Smad7, a negative feedback regulator of the
TGF-β1/Smad signaling pathway that protects against fibrosis by
inducing receptor degradation, which halts the recruitment and
phosphorylation of Smad3 (5,6). A
previous study by Liu et al (7) revealed that the level of Smad7
protein was dynamically modulated by the ubiquitin-proteasome
degradation pathway, and this was mainly attributed to the E3
ubiquitin ligase Arkadia (also referred to as ring finger protein
111). However, whether this process occurs in DN and the mechanisms
involved have yet to be determined.
The purpose of the present study was to investigate
the protective mechanism of ACE2 in DN. To this end, renal tissue
from patients with DN and primary human renal proximal tubular
epithelial cells (HRPTEpiC) stimulated with a high glucose
concentration were studied. In addition, the serum ACE2 expression
was measured in DN patients with microalbuminuria.
Materials and methods
Blood samples and renal tissue
collection
Blood samples were collected from 10 patients with
DN and 10 non-diabetic volunteers from December 2018 to March 2019.
Diabetic renal tissue specimens were obtained from five patients
with a pathological diagnosis of DN and control specimens were
obtained from five patients with minimal change in nephropathy
without renal interstitial fibrosis. None of the recruited
participants had liver dysfunction, heart failure, malignancy and
none were pregnant. The non-diabetic volunteers were those who
visited the hospital for routine physical examination and had no
history of diabetes or nephropathy with normal blood glucose and
serum creatinine concentrations. The blood samples and renal
tissues were obtained from Beijing Friendship Hospital, following a
protocol approved by the Ethics Committee of Beijing Friendship
Hospital, Capital Medical University. Each participant provided
their written informed consent before being included in the
study.
Serum ACE2 measurements
Venous blood was placed in disposable sterile test
tubes and centrifuged at 1,000 × g, 25°C for 10 min to isolate
serum. The serum samples were then transferred to cryovials and
frozen at −80°C until assayed.
The serum ACE2 expression levels were measured using
a commercial ELISA kit (cat. no. E01A0499; Shanghai BlueGene
Biotech Co., Ltd.), according to the manufacturer's
instructions.
Histopathological examination
Renal sections were fixed in 4% neutral-buffered
paraformaldehyde for 24 h at 25°C, embedded in paraffin, and cut
into 4 µm-thick sections, which were then prepared for Periodic
acid-Schiff (PAS), Periodic Schiff-Methenamine Silver (PASM) or
Masson's modified trichrome histological staining. The stained
renal tissue sections were scored as previously described (8). All the scoring was performed by one
observer who was blinded to the group from which the tissue
specimens originated. The data were recorded and used to compare
the diabetic with the non-diabetic groups.
Immunohistochemistry
Paraffin-embedded sections of renal tissues were
deparaffinized in xylene, rehydrated through an alcohol gradient
from 99.5% to 70% alcohol, and underwent antigen retrieval in
citrate buffer (cat. no. P0081; Beyotime Institute of
Biotechnology, Inc.). Endogenous peroxidase activity was blocked
with 3% H2O2, and then the sections were
incubated with 5% normal goat serum at 37°C for 30 min. Sections
were then incubated overnight at 4°C with primary antibodies
targeting ACE2 (1:100; cat. no. ab87436; Abcam) or Arkadia (1:100;
cat. no. OM290104; Omnimabs), followed by incubation with a
biotinylated secondary antibody (1:2,000; cat. no. SP-9000; OriGene
Technologies, Inc.) for 30 min at room temperature. After thorough
rinsing, the antigens were detected by 3,3′-diaminobenzidine
staining (OriGene Technologies, Inc.). Sections were counterstained
with hematoxylin, dehydrated and cover-slipped. The stained renal
tissue sections were scored as aforementioned.
Cell culture and treatments
The HRPTEpiCs were purchased from ScienCell Research
Laboratories, Inc., (cat. no. 4100). Cells were cultured in
epithelial cell medium (EpiCM) supplemented with 2% fetal bovine
serum and epithelial cell growth supplement (EpiCGS; all from
ScienCell Research Laboratories, Inc.), benzylpenicillin (100
U/ml), and streptomycin (100 µg/ml) at 37°C in a humidified
atmosphere containing 5% CO2 as described in previous
studies (9,10). Actively growing cells in their
second to third passage were used for experiments. Cells were
seeded at a density of 2.5×105 cells/well in 6-well
plates. When cell confluency reached 70–80%, the medium was
replaced with serum-free epithelial cell medium. After serum
starvation for 12 h, cells were incubated in medium containing a
normal (5.5 mmol/l D-glucose) or high glucose concentration (30
mmol/l D-glucose; Sigma-Aldrich; Merck KGaA) for 4, 12, 24, 48 or
72 h (9). Subsequently, the cells
were stimulated with a high glucose concentration for 48 h in the
presence or absence of adenoviruses overexpressing ACE2. For
treatment with an ACE2 inhibitor, the ACE2 inhibitor DX600 (0.5
mmol/l; BioVision, Inc.) was added to high glucose medium for 48 h.
Finally, the cells were treated with small interfering (si)RNA
targeting Arkadia or scramble siRNA for 48 h while remaining in
high glucose medium. The cells were then collected by trypsin
digestion for subsequent analysis.
Adenovirus transduction and siRNA
transfection
Adenoviruses overexpressing ACE2 (pHBAd-BHG; Hanbio
Biotechnology Co., Ltd), siRNA targeting Arkadia
(5′-TTCCATGCAGAAAGAGATTTGTAAA-3′) and a scrambled siRNA
(5′-TTCTCCGAACGTGTCACGTAA-3′) were purchased from Hanbio
Biotechnology Co., Ltd. HRPTEpiCs were either infected with
adenoviruses using the polybrene method at an MOI of 100 or
transfected with 100 nmol/l siRNA at 37°C and in 5% CO2
for 6 h. All transfections were carried out using Transfection
Reagent (Hanbio Biotechnology Co., Ltd.,). The medium was then
replaced with fresh epithelial cell medium after 6-h incubation.
Empty adenovirus and scrambled siRNA were used as negative
controls. HRPTEpiCs were used for experiments 48 h after
transfection.
Western blot analysis
Western blotting was performed as previously
described (9). Briefly, after
blocking nonspecific binding with 5% BSA (Sigma-Aldrich; Merck
KGaA), the membranes were incubated overnight at 4°C with primary
antibodies. Primary antibodies against ACE2 (1:1,000; cat. no.
4355), Smad3 (1:1,000; cat. no. 9523), phosphorylated (p)-Smad3
(1:1,000; cat. no. 9520), E-cadherin (1:1,000; cat. no. 14472),
α-smooth muscle actin (α-SMA; 1:1,000; cat. no. 19245) were
purchased from Cell Signaling Technology, Inc. Antibodies targeting
Arkadia (1:500; cat. no. ab174624), Smad7 (1:1,000; cat. no.
ab190987), TGF-β (1:1,000; cat. no. ab92486) or β-actin (1:1,000;
cat. no. ab8226) were purchased from Abcam. After the first
incubation, the membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies (1:2,000; cat. no.
ZB-2301 or ZB-2305; ZSGB-BIO; OriGene Technologies, Inc.). Target
proteins were detected and analyzed using Immobilon Western
Chemiluminescent HRP Substrate (EMD Millipore). Densitometry
analysis was performed using ImageJ software (v1.52; National
Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA was extracted from cells with TRIzol®
Reagent (Invitrogen; Thermo Fisher Scientific, Inc.), following the
manufacturer's protocol. Complementary DNA (cDNA) was generated
using a RevertAid cDNA Synthesis kit (Fermentas; Thermo Fisher
Scientific, Inc.). RT-qPCR was performed using a SYBR1 Taq™ kit
(Takara Bio, Inc.,) based on the ABI PRISM 7000 system. The
thermocycling conditions consisted of an initial denaturation at
95°C for 5 min, followed by 40 cycles at 95°C for 20 sec, 55°C for
20 sec, and 72°C for 20 sec. β-actin was used as the
reference gene. The results were analyzed using the
2−ΔΔCq method (9). The
primer sequences were as follows: ACE2 forward,
5′-CATTGGAGCAAGTGTTGGATCTT-3′ and reverse
5′-GAGCTAATGCATGCCATTCTCA-3′; Arkadia forward,
5′-CCACATAGGATGCACCCAAAC-3′ and reverse, 5′-AATTCCCAGTTCCCAGGCA-3′;
Smad7 forward, 5′-CCTTAGCCGACTCTGCGAACTA-3′ and reverse,
5′-CCAGATAATTCGTTCCCCCTGT-3′; E-cadherin forward,
5′-GAGAACGCATTGCCACATACAC-3′ and reverse,
5′-GCACCTTCCATGACAGACCC-3′; α-SMA forward,
5′-ACTGGGACGACATGGAAAAG-3′ and reverse, 5′-CATCTCCAGAGTCCAGCACA-3′;
and β-actin forward, 5′-TGGGTCAGAAGGACTCCTATG-3′ and
reverse, 5′-CAGGCAGCTCATAGCTCTTCT-3′.
Statistical analysis
Data were expressed as the means ± standard
deviations (SDs). Comparisons between groups were performed using
one-way analysis of variance, followed by the Student-Newman-Keuls
test. Statistical analyses were performed using SPSS v17.0 software
(SPSS, Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
Clinical characteristics of and serum
ACE2 expression levels in diabetic and non-diabetic
participants
In total, 20 participants were recruited to the
present study (10 diabetic and 10 non-diabetic). The
characteristics of the study participants are presented in Table I. The age of the diabetic patients
(59.0±14.7 years) and non-diabetic volunteers (62.6±17.0 years)
were similar (P=0.248). There were no significant differences in
serum creatinine or systolic and diastolic blood pressure. However,
the serum ACE2 level was significantly higher in patients with
diabetes compared with non-diabetic volunteers (10.2±2.6 vs.
24.7±3.9 ng/ml, P=0.007).
 | Table I.Clinical characteristics of study
subjects. |
Table I.
Clinical characteristics of study
subjects.
|
Characteristics | Non-diabetic
(n=10) | Diabetic
(n=10) | P-value |
|---|
| Age, years | 59.0±14.7 | 62.6±17.0 | 0.248 |
| Sex,
male/female | 5/5 | 5/5 | – |
| SBP, mmHg | 135.2±15.4 | 138.1±14.3 | 0.423 |
| DBP, mmHg | 78.5±9.8 | 80.2±9.7 | 0.527 |
| Glucose,
mmol/l | 4.8±0.4 | 9.5±2.3 | 0.002 |
| Serum creatinine,
µmol/l | 56.6±7.5 | 57.1±10.7 | 0.454 |
| UACR, mg/g | – | 112.5±11.8 | – |
| ACE2, ng/ml | 10.2±2.6 | 24.7±3.9 | 0.007 |
Expression of ACE2 is lower and
Arkadia expression is higher in diabetic human kidneys
Since tubulointerstitial fibrosis is one of the
major pathological alterations in DN (11), renal fibrosis was evaluated by
Masson's trichrome staining in diabetic and non-diabetic human
kidneys. Furthermore, immunohistochemistry was used to examine the
expression of ACE2 and Arkadia in diabetic and non-diabetic human
kidneys. Histologically, PAS and PASM staining demonstrated
profound extracellular matrix deposition and Masson's trichrome
staining revealed more severe renal fibrosis in diabetic than in
non-diabetic human kidneys (Fig. 1A
and B). Immunohistochemistry demonstrated significantly lower
ACE2 expression in diabetic samples compared with the control
kidneys, whereas the expression of Arkadia was higher (Fig. 1C and D).
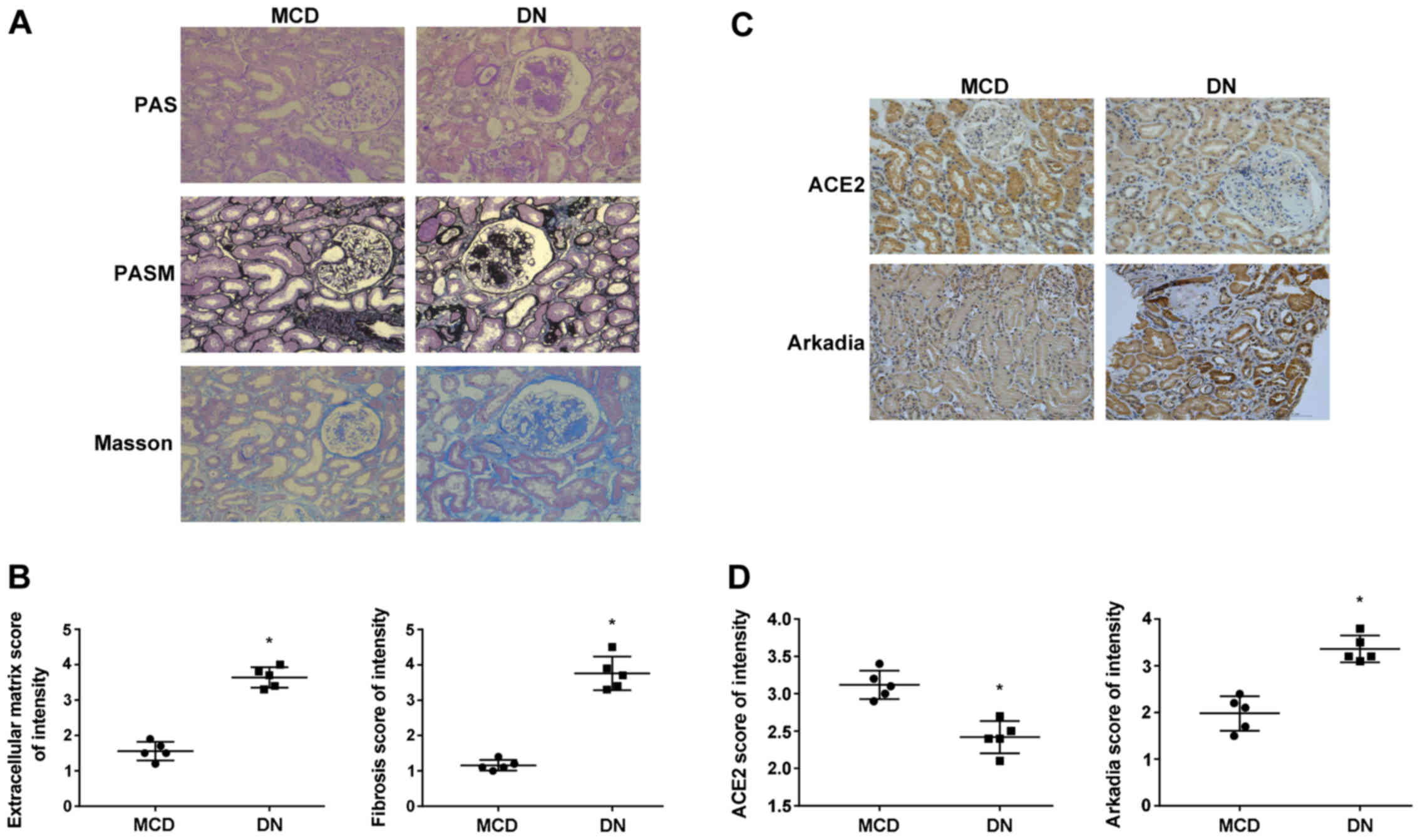 | Figure 1.Deposition of extracellular matrix
and collagen, and the expression of ACE2 and Arkadia in diabetic
and non-diabetic human kidneys. (A) Periodic acid-Schiff staining,
Periodic Schiff-Methenamine Silver staining and Masson's trichrome
staining of renal sections, Scale bar, 100 µm. (B) Scored results
of the PASM and Masson staining (1 = weakest; 4 = strongest). Each
dot represents an individual specimen. (C) Immunohistochemical
staining for ACE2 and Arkadia, Scale bar, 100 µm. (D) Scored
results of the immunohistochemical staining (1 = weakest; 4 =
strongest). Each dot represents an individual specimen. *P<0.05
compared with the minimal change nephropathy samples. ACE2,
angiotensin-converting enzyme 2; MCD, minimal change nephropathy;
DN, diabetic nephropathy; PAS, Periodic acid-Schiff; PASM, Periodic
Schiff-Methenamine Silver. |
High glucose reduces ACE2 and induces
Arkadia expression and increases α-SMA and decreases E-cadherin
expression in HRPTEpiCs
The present study subsequently investigated the
effects of high glucose on the expression of ACE2, Arkadia and EMT
in HRPTEpiCs. As revealed in Fig.
2, both the protein (Fig. 2A and
B) and mRNA (Fig. 2C)
expression levels of ACE2 decreased with time, while the mRNA and
protein expression levels of Arkadia were increased with time in
HRPTEpiCs incubated with a high glucose media. Furthermore, the
expression of α-SMA, a myofibroblast marker, increased, whereas
that of E-cadherin, an epithelial marker that plays a key role in
maintaining the integrity of epithelial cells, decreased.
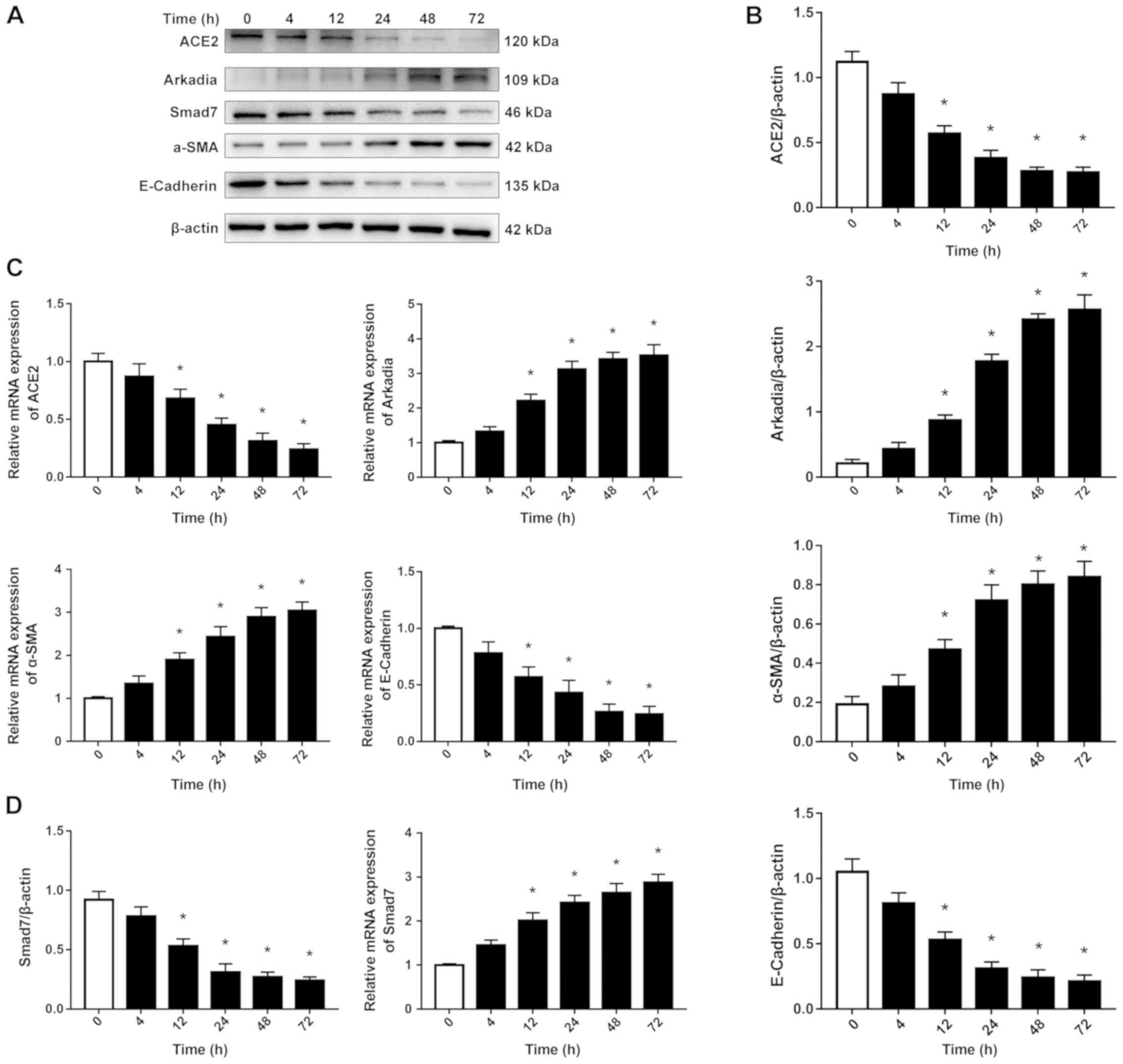 | Figure 2.ACE2, Arkadia, Smad7, α-SMA and
E-cadherin expression in HRPTEpiCs stimulated with high glucose.
HRPTEpiCs were incubated in a high glucose medium (30 mmol/l
D-glucose) for the indicated periods of time. (A) Western blotting
and (B) its semi-quantification demonstrated that high glucose
increased the expression of Arkadia and α-SMA, and decreased the
expression of ACE2 and E-cadherin in a time-dependent manner. (C)
RT-qPCR revealed that the mRNA expression levels of Arkadia and
α-SMA increased, while that of ACE2 and E-cadherin decreased with
time. Results were normalized to β-actin expression. (D) Semi
-quantification of western blotting results revealed a gradual
reduction in Smad7 protein expression level and RT-qPCR revealed
that Smad7 mRNA expression level was increased in HRPTEpiCs
stimulated with high glucose. *P<0.05 compared with time 0 h.
ACE2, angiotensin-converting enzyme 2; α-SMA, α-smooth muscle
actin; HRPTEpiCs, human renal proximal tubular epithelial cells;
RT-qPCR, reverse transcription-quantitative PCR; Smad7, SMAD Family
member 7. |
Smad7 mRNA increases while Smad7
protein expression level decreases in HRPTEpiCs stimulated with
high glucose
Western blot analysis was used to determine the
expression levels of Smad7 protein in HRPTEpiCs at different
time-points during the stimulation of high glucose (Fig. 2A and D). High glucose gradually but
significantly reduced the expression of Smad7 protein. To determine
whether the decrease in Smad7 protein expression resulted from
downregulation of Smad7 mRNA expression, mRNA expression
levels were assessed by RT-qPCR. In contrast to the significant
decrease in Smad7 protein, the expression levels of Smad7
mRNA increased with time in HRPTEpiCs stimulated with high glucose.
These results indicated that Smad7 expression was downregulated at
the protein level in the presence of a high glucose
concentration.
ACE2 overexpression inhibits Arkadia
expression and alleviates high-glucose-induced EMT in
HRPTEpiCs
Next, ACE2 was overexpressed or inhibited in
HRPTEpiCs stimulated with high glucose. Ad-ACE2 infection
significantly increased the expression of ACE2 mRNA (Fig. S1A). The RT-qPCR and western blot
analysis revealed that ACE2 overexpression significantly reduced
both the mRNA and protein expression of Arkadia (Fig. 3A and B), whereas ACE2 inhibition
increased Arkadia mRNA and protein levels (Fig. 4A and B) in HRPTEpiCs stimulated
with high glucose. Accordingly, high-glucose-induced EMT, indicated
by upregulation of α-SMA and downregulation of E-cadherin
expression, was alleviated when ACE2 was overexpressed (Fig. 3A and C) and aggravated when ACE2
was inhibited (Fig. 4A and D).
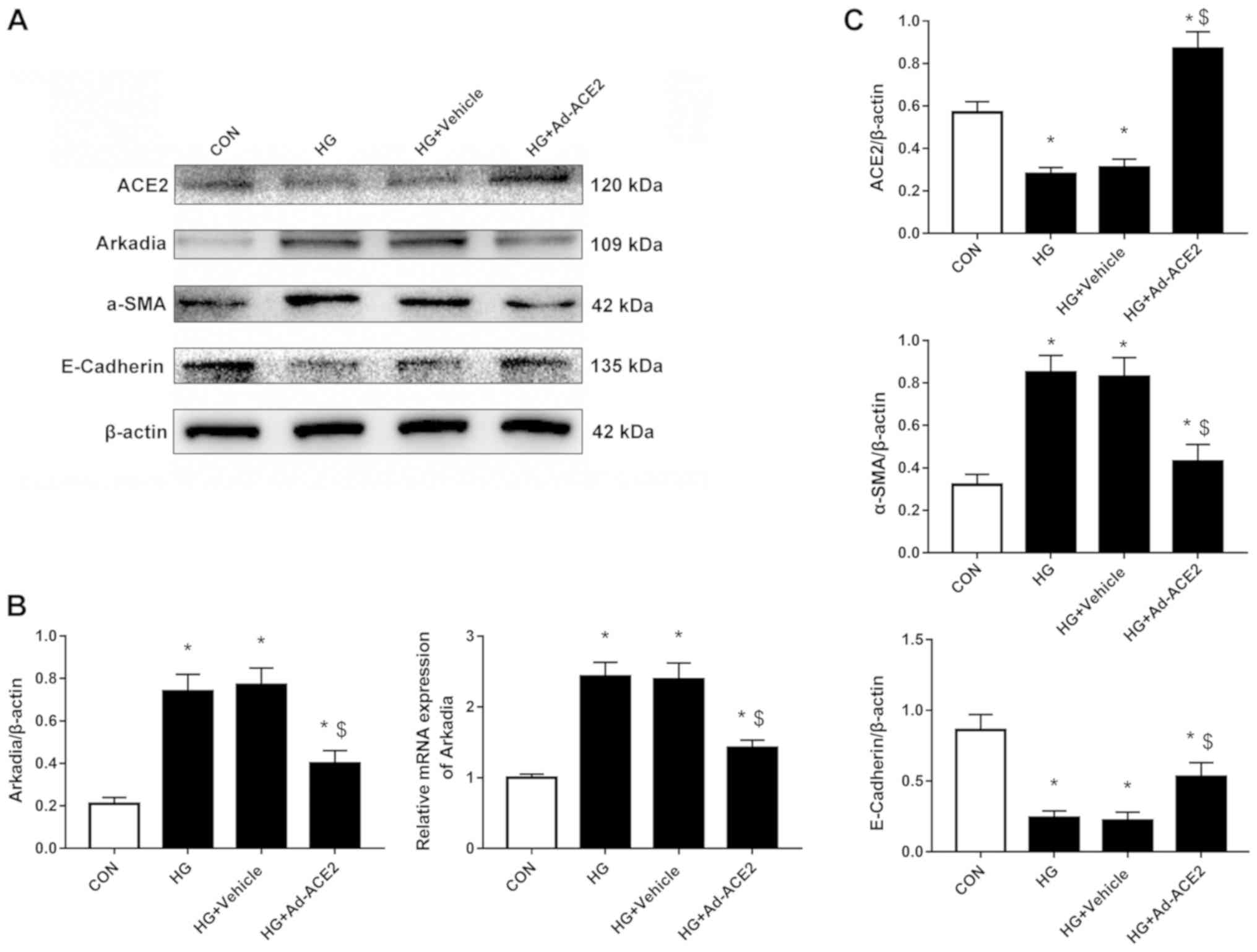 | Figure 3.ACE2, Arkadia, α-SMA and E-cadherin
expression when ACE2 is overexpressed. HRPTEpiCs were infected with
Ad-ACE2 or empty adenovirus for 48 h. (A) Western blotting
demonstrated that the expression of ACE2 was significantly induced
by the specific adenovirus and reduced in high glucose conditions.
ACE2 overexpression alleviated the effects of high glucose on the
expression of Arkadia, α-SMA and E-cadherin. (B)
Semi-quantification of the Arkadia protein and mRNA expression
levels when ACE2 was overexpressed as detected by western blotting
and reverse transcription-quantitative PCR, respectively. (C)
Semi-quantification of ACE2, α-SMA and E-cadherin protein
expression levels when ACE2 was overexpressed. Results were
normalized to β-actin expression. *P<0.05 compared with control
cells; $P<0.05 compared with high glucose-treated
cells. ACE2, angiotensin-converting enzyme 2; α-SMA, α-smooth
muscle actin; CON, control; HG, high glucose; HRPTEpiCs, human
renal proximal tubular epithelial cells; Ad-ACE2, adenoviruses
overexpressing ACE2. |
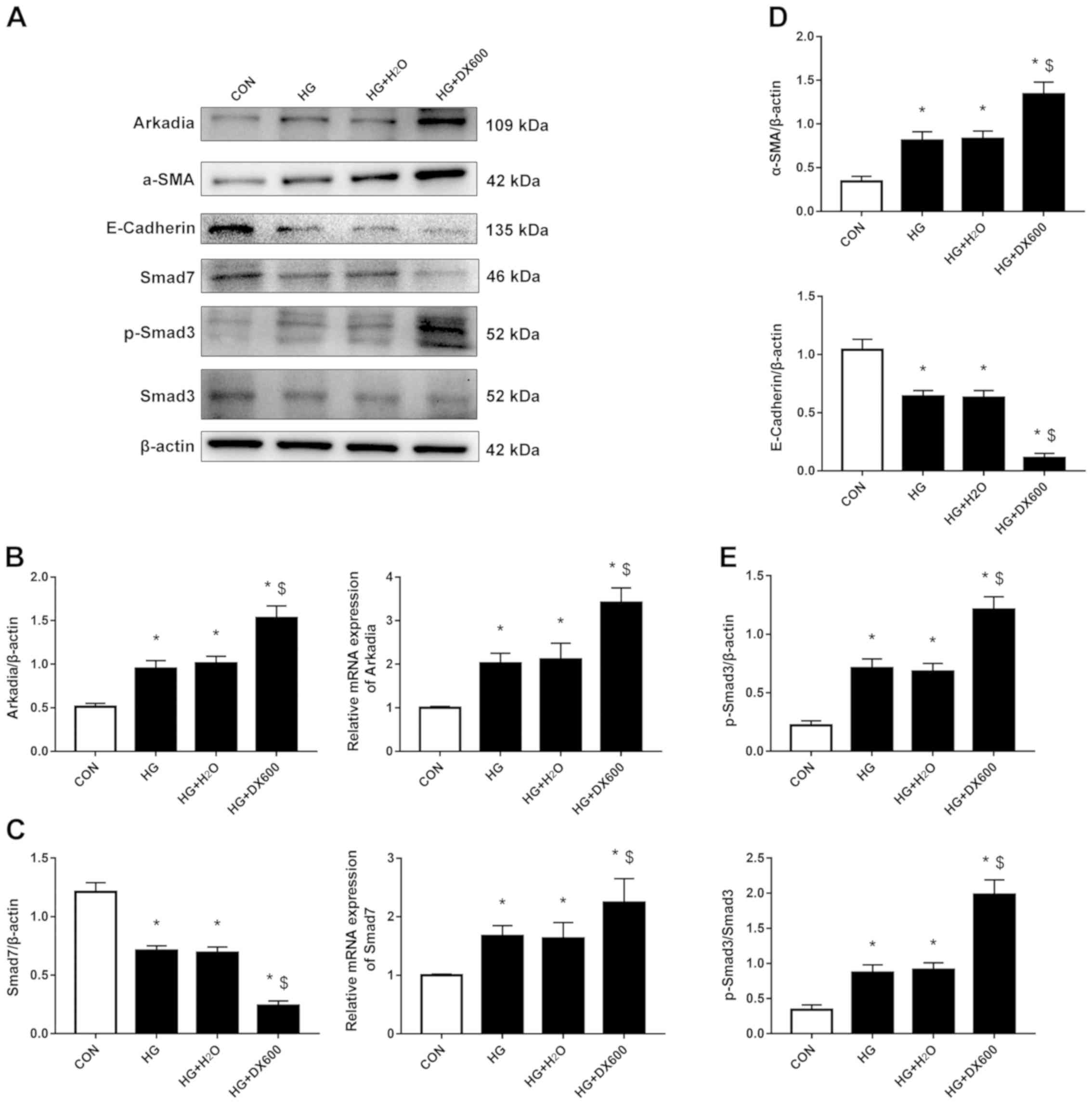 | Figure 4.ACE2, Arkadia, Smad7, p-Smad3, α-SMA
and E-cadherin expression when ACE2 was inhibited. HRPTEpiCs were
treated with the ACE2 inhibitor DX600 for 48 h. (A) Western
blotting demonstrated that ACE2 inhibition aggravated the effects
of high glucose on the expression of Arkadia, α-SMA and E-cadherin.
Smad7 protein expression levels were decreased in HRPTEpiCs
stimulated with high glucose and further reduced by DX600
treatment, while p-Smad3 exhibited the opposite changes. (B)
Semi-quantification of Arkadia protein and mRNA expression levels
after ACE2 inhibition as detected by western blotting and RT-qPCR,
respectively. (C) Semi-quantification of Smad7 protein and mRNA
expression levels after ACE2 inhibition as detected by western
blotting and RT-qPCR, respectively. (D) Semi-quantification of the
expression levels of α-SMA and E-cadherin protein when ACE2 was
inhibited. (E) Semi-quantification of p-Smad3 protein when ACE2 was
inhibited. Results were normalized to β-actin expression.
*P<0.05 compared with control cells; $P<0.05
compared with high glucose-treated cells. ACE2,
angiotensin-converting enzyme 2; α-SMA, α-smooth muscle actin; CON,
control; HG, high glucose; HRPTEpiCs, human renal proximal tubular
epithelial cells; p-, phosphorylat |
ACE2 alleviates high-glucose-induced
EMT in HRPTEpiCs by downregulating Arkadia-dependent ubiquitin
degradation of Smad7 and inhibiting the TGF-β/Smad pathway
HRPTEpiCs were stimulated with high glucose in the
presence of an ACE2 inhibitor and/or Arkadia siRNA to
explore the mechanism by which ACE2 deactivation promotes EMT when
exposed to a high glucose concentration. Transfection with
siArkadia significantly decreased Arkadia expression (Fig. S1A). ACE2 inhibition was found to
reduce Smad7 protein expression, but induced Smad7 mRNA
expression, and substantially activated the TGF-β/Smad signaling
pathway, as detected by p-Smad3, in high-glucose-stimulated
HRPTEpiCs (Fig. 4A, C and E). To
further confirm the effects of the ACE2/Arkadia/Smad7 axis on EMT,
high-glucose-stimulated HRPTEpiCs were cotreated with an ACE2
inhibitor and Arkadia siRNA. All the effects of ACE2
deactivation observed were reversed following depletion of Arkadia
(Fig. 5). Moreover, the expression
of TGF-β, another crucial component of the TGF-β/Smad signaling
pathway, was detected and exhibited a similar trend to that of
p-Smad3 expression (Fig. S1B and
C). Collectively, these findings indicated that ACE2 protected
against high-glucose-induced renal fibrosis by inhibiting EMT,
which may be at least in part due to inhibition of
Arkadia-dependent ubiquitin degradation of Smad7 and the TGF-β/Smad
signaling pathway.
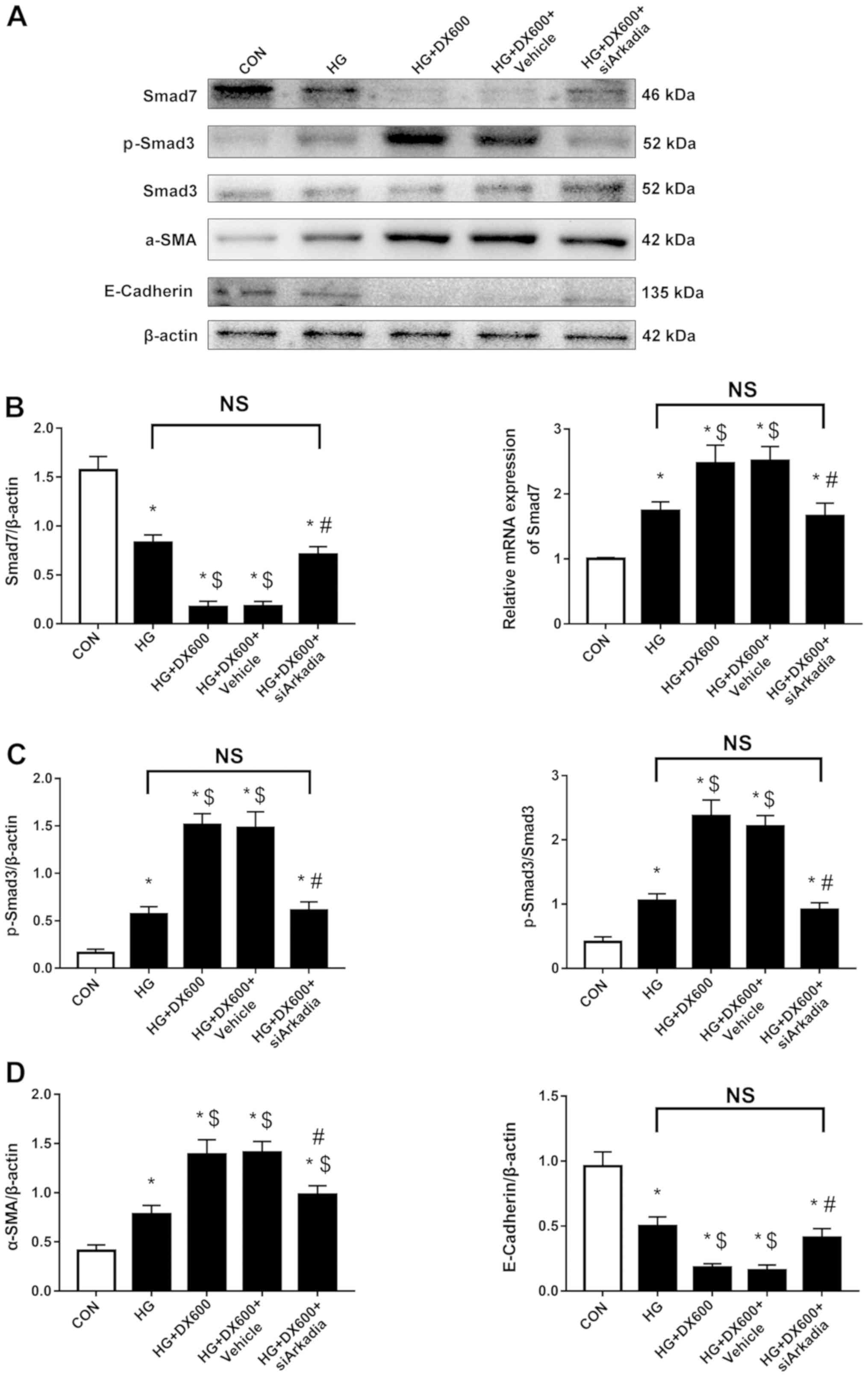 | Figure 5.Arkadia knockdown increases Smad7
protein and inhibits activation of the TGF-β/Smad signaling pathway
and epithelial to mesenchymal cell transition in HRPTEpiCs. (A)
Western blotting demonstrated that Smad7 and E-cadherin protein
expression levels were decreased in HRPTEpiCs co-treated with high
glucose and DX600, but increased following transfection with
siArkadia, while p-Smad3 and α-SMA exhibited the opposite changes.
(B) Semi-quantification of Smad7 protein and mRNA expression levels
after Arkadia depletion as detected by western blotting and reverse
transcription-quantitative PCR, respectively. (C)
Semi-quantification of p-Smad3 protein after Arkadia knockdown. (D)
Semi-quantification of α-SMA and E-cadherin protein after Arkadia
knockdown. Results were normalized to β-actin expression.
*P<0.05 compared with control cells; $P<0.05
compared with high glucose-treated cells; #P<0.05
compared with cells co-treated with high glucose and DX600. α-SMA,
α-smooth muscle actin; CON, control; HG, high glucose; HRPTEpiCs,
human renal proximal tubular epithelial cells; siArkadia, small
interfering RNA targeting Arkadia; p-, phosphorylated; Smad, SMAD
family member; NS, not significant |
Discussion
The present study has demonstrated the relationship
between ACE2 and the ubiquitination pathway in DN. Inhibition of
ACE2 appeared to suppress EMT in vitro and in vivo
via an Arkadia-dependent mechanism. This process would lead to an
increase in TGF-β/Smad signaling, the key regulatory pathway in
EMT, by targeting Smad7 for ubiquitin-mediated degradation.
Moreover, the present study also revealed that serum ACE2 was
higher in patients with DN with microproteinuria.
It is widely accepted that the activation of RAS,
particularly the intrarenal RAS, is important for the progression
of DN (12). ACE2 is an ACE
homolog that counter-regulates the RAS. It exists both as a
tissue-bound enzyme, which is highly expressed in the proximal
tubule brush border, and as a soluble protein that has enzymatic
effects in the circulatory system (13,14).
ACE2 expression is downregulated in the kidneys of patients with DN
(15). More recently, ACE2
knockout was revealed to be associated with EMT and renal fibrosis
in mouse models of obstructive, hypertensive and diabetic
nephropathy, which feature α-SMA accumulation and E-cadherin
deficiency (3,16,17).
In contrast, ACE2 overexpression ameliorated renal fibrosis and
albuminuria in diabetic mice (18). ACE2 is generally conceived as a
renoprotective factor that works by converting Ang II to Ang(1–7),
which has its cellular effects via the G protein-coupled receptor
Mas (19,20). The cross talk between renal ACE and
ACE2, as well as the balance between the ACE/Ang II/AT1R and
ACE2/Ang(1–7)/Mas axes have also been emphasized in vivo and
in vitro (21–24). However, it appears that ACE2 has
effects that are far more complex than RAS regulation (3,25).
This led to the present investigation to decipher the detailed
mechanism by which ACE2 exerts its renoprotective effects in
patients with DN and HRPTEpiCs in a high glucose environment.
In the present study, ACE2 expression was
downregulated, whereas the expression of Arkadia, an E3 ubiquitin
ligase critical for the TGF-β/Smad signaling pathway during EMT
(26,27), was upregulated in both diabetic
human kidneys and HRPTEpiCs stimulated with high glucose. These
results indicated that ACE2 affected EMT by modulating Arkadia
expression. Accumulating evidence suggests that Smad7 is a major
regulator of the intensity and duration of Smad signals (28–31).
In the present study, Smad7 protein expression level was decreased
while its mRNA expression level was increased. Therefore, it was
hypothesized that the decrease in Smad7 protein expression was
caused by greater degradation rather than lower expression. Arkadia
has been reported to aggravate organ damage by inducing Smad
signals via the degradation of Smad7 in pulmonary fibrosis, atrial
fibrillation and aristolochic acid nephropathy (32–34).
In fact, inhibition or overexpression of ACE2 caused upregulation
and downregulation of Arkadia expression, respectively, which
resulted in changes in Smad7 protein and mRNA expression levels in
high-glucose-stimulated HRPTEpiCs. Consistently, Arkadia knockdown
increased Smad7 protein expression levels and attenuated
high-glucose-induced EMT when ACE2 was inhibited. Therefore, it was
hypothesized that ACE2 was able to suppress EMT by preventing
Arkadia-dependent ubiquitination, which degrades Smad7 protein in
high-glucose-stimulated HRPTEpiCs. This process is essential for
renal fibrosis in DN and had not been reported in previous
studies.
Another finding of the present study was that
patients with DN with microalbuminuria displayed a significantly
higher serum ACE2 expression level compared with the controls. Few
human studies have previously measured circulating ACE2 in the
context of DN. In experimental studies, circulating ACE2 activity
has been reported to be higher in mouse models of diabetes
(35–38). To date, only one study of patients
with type I diabetes with vascular complications demonstrated
higher ACE2 activity in the circulation (39). In concordance, the present study
revealed a high serum ACE2 level in patients with DN with
microalbuminuria. Numerous previous studies have linked high
circulating ACE2 expression levels with an increased risk of
cardiovascular disease, which is common in patients with DN
(38,40–42).
The increased circulating ACE2 expression level in these patients
may stem from cardiovascular injury or occur to compensate for the
downregulated renal ACE2. However, this result was difficult to
confirm due to the small sample size. Nevertheless, the present
findings still suggest a possible diagnostic role for the
measurement of circulating ACE2 in DN, but further larger-scale
clinical studies are still required to confirm this finding.
In conclusion, the present study has demonstrated
that intrarenal ACE2 had a protective effect in DN. ACE2 suppressed
Arkadia-dependent ubiquitin degradation of Smad7, which may be an
essential mechanism by which TGF-β/Smad-mediated EMT was
ameliorated in high-glucose-stimulated HRPTEpiCs.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the Beijing Municipal
Administration of Hospitals Clinical Medicine Development of
Special Funding Support, Grant/Award no. ZYLX201824 and the
National Natural Science Foundation of China, Grant/Award no.
81570660.
Availability of data and materials
The datasets used and analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
WL and ZC provided the concept and designed the
study. ZC and XC performed the experiments, and the results were
then interpreted by ZC, ZD and YB. ZC prepared the figures and
drafted the manuscript. ZC, ZD and WL edited and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures were approved by the Ethics Committee
of Beijing Friendship Hospital, Capital Medical University. Each
participant provided their written informed consent before being
included in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ACE2
|
angiotensin-converting enzyme 2
|
|
DN
|
diabetic nephropathy
|
|
HRPTEpiCs
|
Human renal proximal tubular
epithelial cells
|
|
EMT
|
epithelial to mesenchymal cell
transition
|
|
RAS
|
renin-angiotensin system
|
|
TGF-β
|
transforming growth factor-β
|
References
|
1
|
Giacco F, Du X, D'Agati VD, Milne R, Sui
G, Geoffrion M and Brownlee M: Knockdown of glyoxalase 1 mimics
diabetic nephropathy in nondiabetic mice. Diabetes. 63:3008–299.
2014. View Article : Google Scholar
|
|
2
|
Rahimi Z: The role of renin angiotensin
aldosterone system genes in diabetic nephropathy. Can J Diabetes.
40:178–183. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Culver S, Li C and Siragy HM: Intrarenal
angiotensin-converting enzyme: The old and the new. Curr Hypertens
Rep. 19:802017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Batlle D, Wysocki J, Soler MJ and
Ranganath K: Angiotensin-converting enzyme 2: Enhancing the
degradation of angiotensin II as a potential therapy for diabetic
nephropathy. Kidney Int. 81:520–528. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen L, Yang T, Lu DW, Zhao H, Feng YL,
Chen H, Chen DQ, Vaziri ND and Zhao YY: Central role of
dysregulation of TGF-β/Smad in CKD progression and potential
targets of its treatment. Biomed Pharmacother. 101:670–681. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meng XM, Tang PM, Li J and Lan HY:
TGF-β/Smad signaling in renal fibrosis. Front Physiol. 6:822015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu FY, Li XZ, Peng YM, Liu H and Liu YH:
Arkadia regulates TGF-β signaling during renal tubular epithelial
to mesenchymal cell transition. Kidney Int. 73:588–594. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen X, Wu Y, Diao Z, Han X, Li D, Ruan X
and Liu W: C1q/tumor necrosis factor-related protein-3 improves
renal fibrosis via inhibiting notch signaling pathways. J Cell
Physiol. 234:22352–22364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li X, Diao Z, Ding J, Liu R, Wang L, Huang
W and Liu W: The downregulation of SnoN expression in human renal
proximal tubule epithelial cells under high-glucose conditions is
mediated by an increase in Smurf2 expression through TGF-β1
signaling. Int J Mol Med. 37:415–422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang LY, Diao ZL, Zheng JF, Wu YR, Zhang
QD and Liu WH: Apelin attenuates TGF-β1-induced epithelial to
mesenchymal transition via activation of PKC-ε in human renal
tubular epithelial cells. Peptides. 96:44–52. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu SM and Bonventre JV: Acute kidney
injury and progression of diabetic kidney disease. Adv Chronic
Kidney Dis. 25:166–180. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Williams VR and Scholey JW:
Angiotensin-converting enzyme 2 and renal disease. Curr Opin
Nephrol Hypertens. 27:35–41. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Koka V, Huang XR, Chung AC, Wang W, Truong
LD and Lan HY: Angiotensin II up-regulates angiotensin I-converting
enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP
kinase pathway. Am J Pathol. 172:1174–1183. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wysocki J, Ye M, Khattab AM, Fogo A,
Martin A, David NV, Kanwar Y, Osborn M and Batlle D:
Angiotensin-converting enzyme 2 amplification limited to the
circulation does not protect mice from development of diabetic
nephropathy. Kidney Int. 91:1336–1346. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Reich HN, Oudit GY, Penninger JM, Scholey
JW and Herzenberg AM: Decreased glomerular and tubular expression
of ACE2 in patients with type 2 diabetes and kidney disease. Kidney
Int. 74:1610–1616. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu Z, Huang XR, Chen HY, Penninger JM and
Lan HY: Loss of angiotensin-converting enzyme 2 enhances
TGF-β/Smad-mediated renal fibrosis and NF-κB-driven renal
inflammation in a mouse model of obstructive nephropathy. Lab
Invest. 92:650–661. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu Z, Huang XR, Chen HY, Fung E, Liu J
and Lan HY: Deletion of angiotensin-converting enzyme-2 promotes
hypertensive nephropathy by targeting Smad7 for ubiquitin
degradation. Hypertension. 70:822–830. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lo CS, Shi Y, Chang SY, Abdo S, Chenier I,
Filep JG, Ingelfinger JR, Zhang SL and Chan JS: Overexpression of
heterogeneous nuclear ribonucleoprotein F stimulates renal Ace-2
gene expression and prevents TGF-β1-induced kidney injury in a
mouse model of diabetes. Diabetologia. 58:2443–2454. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Santos RA, Simoes e Silva AC, Maric C,
Silva DM, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SV,
Lopes MT, Bader M, et al: Angiotensin-(1–7) is an endogenous ligand
for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA.
100:8258–8263. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Simões E Silva AC and Teixeira MM and
Teixeira MM: ACE inhibition, ACE2 and angiotensin-(1–7) axis in
kidney and cardiac inflammation and fibrosis. Pharmacol Res.
107:154–162. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oudit GY, Liu GC, Zhong J, Basu R, Chow
FL, Zhou J, Loibner H, Janzek E, Schuster M, Penninger JM, et al:
Human recombinant ACE2 reduces the progression of diabetic
nephropathy. Diabetes. 59:529–538. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Márquez E, Riera M, Pascual J and Soler
MJ: Albumin inhibits the insulin-mediated ACE2 increase in cultured
podocytes. Am J Physiol Renal Physiol. 306:F1327–F1334. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Callera GE, Antunes TT, Correa JW, Moorman
D, Gutsol A, He Y, Cat AN, Briones AM, Montezano AC, Burns KD, et
al: Differential renal effects of candesartan at high and
ultra-high doses in diabetic mice-potential role of the
ACE2/AT2R/Mas axis. Biosci Rep. 36:362016. View Article : Google Scholar
|
|
24
|
Lin M, Gao P, Zhao T, He L, Li M, Li Y,
Shui H and Wu X: Calcitriol regulates angiotensin-converting enzyme
and angiotensin converting-enzyme 2 in diabetic kidney disease. Mol
Biol Rep. 43:397–406. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin HY, Chen LJ, Zhang ZZ, Xu YL, Song B,
Xu R, Oudit GY, Gao PJ, Zhu DL and Zhong JC: Deletion of
angiotensin-converting enzyme 2 exacerbates renal inflammation and
injury in apolipoprotein E-deficient mice through modulation of the
nephrin and TNF-alpha-TNFRSF1A signaling. J Transl Med. 13:2552015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Koinuma D, Shinozaki M, Komuro A, Goto K,
Saitoh M, Hanyu A, Ebina M, Nukiwa T, Miyazawa K, Imamura T, et al:
Arkadia amplifies TGF-beta superfamily signalling through
degradation of Smad7. EMBO J. 22:6458–6470. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Birkou M, Chasapis CT, Marousis KD,
Loutsidou AK, Bentrop D, Lelli M, Herrmann T, Carthy JM, Episkopou
V and Spyroulias GA: A Residue Specific Insight into the Arkadia E3
Ubiquitin Ligase Activity and Conformational Plasticity. J Mol
Biol. 429:2373–2386. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen HY, Huang XR, Wang W, Li JH, Heuchel
RL, Chung AC and Lan HY: The protective role of Smad7 in diabetic
kidney disease: Mechanism and therapeutic potential. Diabetes.
60:590–601. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Henique C and Tharaux PL: Targeting
signaling pathways in glomerular diseases. Curr Opin Nephrol
Hypertens. 21:417–427. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lai JY, Luo J, O'Connor C, Jing X, Nair V,
Ju W, Randolph A, Ben-Dov IZ, Matar RN, Briskin D, et al:
MicroRNA-21 in glomerular injury. J Am Soc Nephrol. 26:805–816.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luo M, Tan X, Mu L, Luo Y, Li R, Deng X,
Chen N, Ren M, Li Y, Wang L, et al: MiRNA-21 mediates the
antiangiogenic activity of metformin through targeting PTEN and
SMAD7 expression and PI3K/AKT pathway. Sci Rep. 7:434272017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He X, Gao X, Peng L, Wang S, Zhu Y, Ma H,
Lin J and Duan DD: Atrial fibrillation induces myocardial fibrosis
through angiotensin II type 1 receptor-specific Arkadia-mediated
downregulation of Smad7. Circ Res. 108:164–175. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tian Y, Liao F and Wu G, Chang D, Yang Y,
Dong X, Zhang Z, Zhang Y and Wu G: Ubiquitination and regulation of
Smad7 in the TGF-β1/Smad signaling of aristolochic acid
nephropathy. Toxicol Mech Methods. 25:645–652. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Elkouris M, Kontaki H, Stavropoulos A,
Antonoglou A, Nikolaou KC, Samiotaki M, Szantai E, Saviolaki D,
Brown PJ, Sideras P, et al: SET9-Mediated Regulation of TGF-β
Signaling Links Protein Methylation to Pulmonary Fibrosis. Cell
Rep. 15:2733–2744. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tikellis C, Bialkowski K, Pete J, Sheehy
K, Su Q, Johnston C, Cooper ME and Thomas MC: ACE2 deficiency
modifies renoprotection afforded by ACE inhibition in experimental
diabetes. Diabetes. 57:1018–1025. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamaleyeva LM, Gilliam-Davis S, Almeida I,
Brosnihan KB, Lindsey SH and Chappell MC: Differential regulation
of circulating and renal ACE2 and ACE in hypertensive mRen2.Lewis
rats with early-onset diabetes. Am J Physiol Renal Physiol.
302:F1374–F1384. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Riera M, Anguiano L, Clotet S, Roca-Ho H,
Rebull M, Pascual J and Soler MJ: Paricalcitol modulates ACE2
shedding and renal ADAM17 in NOD mice beyond proteinuria. Am J
Physiol Renal Physiol. 310:F534–F546. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Anguiano L, Riera M, Pascual J and Soler
MJ: Circulating ACE2 in cardiovascular and kidney diseases. Curr
Med Chem. 24:3231–3241. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Soro-Paavonen A, Gordin D, Forsblom C,
Rosengard-Barlund M, Waden J, Thorn L, Sandholm N, Thomas MC and
Groop PH; FinnDiane Study Group, : Circulating ACE2 activity is
increased in patients with type 1 diabetes and vascular
complications. J Hypertens. 30:375–383. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Anguiano L, Riera M, Pascual J,
Valdivielso JM, Barrios C, Betriu A, Mojal S, Fernández E and Soler
MJ; NEFRONA study, : Circulating angiotensin-converting enzyme 2
activity in patients with chronic kidney disease without previous
history of cardiovascular disease. Nephrol Dial Transplant.
30:1176–1185. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Anguiano L, Riera M, Pascual J,
Valdivielso JM, Barrios C, Betriu A, Clotet S, Mojal S, Fernández E
and Soler MJ; Investigators from the NEFRONA Study, : Circulating
angiotensin converting enzyme 2 activity as a biomarker of silent
atherosclerosis in patients with chronic kidney disease.
Atherosclerosis. 253:135–143. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen YY, Zhang P, Zhou XM, Liu D, Zhong
JC, Zhang CJ, Jin LJ and Yu HM: Relationship between genetic
variants of ACE2 gene and circulating levels of ACE2 and its
metabolites. J Clin Pharm Ther. 43:189–195. 2018. View Article : Google Scholar : PubMed/NCBI
|














