Introduction
Usher syndrome is a group of genetically and
clinically heterogeneous autosomal recessive diseases with
progressive retinitis pigmentosa (RP) and sensorineural hearing
deficiencies, including Usher syndrome types I, II and III
(1–4). Patients with Usher syndrome type II
(USH2) present with mild hearing impairments but normal vestibular
responses, and type II is the most common type amongst the three
types (1). Usher syndrome type II
is genetically heterogeneous, including USH2A, USH2C and USH2D.
Usher syndrome type IIA (USH2A locus) (OMIM, 276901) is the result
of mutations of the USH2A gene (OMIM, 608400) (5), whereas USH2C (OMIM, 605472) is the
result of mutations of the ADGRV1 gene (OMIM, 602851)
(1,6) or by digenic mutations of both
ADGRV1 and PDZD7 genes (OMIM, 612971) (7) and USH2D (OMIM, 611383) can be caused
by mutations of the whirlin (WHRN) gene (OMIM, 607928)
(8). As Hmani-Aifa et al
(9) identified mutations of the
adhesion G-protein coupled receptor V1 gene in an Usher II syndrome
pedigree which had been previously mapped to the USH2B locus of
chromosome 3p23-p24.2, the designation for the USH2B locus was
withdrawn.
Aliases for the USH2A gene include
Usherin, USH2, US2, Usher Syndrome 2A, Usher Syndrome Type-2A
Protein, Usher Syndrome Type IIa Protein, DJ1111A8.1 and
RP39. This gene maps to chromosome 1q41, encoding a protein
5,202 amino acids in length that is comprised of a pentaxin motif,
laminin epidermal growth factor (EGF) motifs and numerous
fibronectin type III domains (10). Different isoforms have also been
identified (5,10–12).
The protein is localized in the basement membrane and serves a
vital role in the development and homeostasis of the inner ear and
retina (2). Mutations of the
USH2A gene have been identified in patients with Usher
syndrome type IIA and non-syndromic RP (13). Eudy et al (5) was the first to identify USH2A
mutations among patients with Usher syndrome type IIA.
The association between the variants in the Usher
syndrome-causative genes and the resultant Usher syndrome
phenotypes in patients are highly variable (1,3,14).
To the best of our knowledge, novel USH2A mutations in
patients with Usher syndrome type IIA and association between the
genotypes and phenotypes have not been well documented in the
Chinese population. In the present study, by using targeted
next-generation sequencing (TGS), Sanger sequencing and segregation
analysis, novel nonsense, compound heterozygous mutations in the
USH2A gene were identified in a Chinese pedigree with Usher
syndrome.
Materials and methods
Pedigree and clinical assessment,
blood collection, and DNA isolation
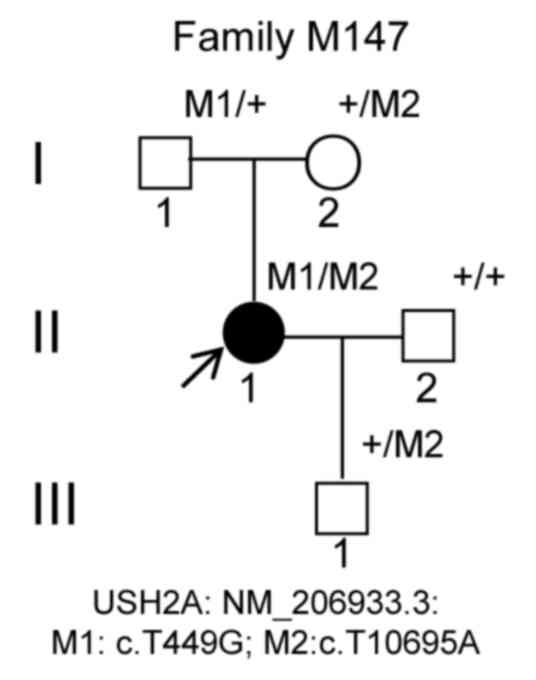
The M147 pedigree consisted of a proband and four
family members from Sichuan, China (Fig. 1A). The proband was a 35-year-old
female, and her parents, husband and son were 71, 60, 36 and
5-years-old, respectively. The blood samples of the proband were
collected in June 2013, and TGS analysis was performed in December
2016. The blood samples of the four family members were collected
in March 2019 for segregation analysis in our laboratory. For
detailed clinical assessments, clinical history and ophthalmic
examinations were performed with the proband, including fundoscopy,
fundus photographs and fundus fluorescent photographs as previously
described (15). Audiometric
testing in the proband was also performed using pure-tone
audiometry with different frequencies (0.25, 0.5, 1, 2, 4 and 8
kHz) (1). Written informed consent
from the participants or guardians was obtained, and the study
conformed to the Declaration of Helsinki. Blood samples were taken,
and DNA was isolated from the family members (16,17).
DNA from blood samples was also taken from 200 healthy controls
with an average age of 40.6 years-old (age ranges: 20–66; sex
distribution: 2:3 female: Male) from Luzhou city between December
2016 and February 2017 without hearing and vision problems. The DNA
integrity was verified by running the DNA sample on a 0.8% agarose
gel. Written informed consent was obtained from the patients and
healthy controls.
TGS
TGS analysis was performed on the proband M147 DNA,
according to the manufacturer's protocol (Illumina, Inc.) as
described previously (18–20). Library construction, TGS and data
analysis were performed according to the manufacturer's protocols
and as described previously (19,21).
Sanger validation and segregation
analysis
PCR amplification was performed for mutation
validation. The primer pairs M147-USH-T10695A and M147-M201-USH
were designed using the Primer3 program (http://bioinfo.ut.ee/primer3-0.4.0/) with genomic DNA
sequences containing the NM_206933.3: c.T449G and c.T10695A,
respectively, of the USH2A gene (Table I). PCR amplification was performed
using the aforementioned primer pairs and the amplified PCR
products were then sequenced using the Sanger method on an
ABI-3500DX sequencer (Thermo Fisher Scientific, Inc.) (22) using M147-USH-T10695A-L and
M147-M201-USH-L primers (Table I).
Control samples from unrelated, ethnically matched individuals were
also amplified using the primer pairs M147-USH-T10695A and
M147-M201-USH and sequenced using the primers M147-USH-T10695A-L
and M147-M201-USH-L. Segregation analysis in the M147 family was
performed based on the sequencing results.
 | Table I.Sequences of USH2A PCR
primers. |
Table I.
Sequences of USH2A PCR
primers.
| Primer name | Sequence,
5′-3′ | Size, bp | Mutation site |
|---|
|
M147-USH-T10695A | F:
AGGAACTGCTTGAGACAGCAA | 346 | c.T10695A |
|
| R:
CTGAACCCCTTTTCCCAGAG |
|
|
| M147-M201-USH | F:
AGGCCTCAGTAGCTGCATCA | 321 | c.T449G |
|
| R:
TTGGGGAAACAACTGGAAGA |
|
|
| RT-ush2a | F:
CGCTCTGCCTCTCCTCTCTA | 320 | N/A |
|
| R:
TTTATTGGAGGCTGCAAACC |
|
|
| RT-actin-m | F:
TGTTACCAACTGGGACGACA | 392 | N/A |
|
| R:
TCTCAGCTGTGGTGGTGAAG |
|
|
Prediction of protein structure and
bioinformatics analysis
The homologs of the USH2A gene (NM_206933.3)
in humans were analyzed using HomoloGene (ncbi.nlm.nih.gov/homologene?Db=homologene&Cmd=Retrieve&list_uids=66151).
Conserved domains of the protein structure of USH2A
(NP_996816.2) were searched using the NCBI conserved domain
database (ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) (23–25).
RNA-sequencing (RNA-seq) profile, RNA
isolation and reverse transcription-PCR (RT-PCR)
To determine tissue specificity in humans,
USH2A gene expression profiles were analyzed using RNA-seq
data which was performed on normal samples from 95 human
individuals representing 27 different tissues using: https://www.ncbi.nlm.nih.gov/gene/7399/?report=expression.
The above RNA-seq data were collected from a project called HPA
RNA-seq normal tissues (BioProject no. PRJEB4337).
Total RNA was extracted from tissues in mice using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Experiments involving mice followed the international,
national and institutional guidelines for the care and use of
laboratory animals (26,27,28)
and was reviewed and approved by the Ethics Committee of Southwest
Medical University, Sichuan, China. Male and female BALB/c mice (8
weeks old, ~21 g), were purchased from SPF (Beijing) Biotechnology
Co., Ltd, China, and housed individually at room temperatures
(18–22°C) with 40–60% and with a 12-h light/dark cycle. All mice
were negative for pathogens. Mice received food and water ad
libitum. At least five healthy mice for eye tissues and one
healthy mouse for other tissues were used for RNA sampling. Retinal
tissue at developmental stages, including whole embryo eye at 12.5
and 20.5 days before birth, and 2 weeks, 1 month, 2 and 3 months
after birth, were used. Pentobarbital sodium (200 mg/kg of body
weight) was intraperitoneally injected to mice for euthanasia.
Death was verified as absence of vital signs, including no
heartbeat, dilated pupils or cervical dislocation after anesthesia.
First strand cDNA was then synthesized from 1 µg total RNA with the
ReverTra Ace® qPCR RT kit (cat. no. FSQ-201; Toyobo Life
Science) according to the manufacturer's instructions and
semi-quantitative RT-PCR was performed as described in our previous
study (29,30). Briefly, the PCR reaction system
consisted of 5 µl 2X Taq PCR MasterMix (Tiangen Biotech Co.), 1 µl
2.5 µM RT primers, 1 µl RT products and 3 µl double distilled
H2O. Amplification reactions were performed using the
Applied Biosystems® Veriti® 96-Well Thermal
Cycler PCR machine (Thermo Fisher Scientific, Inc.) with the
following steps: Initial denaturation at 95°C for 90 sec, 28–30
cycles of denaturation at 94°C for 30 sec, annealing at 72°C for 30
sec, extension at 72°C for 30 sec and a final extension step at
72°C for 5 min. Primer pairs for RT-ush2a (RT-ush2a-L and
RT-ush2a-R) targeting the mouse Ush2a gene, and the mouse
β-actin gene which served as a control, are listed in
Table I. RT-PCR products were
separated on a 1.2% agarose gel in triplicate, and the gels were
visualized by 0.5 µg/ml ethidium bromide (EB) staining.
Densitometry was performed using ChemiDoc XR (version 5.2, Bio-Rad
Laboratories, Inc.) (17,31).
Results
Proband and clinical
characteristics
The patient (Fig.
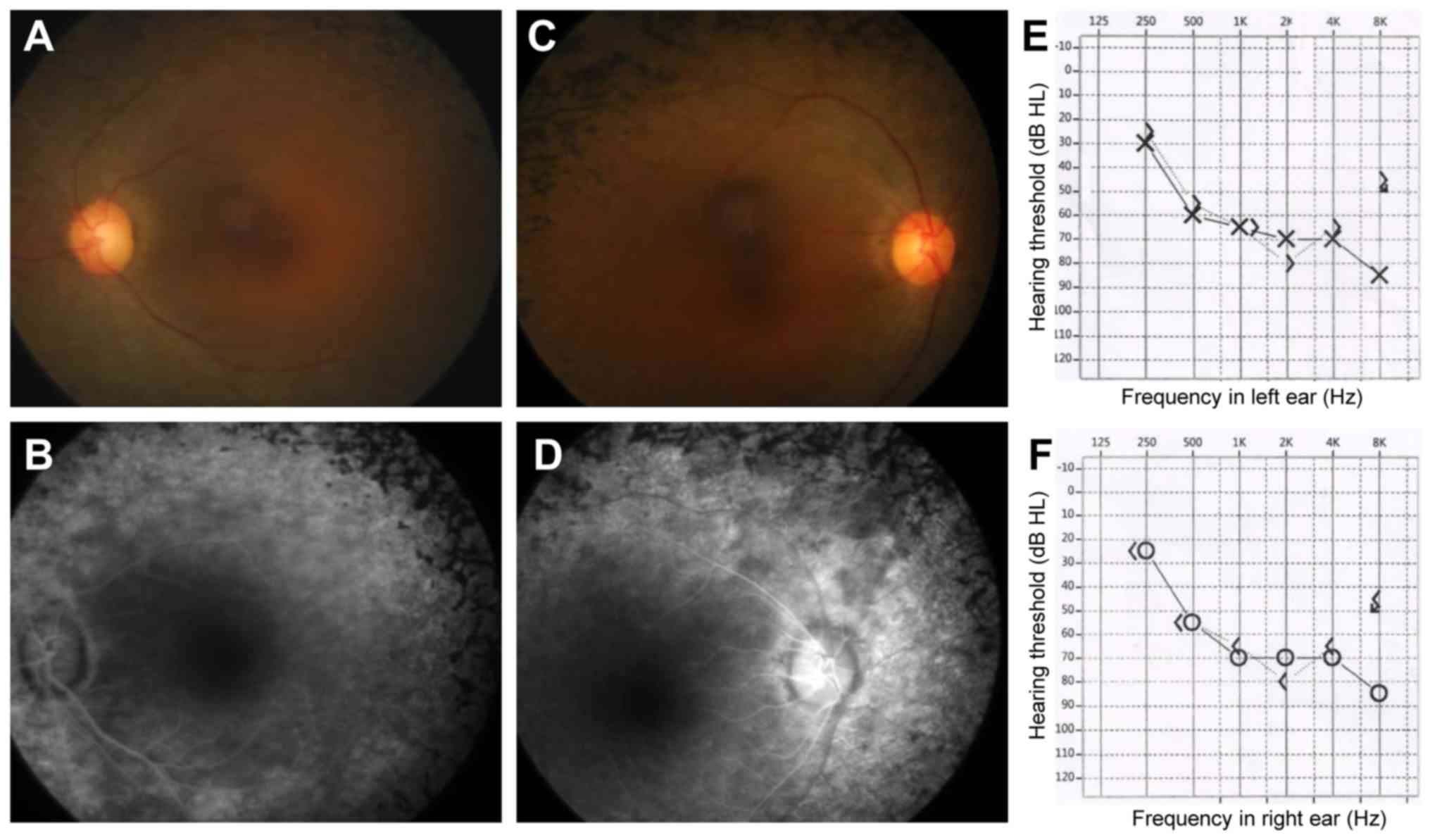
1A, II: 1) was a 35-year-old Chinese female. She suffered from
night blindness since adolescence at age 12 and was first diagnosed
with RP at the Yibin local county hospital 7 years ago. Fundus
examinations showed bony spicule pigmentation and attenuated
retinal vessels in both eyes, characteristic of a typical RP
phenotype (Fig. 2A-D). The retinal
pigment epitheliums were atrophied, and electroretinography results
displayed no amplitude reactions (data not shown). The proband had
a right naked eye vision of 0.6 and a left naked eye vision of 0.7,
and a visual acuity with correction of 0.8 in the right eye and
0.8+ in the left eye. The logMAR or Snellen measurements were not
available for this patient. The proband did not claim to exhibit
hearing loss. The proband was characterized as RP. Based on genetic
diagnostic results, pure tone audiometry testing was performed and
found 65 dB hearing losses of both ears, indicating a
moderate-to-severe binaural sensorineural hearing loss across all
frequencies (Fig. 2E, left ear;
Fig. 2F, right ear). Both the
proband's parents and her son was normal. Thus, the proband was
diagnosed with possible Usher syndrome type IIA.
Results of TGS and co-segregation
analysis
Compound heterozygous mutations (c.T449G) in exon 2
and (c.T10695A) in exon 54 in the USH2A gene (NM_206933.3)
were identified, leading to the presence of early stop codons (from
TTA to TGA and from TAT to TAA, respectively) at amino acid
positions 150 (p.L150*) and 3565 (p.Y3565*), respectively, in the
USH2A protein (NP_996816.2; Fig.
1A, II: 1). Thus, both USH2A variants (c.T449G, p.L150*) and
(c.T10695A, p.Y3565*) resulted in the production of truncated
proteins, which were hypothesized to affect protein function. The
characteristics of the USH2A variants and disease-causing
effects of the proband are shown in Table II. Hence, these nonsense mutations
(c.T449G, p.L150*) and (c.T10695A, p.Y3565*) in the USH2A
gene affected protein function and supported the diagnosis of Usher
syndrome type IIA. Both variants were revealed to be novel by
searching the databases for Exome Aggregation Consortium
(https://gnomad.broadinstitute.org/)
and HGMD (http://www.hgmd.cf.ac.uk/ac/index.php) (Table II).
| Table II.Characteristics of USH2A
variants and disease-causing effects in the proband. |
Table II.
Characteristics of USH2A
variants and disease-causing effects in the proband.
|
|
| Variation |
|
|---|
|
|
|
|
|
|---|
| Gene | Exon | Nucleotide | Protein | Type of
mutation | Status | ExAC status |
|---|
| USH2A | 2 | c.T449G | p.L150* | Nonsense | Heterozygous | Novel |
| USH2A | 54 | c.T10695A | p.Y3565* | Nonsense | Heterozygous | Novel |
Sanger sequencing was performed to confirm the
variants and analyze co-segregation (Fig. 3). The mutations (c.T449G,
c.T10695A) in the USH2A gene were validated to be compound
heterozygous in the proband (Fig. 3C
and H; pedigree II: 1), of which c.T449G was inherited from her
father (Fig. 3A and F; pedigree I:
1) and c.T10695A was inherited from her mother (Fig. 3B and G; pedigree I: 2). The
proband's son was revealed to be heterozygous c.T10695A with a
normal phenotype (pedigree III: 1; Fig. 3D and I), and the proband's husband
had a normal phenotype and with wild-type alleles (Fig. 3E and J; pedigree II: 2). Therefore,
these mutations in the USH2A gene and the mutations were
co-segregated with this clinical phenotype in this family. Both
c.T449G and c.T10695A variants were absent in the blood samples of
200 normal ethnically matched controls. Taken together, this
discovery highlighted co-segregation of the variants in this family
and pinpointed their roles in the pathogenesis of Usher syndrome
type IIA.
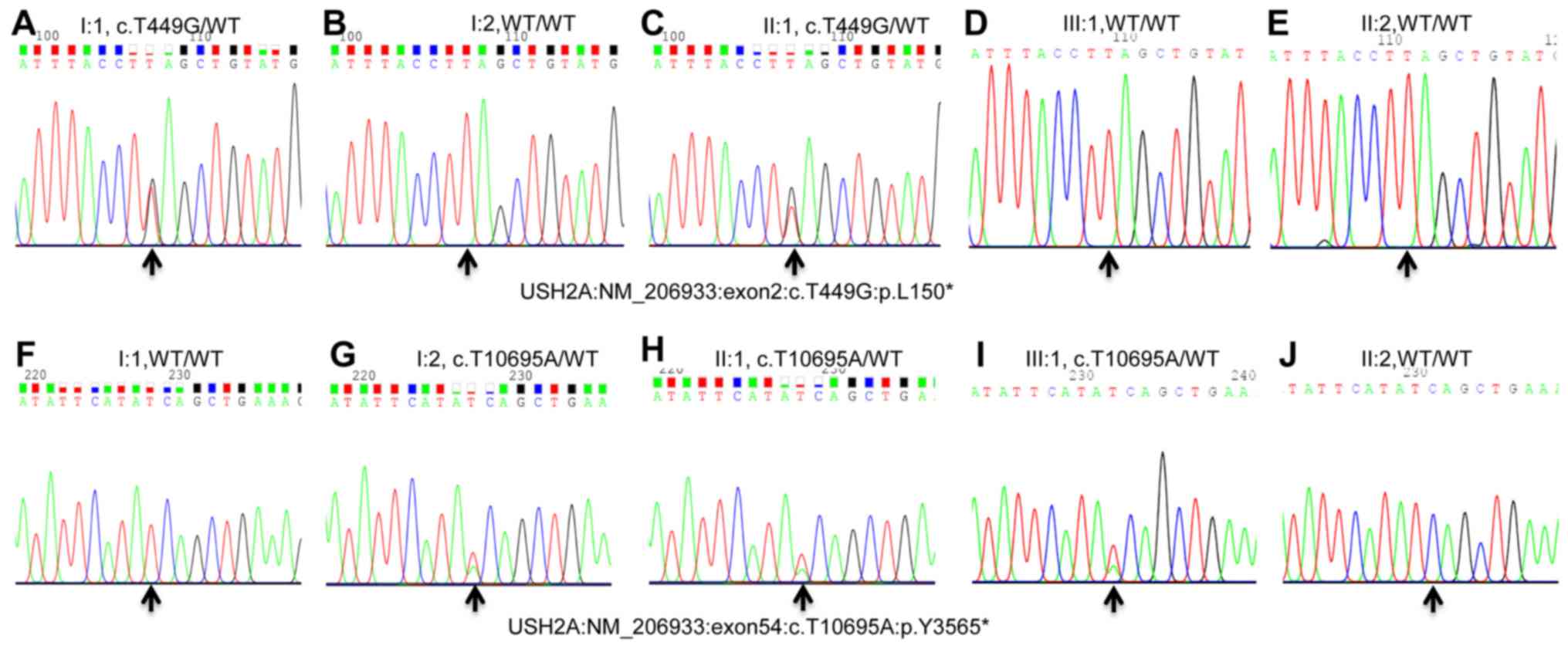 | Figure 3.Pyrogram profiles for variant
verification using Sanger sequencing. (A-E) Sequencing results pf
I: 1 (heterozygous mutant type), I: 2 (WT), II: 1 (heterozygous
mutant type), III: 1 (WT), and II: 2 (WT) of variant c.T449G. (F-J)
Sequencing results of I: 1 (WT), I: 2 (heterozygous mutant type),
II: 1 (heterozygous mutant type), III: 1 (heterozygous), and II: 2
(WT) of variant c.T10695A. The arrows indicate the mutation
position of NM_206933.3: c.T449G or c.T10695A in the USH2A
gene. USH2A, usherin; WT, wild-type. |
Functional effects of the variants
c.T449G (p.L150*) and c.T10695A (p.Y3565*) in USH2A
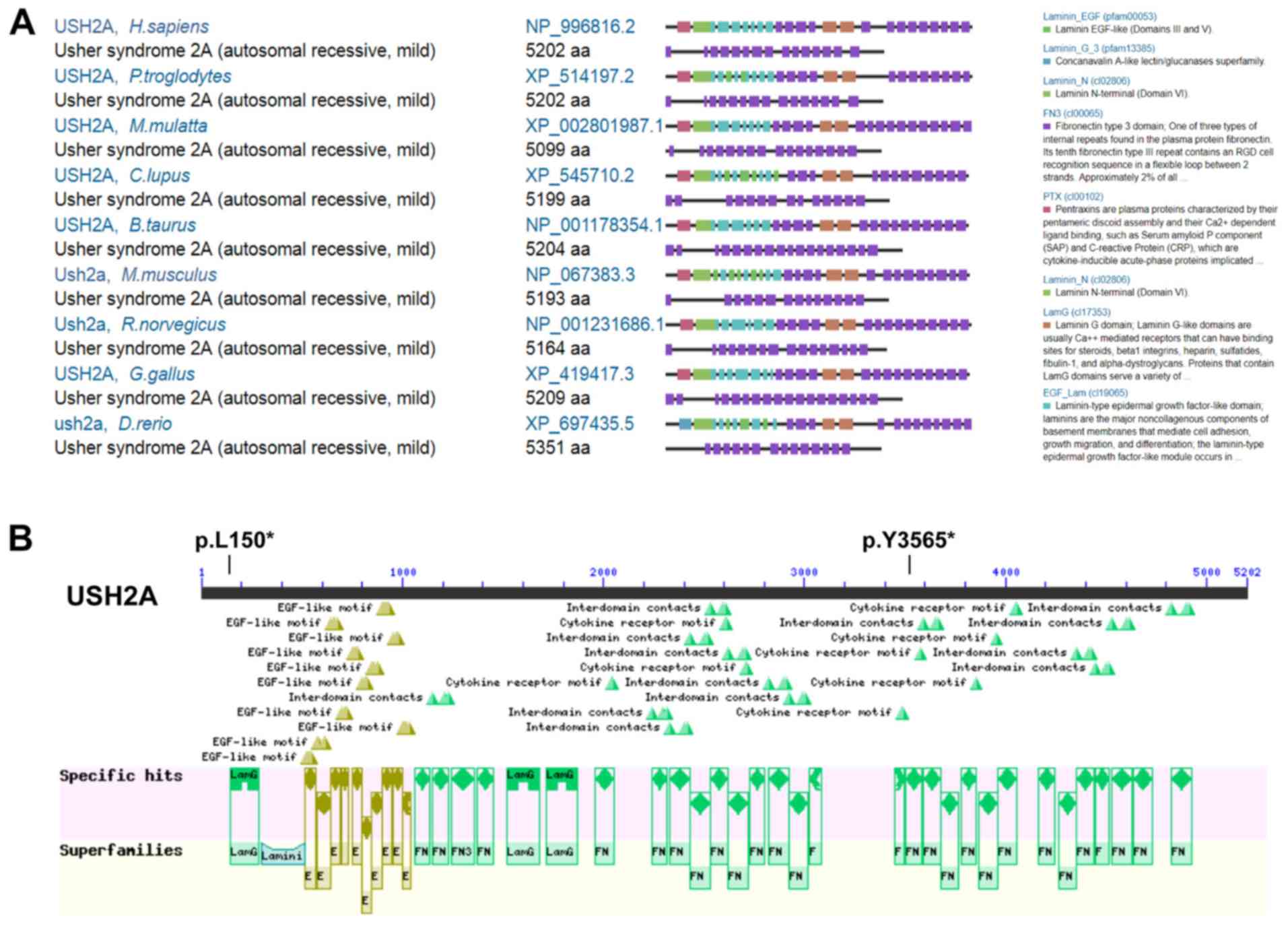
By comparing of Homo sapiens (H. sapiens)
USH2A protein to eight other species, including Pan troglodytes,
Macaca mulatta, Canis lupus, Bos taurus, Rattus norvegicus, Mus
musculus, Gallus gallus and Danio rerio, it was shown
that USH2A is highly orthologously conserved (Fig. 4A). USH2A protein in H.
sapiens contains a lamG-like jellyroll fold domain, laminin EGF
domains, laminin-type EGF-like domains, laminin G domains, laminin
N-terminal (Domain VI) and many fibronectin type 3 domains
(Fig. 4A and B). The c.T449G
(p.L150*) variant is located in the lamG-like jellyroll fold domain
(aa.146-aa.283), leading to the production of a truncated protein
which had lost almost all the functional domains (Fig. 4B); whereas the c.T10695A (p.Y3565*)
variant is located in one of the fibronectin type 3 domains (aa.
3503-aa. 3586), leading to the production of a truncated protein,
that lost several fibronectin type 3 domains at the C-terminus of
USH2A (Fig. 4B). Together, these
results showed that the USH2A pathogenic compound
heterozygous variants c.T449G (p.L150*) and c.T10695A (p.Y3565*)
may have caused Usher syndrome type IIA disease.
mRNA expression profiles of USH2A and
Ush2a
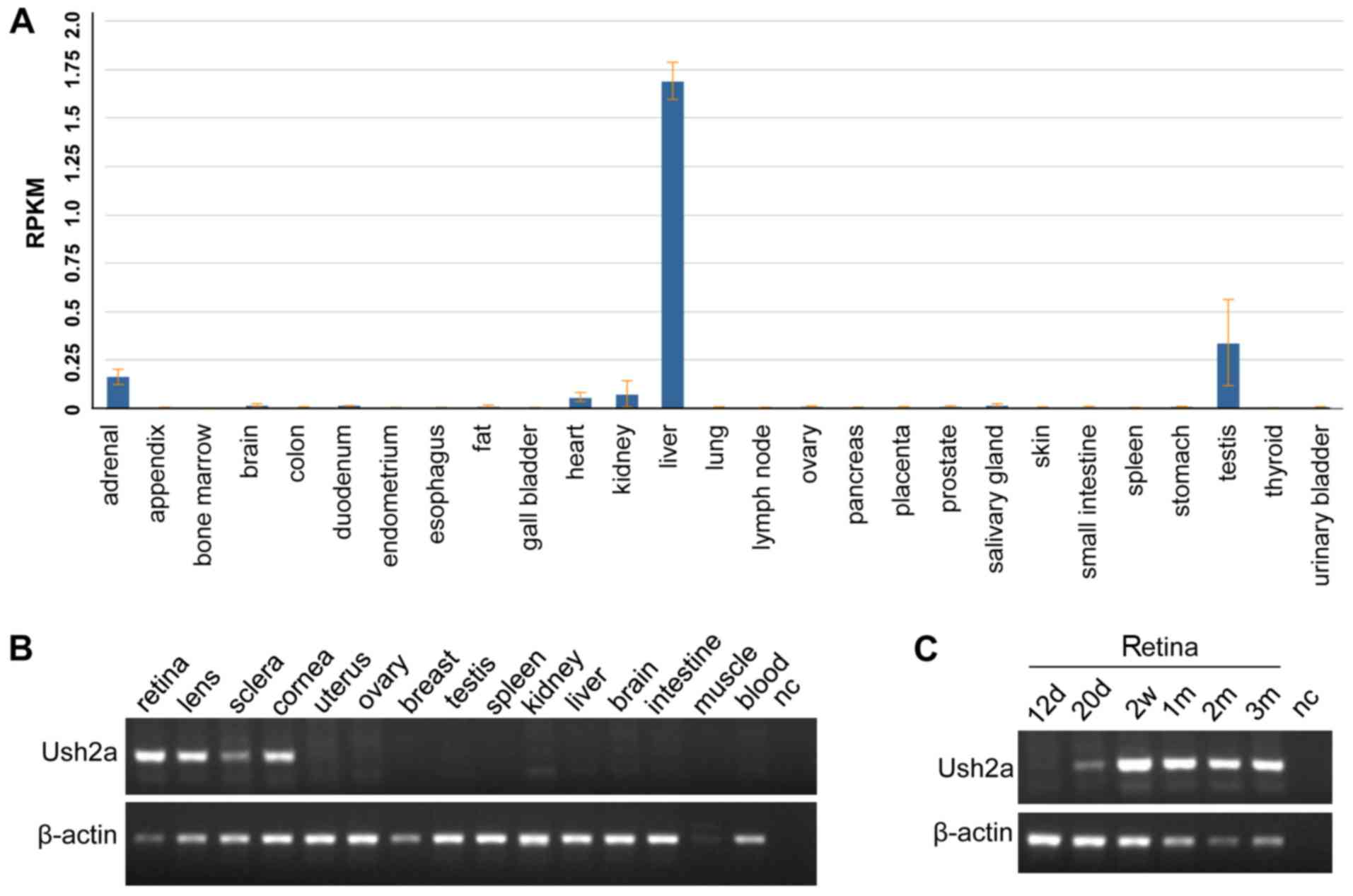
USH2A expression analysis in humans indicated
that USH2A mRNA expression was highest in the liver, with a
reads per kilobase of transcript per million mapped reads value of
1.691±0.096, followed by the testis, and was low or very low in
other tissues (Fig. 5A). RNA-seq
values in different tissues are presented in Table III. No human eye tissues and
developmental retinal stage tissues were available. Thus, the
Ush2a expression in mice was studied. Ush2a mRNA
expression levels were highly expressed in the retina and other
indicated tissues including the lens, sclera and cornea (Fig. 5B); whereas expression was not
detected in the tissues, uterus, ovary, testis, breast, spleen,
kidney, liver, brain, intestine, skeletal muscle, and blood.
Ush2a was also highly expressed in the latter four different
developmental retinal stages following birth (Fig. 5C). These results suggested that
USH2A may serve a vital role in retinal/eye function based on the
very high levels of Ush2a2 expression in the retinal tissue
and its highly ubiquitous expression in other eye tissues.
| Table III.Expression of USH2A mRNA in
human tissues. |
Table III.
Expression of USH2A mRNA in
human tissues.
| Tissue | Number of
samples | RPKM values | Counts |
|---|
| Adrenal | 3 | 0.163±0.040 | 37,820 |
| Appendix | 3 | 0.005±0.002 | 984 |
| Bone marrow | 4 | 0.001±0.001 | 355 |
| Brain | 3 | 0.017±0.008 | 4,671 |
| Colon | 5 | 0.005±0.004 | 4,064 |
| Duodenum | 2 | 0.015±0.001 | 2,072 |
| Endometrium | 3 | 0.005±0.0020 | 1,114 |
| Esophagus | 3 | 0.004±0.002 | 1,592 |
| Fat | 3 | 0.011±0.007 | 2,513 |
| Gall bladder | 3 | 0.004±0.002 | 1,546 |
| Heart | 4 | 0.06±0.0230 | 30,633 |
| Kidney | 4 | 0.076±0.068 | 21,041 |
| Liver | 3 | 1.691±0.096 | 469,141 |
| Lung | 5 | 0.006±0.004 | 2,714 |
| Lymph node | 5 | 0.003±0.004 | 1,827 |
| Ovary | 2 | 0.009±0.004 | 2,706 |
| Pancreas | 2 | 0.006±0.002 | 1,498 |
| Placenta | 4 | 0.005±0.005 | 2,596 |
| Prostate | 4 | 0.009±0.006 | 2,782 |
| Salivary gland | 3 | 0.015±0.009 | 7,066 |
| Skin | 3 | 0.006±0.003 | 2,279 |
| Small
intestine | 4 | 0.007±0.004 | 2,300 |
| Spleen | 4 | 0.003±0.003 | 1,410 |
| Stomach | 3 | 0.009±0.003 | 2,563 |
| Testis | 7 | 0.341±0.222 | 348,396 |
| Thyroid | 4 | 0.003±0.002 | 1,543 |
| Urinary
bladder | 2 | 0.006±0.004 | 1,432 |
Discussion
The association between the variations in Usher
syndrome-causative genes and the resultant Usher syndrome diseases
or phenotypes in patients are highly variable; the
genotype/phenotype associations are also divergent (1,3,4).
Since Eudy et al (5) first
identified three mutations in the USH2A gene in patients
with Usher syndrome type IIA with RP and hearing loss, additional
USH2A mutations have been shown to be associated with Usher
syndrome type IIA (10–12,32–39).
Patients with autosomal recessive RP (arRP; RP39; OMIM, 613809)
without hearing loss were also identified to possess USH2A
mutations, highlighting the complexity of genotype/phenotype
associations in this disease (4).
For example, Rivolta et al (40) identified USH2A mutations in
patients with arRP without hearing loss. Zlotogora (41) reviewed examples and identified arRP
patients without hearing loss who possessed mutations in the
USH2A gene. Compound pathogenic mutations in the
USH2A gene in Chinese RP families were also recently
identified (42).
In the present study, using TGS, Sanger sequencing
and co-segregation analysis, compound heterozygous pathogenic
nonsense mutations, c.T449G (p.L150*) and c.T10695A (p.Y3565*),
were identified in the USH2A gene in a Chinese pedigree.
USH2A, very large G-protein coupled receptor 1 (VLGR1), USH1 and
electroneutral sodium bicarbonate exchanger 1 (NBC3) are
co-expressed in the synaptic terminals in both retinal
photoreceptors and inner ear hair cells of mice and rats. The
scaffold proteins harmonin, USH2A, VLGR1 and NBC3 interact with
each other to assemble a multiprotein complex (43). USH2 and NBC3 proteins are
interaction partners in a network in the retina and inner ear
(43,44). These p.L150* and p.Y3565* truncated
USH2A mutations are hypothesized to affect complex formation
(45), thereby elucidating the
genetic roles of these USH2A mutant alleles in Usher
syndrome type IIA.
The patient examined in the present study was first
diagnosed as a nonsyndromic RP as she did not claim to suffer from
hearing loss. Based on gene diagnostic results, pure tone
audiometry testing was performed which presented binaural
sensorineural deafness. Thus, USH2A mutations with Usher
syndrome type IIA and an association of genotype/phenotype have
been successfully linked in the patient of the studied family.
Pater et al (46) recently
reassigned the diagnosis of Usher syndrome by identifying novel
USH2A splicing variants. To the best of our knowledge, the
USH2A variants c.T449G (p.L150*), c.T10695A (p.Y3565*) are
novel, thereby extending the spectrum of known mutations associated
with this disease.
By comparing the of H. sapiens USH2A to eight
other species, it was demonstrated that the USH2A protein is highly
conserved. Ush2a mRNA expression levels in mice were
demonstrated to only be highly expressed in the retina, lens,
sclera and cornea from the eye tissues, consistent with a previous
study (5), suggesting that USH2A
serves vital roles in the functions of retina/eye. In addition to
its high expression in the retina, USH2A in humans, rat and mice is
also expressed in the cochlea (5,47).
USH2A was found to a likely component of the interstereocilia ankle
which links the inner ear sensory cells (12). Taken together, the present study
showed that the USH2A compound heterozygous variant, c.T449G
(p.L150*) and c.T10695A (p.Y3565*), resulted in Usher syndrome type
IIA.
As a rare disorder and the most common inherited
form of combined visual and hearing impairment, up to 14 genes
(MYO7A, CDH23, USH1C, PCDH15, USH1G and CIB2 for USH
I, USH2A, ADGRV1 and WHRN for USH II, CLRN1
and HARS for USH III, and PDZD7, CEP250 and C2orf71
for either type I, II, or III of USH) are associated with Usher
syndrome, with USH2A being the most prevalent worldwide (2,48,49).
With genetic diagnosis for Usher syndrome, repairing specific
mutations using CRISPR/Cas9 editing system may now be possible. For
example, Fuster-García et al (48) investigated gene editing to target
the mutation in fibroblasts of a USH patient bearing the c.2299delG
homozygous variation, highlighting the potential of the CRISPR
editing system for the treatment of Usher syndrome.
In conclusion, the present study is the first to
identify the novel compound heterozygous variants c.T449G (p.L150*)
and c.T10695A (p.Y3565*) in the USH2A gene, which caused
Usher syndrome type IIA in the examined patient. These mutations
expand upon the library of known mutations associated with Usher
syndrome. TGS provides a useful gene diagnostic approach (50). The present discovery may assist in
understanding the molecular pathogenesis of RP and Usher syndrome
type IIA, and in the development of strategies for the prevention,
diagnosis, therapy and genetic counseling of patients with Usher
syndrome type IIA. Additionally, the recruitment of more patients
with Usher syndrome is an aim of future studies.
Acknowledgements
The authors would like to thank Professor Rui Chen
at the Baylor College of Medicine for technical assistance and Ms.
Shelly Fu at the University of Houston and Baylor College of
Medicine for assistance and reading the manuscript.
Funding
The project was funded by the National Natural
Science Foundation of China (grant nos. 31701087, 30371493 and
81672887), the Joint Research Foundation of Luzhou City and
Southwest Medical University (grant no. 2018LZXNYD-YL01), and The
Translational Medicine Foundation of Southwest Medical University
(grant nos. 00031476 and 00031477).
Availability of data and materials
The data generated using high-throughput sequencing
were not submitted to any public databases due to the containing
information that may compromise the proband's privacy but are
available from the corresponding author on reasonable request.
Authors' contributions
JuF designed and conceptualized the study. JiF and
JC performed DNA extraction, PCR amplification, sequencing and data
analysis. HL, QZ and CD recruited the clinical patients and were in
charge of the clinical assessments. JuF, MAK and JP analyzed the
data, wrote and revised the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Southwest Medical University. The protocol and
procedures employed for mice experiments were ethically reviewed
and approved by the Ethics Committee of Southwest Medical
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wei C, Yang L, Cheng J, Imani S, Fu S, Lv
H, Li Y, Chen R, Leung ELH and Fu J: A novel homozygous variant of
GPR98 causes Usher syndrome type IIC in a consanguineous Chinese
family by next generation sequencing. BMC Med Genet. 19:34642018.
View Article : Google Scholar
|
|
2
|
Mathur P and Yang J: Usher syndrome:
Hearing loss, retinal degeneration and associated abnormalities.
Biochim Biophys Acta. 1852:406–420. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fu J, Shen S, Cheng J, Lv H and Fu J: A
case of Usher syndrome type IIA caused by a rare USH2A homozygous
frameshift variant with maternal uniparental disomy (UPD) in a
Chinese family. J Cell Mol Med. 24:7743–7750. 2020. View Article : Google Scholar
|
|
4
|
Zhu X, Li X, Tian W, Yang Y, Sun K, Li S
and Zhu X: Identification of novel USH2A mutations in patients with
autosomal recessive retinitis pigmentosa via targeted
nextgeneration sequencing. Mol Med Rep. 22:193–200. 2020.PubMed/NCBI
|
|
5
|
Eudy JD, Weston MD, Yao S, Hoover DM, Rehm
HL, Ma-Edmonds M, Yan D, Ahmad I, Cheng JJ, Ayuso C, et al:
Mutation of a gene encoding a protein with extracellular matrix
motifs in Usher syndrome type IIa. Science. 280:1753–1757. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Besnard T, Vaché C, Baux D, Larrieu L,
Abadie C, Blanchet C, Odent S, Blanchet P, Calvas P, Hamel C, et
al: Non-USH2A mutations in USH2 patients. Hum Mutat. 33:504–510.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ebermann I, Phillips JB, Liebau MC,
Koenekoop RK, Schermer B, Lopez I, Schäfer E, Roux AF, Dafinger C,
Bernd A, et al: PDZD7 is a modifier of retinal disease and a
contributor to digenic Usher syndrome. J Clin Invest.
120:1812–1823. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ebermann I, Scholl HP, Charbel Issa P,
Becirovic E, Lamprecht J, Jurklies B, Millán JM, Aller E, Mitter D
and Bolz H: A novel gene for Usher syndrome type 2: Mutations in
the long isoform of whirlin are associated with retinitis
pigmentosa and sensorineural hearing loss. Hum Genet. 121:203–211.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hmani-Aifa M, Benzina Z, Zulfiqar F,
Dhouib H, Shahzadi A, Ghorbel A, Rebaï A, Söderkvist P, Riazuddin
S, Kimberling WJ and Ayadi H: Identification of two new mutations
in the GPR98 and the PDE6B genes segregating in a Tunisian family.
Eur J Hum Genet. 17:474–482. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
van Wijk E, Pennings RJ, te Brinke H,
Claassen A, Yntema HG, Hoefsloot LH, Cremers FPM, Cremers CWRJ and
Kremer H: Identification of 51 novel exons of the Usher syndrome
type 2A (USH2A) gene that encode multiple conserved functional
domains and that are mutated in patients with Usher syndrome type
II. Am J Hum Genet. 74:738–744. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weston MD, Eudy JD, Fujita S, Yao S, Usami
S, Cremers C, Greenberg J, Ramesar R, Martini A, Moller C, et al:
Genomic structure and identification of novel mutations in usherin,
the gene responsible for Usher syndrome type IIa. Am J Hum Genet.
66:1199–1210. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Adato A, Lefèvre G, Delprat B, Michel V,
Michalski N, Chardenoux S, Weil D, El-Amraoui A and Petit C:
Usherin, the defective protein in Usher syndrome type IIA, is
likely to be a component of interstereocilia ankle links in the
inner ear sensory cells. Hum Mol Genet. 14:3921–3932. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang L, Mao Y, Yang J, Li Y, Li Y and
Yang Z: Mutation screening of the USH2A gene in retinitis
pigmentosa and USHER patients in a han Chinese population. Eye
(Lond). 32:1608–1614. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang L, Cheng J, Zhou Q, Khan MA and Fu
J, Duan C, Sun S, Lv H and Fu J: Targeted next-generation
sequencing identified novel compound heterozygous variants in the
CDH23 gene causing Usher syndrome type ID in a Chinese patient.
Front Genet. 11:4222020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang J, Fu J, Fu S, Yang L, Nie K, Duan
C, Cheng J, Li Y, Lv H, Chen R, et al: Diagnostic value of a
combination of next-generation sequencing, chorioretinal imaging
and metabolic analysis: Lessons from a consanguineous Chinese
family with gyrate atrophy of the choroid and retina stemming from
a novel OAT variant. Br J Ophthalmol. 103:428–435. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fu J, Li L and Lu G: Relationship between
microdeletion on Y chromosome and patients with idiopathic
azoospermia and severe oligozoospermia in the Chinese. Chin Med J
(Engl). 115:72–75. 2002.PubMed/NCBI
|
|
17
|
Cheng J and Fu J, Zhou Q, Xiang X, Wei C,
Yang L, Fu S, Khan MA, Lv H and Fu J: A novel splicing mutation in
the PRPH2 gene causes autosomal dominant retinitis pigmentosa in a
Chinese pedigree. J Cell Mol Med. 23:3776–3780. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang F, Wang H, Tuan HF, Nguyen DH, Sun V,
Keser V, Bowne SJ, Sullivan LS, Luo H, Zhao L, et al: Next
generation sequencing-based molecular diagnosis of retinitis
pigmentosa: Identification of a novel genotype-phenotype
correlation and clinical refinements. Hum Genet. 133:331–345. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fu Q, Xu M, Chen X, Sheng X, Yuan Z, Liu
Y, Li H, Sun Z, Li H, Yang L, et al: CEP78 is mutated in a distinct
type of Usher syndrome. J Med Genet. 54:190–195. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Q, Xu M, Verriotto JD, Li Y, Wang H,
Gan L, Lam BL and Chen R: Next-generation sequencing-based
molecular diagnosis of 35 Hispanic retinitis pigmentosa probands.
Sci Rep. 6:327922016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koenekoop RK, Wang H, Majewski J, Wang X,
Lopez I, Ren H, Chen Y, Li Y, Fishman GA, Genead M, et al:
Mutations in NMNAT1 cause Leber congenital amaurosis and identify a
new disease pathway for retinal degeneration. Nat Genet.
44:1035–1039. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cheng J, Peng J and Fu J, Khan MA, Tan P,
Wei C, Deng X, Chen H and Fu J: Identification of a novel germline
BRCA2 variant in a Chinese breast cancer family. J Cell Mol Med.
24:1676–1683. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marchler-Bauer A, Bo Y, Han L, He J,
Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR,
et al: CDD/SPARCLE: Functional classification of proteins via
subfamily domain architectures. Nucleic Acids Res. 45:D200–D203.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Imani S, Ijaz I, Shasaltaneh MD, Fu S,
Cheng J and Fu J: Molecular genetics characterization and homology
modeling of the CHM gene mutation: A study on its association with
choroideremia. Mutat Res. 775:39–50. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Imani S, Cheng J, Fu J, Mobasher-Jannat A,
Wei C, Mohazzab-Torabi S, Jadidi K, Khosravi MH, Shasaltaneh MD,
Yang L, et al: Novel splicing variant c. 208+2T>C in BBS5
segregates with bardet-biedl syndrome in an Iranian family by
targeted exome sequencing. Biosci Rep. 39:BSR201815442019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wei C, Xiao T, Cheng J and Fu J, Zhou Q,
Yang L, Lv H and Fu J: Novel compound heterozygous EYS variants may
be associated with arRP in a large Chinese pedigree. Biosci Rep.
40:BSR201934432020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fu J, Zhou B, Zhang L, Balaji KS, Wei C,
Liu X, Chen H, Peng J and Fu J: Expressions and significances of
the angiotensin-converting enzyme 2 gene, the receptor of
SARS-CoV-2 for COVID-19. Mol Biol Rep. 47:4383–4392. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the care and use of laboratory animals. 8th.
National Academies Press (US); Washington, DC: 2011
|
|
29
|
Fu J, Cheng J, Zhou Q, Wei C, Chen H, Lv H
and Fu J: A novel missense variant c.G644A (p.G215E) of the RPGR
gene in a Chinese family causes X-linked retinitis pigmentosa.
Biosci Rep. 39:BSR201922352019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang L and Fu J, Cheng J, Wei C, Zhou Q,
Ijaz I, Lv H and Fu J: A novel variant of the FZD4 gene in a
Chinese family causes autosomal dominant familial exudative
vitreoretinopathy. Cell Physiol Biochem. 51:2445–2455. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu X, Cheng J, Mei Z, Wei C, Khan MA,
Peng J and Fu J: SCAR marker for identification and discrimination
of specific medicinal lycium chinense miller from lycium species
from ramp-PCR RAPD fragments. 3 Biotech. 10:3342020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dreyer B, Tranebjaerg L, Rosenberg T,
Weston MD, Kimberling WJ and Nilssen O: Identification of novel
USH2A mutations: Implications for the structure of USH2A protein.
Eur J Hum Genet. 8:500–506. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aller E, Jaijo T, Beneyto M, Nájera C,
Oltra S, Ayuso C, Baiget M, Carballo M, Antiñolo G, Valverde D, et
al: Identification of 14 novel mutations in the long isoform of
USH2A in Spanish patients with Usher syndrome type II. J Med Genet.
43:e552006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Baux D, Larrieu L, Blanchet C, Hamel C,
Salah SB, Vielle A, Gilbert-Dussardier B, Holder M, Calvas P,
Philip N, et al: Molecular and in silico analyses of the
full-length isoform of usherin identify new pathogenic alleles in
Usher type II patients. Hum Mutat. 28:781–789. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yan D, Ouyang X, Patterson DM, Du LL,
Jacobson SG and Liu XZ: Mutation analysis in the long isoform of
USH2A in American patients with Usher syndrome type II. J Hum
Genet. 54:732–738. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Testa F, Melillo P, Bonnet C, Marcelli V,
de Benedictis A, Colucci R, Gallo B, Kurtenbach A, Rossi S,
Marciano E, et al: Clinical presentation and disease course of
Usher syndrome because of mutations in Myo7a or Ush2a. Retina.
37:1581–1590. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Garcia-Garcia G, Aparisi MJ, Jaijo T,
Rodrigo R, Leon AM, Avila-Fernandez A, Blanco-Kelly F, Bernal S,
Navarro R, Diaz-Llopis M, et al: Mutational screening of the USH2A
gene in Spanish USH patients reveals 23 novel pathogenic mutations.
Orphanet J Rare Dis. 6:652011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ng TK, Tang W, Cao Y, Chen S, Zheng Y,
Xiao X and Chen H: Whole exome sequencing identifies novel USH2A
mutations and confirms Usher syndrome 2 diagnosis in Chinese
retinitis pigmentosa patients. Sci Rep. 9:56282019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
He C, Liu X, Zhong Z and Chen J: Mutation
screening of the USH2A gene reveals two novel pathogenic variants
in Chinese patients causing simplex usher syndrome 2. BMC
Ophthalmol. 20:702020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rivolta C, Berson EL and Dryja TP:
Paternal uniparental heterodisomy with partial isodisomy of
chromosome 1 in a patient with retinitis pigmentosa without hearing
loss and a missense mutation in the Usher syndrome type II gene
USH2A. Arch Ophthalmol. 120:1566–1571. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zlotogora J: Parents of children with
autosomal recessive diseases are not always carriers of the
respective mutant alleles. Hum Genet. 114:521–526. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fu YC, Chen N, Qiu ZL, Liu L and Shen J:
Compound pathogenic mutation in the USH2A gene in Chinese RP
families detected by whole-exome sequencing. Mol Med Rep.
18:5016–5022. 2018.PubMed/NCBI
|
|
43
|
Reiners J, van Wijk E, Märker T,
Zimmermann U, Jürgens K, te Brinke H, Overlack N, Roepman R,
Knipper M, Kremer H and Wolfrum U: Scaffold protein harmonin
(USH1C) provides molecular links between Usher syndrome type 1 and
type 2. Hum Mol Genet. 14:3933–3943. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zou J, Chen Q, Almishaal A, Mathur PD,
Zheng T, Tian C, Zheng QY and Yang J: The roles of USH1 proteins
and PDZ domain-containing USH proteins in USH2 complex integrity in
cochlear hair cells. Hum Mol Genet. 26:624–636. 2017.PubMed/NCBI
|
|
45
|
Sorusch N, Bauß K, Plutniok J, Samanta A,
Knapp B, Nagel-Wolfrum K and Wolfrum U: Characterization of the
ternary Usher syndrome SANS/ush2a/whirlin protein complex. Hum Mol
Genet. 26:1157–1172. 2017.PubMed/NCBI
|
|
46
|
Pater JA, Green J, O'Rielly DD, Griffin A,
Squires J, Burt T, Fernandez S, Fernandez B, Houston J, Zhou J, et
al: Novel Usher syndrome pathogenic variants identified in cases
with hearing and vision loss. BMC Med Genet. 20:682019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang D, Eudy JD, Uzvolgyi E, Davis JR,
Talmadge CB, Pretto D, Weston MD, Lehman JE, Zhou M, Seemayer TA,
et al: Identification of the mouse and rat orthologs of the gene
mutated in Usher syndrome type IIA and the cellular source of USH2A
mRNA in retina, a target tissue of the disease. Genomics.
80:195–203. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fuster-García C, García-García G,
González-Romero E, Jaijo T, Sequedo MD, Ayuso C, Vázquez-Manrique
RP, Millán JM and Aller E: USH2A gene editing Using the CRISPR
system. Mol Ther Nucleic Acids. 8:529–541. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fuster-García C, García-García G, Jaijo T,
Fornés N, Ayuso C, Fernández-Burriel M, Sánchez-De la Morena A,
Aller E and Millán JM: High-throughput sequencing for the molecular
diagnosis of Usher syndrome reveals 42 novel mutations and
consolidates CEP250 as Usher-like disease causative. Sci Rep.
8:171132018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Adams DR and Eng CM: Next-generation
sequencing to diagnose suspected genetic disorders. N Engl J Med.
379:1353–1362. 2018. View Article : Google Scholar : PubMed/NCBI
|