Introduction
Severe craniocerebral trauma is characterized by
high mortality and disability, and has been a focus of
neuroscientific research (1).
Cerebral ischemia is an important pathophysiological feature of
secondary brain injury after trauma (2). Secondary injury related mechanisms
include primary mechanical injury of blood vessels caused by
cerebral tissue ischemia, hematoma and other placeholder lesions
caused by local cerebral ischemia (3). In addition, secondary brain injury
also includes diffuse brain swelling after trauma, increased
intracranial pressure leading to low perfusion in the brain,
cerebral vasospasm, microvascular lesions and alterations in local
cerebral blood flow, which can influence systemic circulations
(3).
Astrocytes are the most widely distributed glial
cell type in the central nervous system and play a crucial role in
protecting neurons, regulating synaptic function, and promoting
nerve regeneration and repair (4,5).
Astrocytes also play an important role in the process of ischemic
brain injury (6). It was
previously shown that astrocytes are regularly distributed in the
brain, and establish positional relationships with neurons via
close connections to exchange information and material between
cells (7). After cerebral
ischemia, the extent of the damaged area of brain tissue is
determined by the degree of damage to neurons and glial cells
(8). The activation of astrocytes
can promote neuron damage repair, axon regeneration and neuron
migration (9). Previous studies
have shown that astrocytes maintain the homeostasis of the
extracellular microenvironment of brain tissue, and can promote
neuron survival after cerebral ischemia by interacting with neurons
and glial cells, and participating in the process of endogenous
nerve protection (10,11). Therefore, understanding how to
activate the neuroprotective mechanisms of astrocytes and inhibit
cell death could facilitate the treatment of ischemia-induced brain
tissue injury.
Necroptosis is a programmed type of necrosis, which
can be regulated and reversed under certain conditions (12,13).
Necroptosis is mediated by the interaction between receptor
interacting protein (RIP) kinase (RIPK) members, such as RIPK1 and
RIPK3 (12,13). After external signal stimulation, a
series of death receptors are activated, such as tumor necrosis
factor receptor (TNFR)1, TNFR2 and Fas ligand (14). RIPK1 is then activated and
interacts with death receptors to form complex-I, which can recruit
RIPK3 to form a necrosome, which is critical for the process of
necroptosis (14). A previous
study showed that the cerebral ischemic environment could lead to
necroptosis of neurons and astrocytes, and increase the area of
brain injury (15). Based on the
importance of astrocytes in the repair of injured brain tissue,
inhibiting astrocytic death and promoting its normal activation is
key to the functional recovery of the nervous system, as well as
the treatment and prognosis of traumatic cerebral ischemia.
N-myc downstream-regulated gene 2 (NDRG2) belongs to
the NDRG family that consists of four identified members (NDRG1-4),
and has been implicated in the regulation of cell differentiation
and proliferation (16,17). NDRG2 is closely related to
neurological diseases (18). In
the brains of patients with Alzheimer's disease, NDRG2 expression
is upregulated (18,19). Overexpression of NDRG2 in nerve
growth factor induced-differentiated PC12 cells promotes axon
regeneration (19). In astrocytes,
NDRG2 is specifically and more widely expressed than glial
fibrillary acidic protein (GFAP), which is a commonly used
astrocyte marker; thus, NDRG2 is considered as a new astrocyte
marker (20). It was previously
shown that NDRG2 could regulate astrocyte activity by promoting
cell differentiation and stabilizing cell morphology (21). The present study generated NDRG2
conditional knockout mice (Ndrg2-/-) to investigate the
association of NDRG2 and astrocytic activity during cerebral
ischemia.
Materials and methods
Animals
NDRG2 conditional knockout (Ndrg2-/-) mice
were generated via cooperation with Shanghai Biomodel Organism
Science & Technology Development Co., Ltd. and maintained on
C57BL6/J background as previously described (22). Briefly, Ndrg2flox/flox
mice were crossed with B6.C-Tg(CMV-cre)1Cgn/J mice (Jackson
Laboratory) to generate Ndrg2-/- mice. The line was backcrossed
with C57BL/6J a minimum of 8 times before use in any experiments in
this study. Heterozygous mice carrying the knockout mutation were
interbred to obtain the homozygous Ndrg2-deficient mice and their
wild-type (WT) littermates. All animals were raised under specific
pathogen-free conditions. The incubator temperature was maintained
at 21±2°C with 45–60% humidity, on a 12/12 h day/night cycle with
food and water ad libitum. WT mice and Ndrg2-/- mice
were age-matched, and all mice used were male. In total, 60 mice
were used in this study and 8 mice were used for each group of
treatment, all experiments were repeated ≥3 times. All animal
experiments were in accordance with the relevant provisions of the
Laboratory Animal Center and were approved by the Experimental
Animal Ethics Committee of the 101 Hospital of PLA of Anhui Medical
University.
Permanent middle cerebral artery (MCA)
occlusion (pMCAO)
pMCAO was performed as previously described
(23). For all experiments, male
C57BL/6 mice (age, 7 weeks; weight, 18–22 g) were used. Following
anesthesia with 2.5% isoflurane in 100% O2, holes were
drilled to place the laser. A nylon filament was advanced from the
right common carotid artery through the internal carotid to the
MCA. Rectal temperature was maintained at 37.0±0.5°C using a
temperature-regulated heating pad during the procedure. Animals
were randomly assigned to different treatment groups (n=8/group).
Infarct volume was assessed with a direct method using ImageJ v2.1
(National Institutes of Health). The grip forces of the mice were
measured as previously described (24).
Primary cortical astrocyte culture and
NDRG2 lentivirus infection
The 1-or 2-day-old neonates (n=12) were used for
primary cortical astrocyte culture as previously described
(25). The cerebral cortexes were
digested with 0.25% trypsin for 10 min at 37°C and filtered with a
sterile nylon cell strainer. The cells were grown at 37°C in
conditions of 5% CO2 and 95% O2 in DMEM/F12
(1:1) supplemented with 10% FBS (both purchased from Gibco; Thermo
Fisher Scientific, Inc.), streptomycin (100 µg/ml), and penicillin
(100 U/ml). For NDRG2 overexpression in astrocytes, cells were
infected with 1×108 TU/ml NDRG2-overexpressing
lentivirus (pHBLV-CMVIE-NDRG2), which was packaged by Hanbio
Biotechnology Co., Ltd., with 5 µg/ml polybrene. At 48 h after
infection, subsequent experiments were performed.
Oxygen-glucose deprivation (OGD)
treatment
OGD was performed as previously described (26). Cells in the OGD group were cultured
in glucose-free DMEM (Gibco; Thermo Fisher Scientific, Inc.) and
kept for 6 h or 12 h at 37°C in a hypoxic incubator chamber
(Billups-Rothenberg, Inc.) filled with 95% N2/5%
CO2. Cells grown in conditions of 5% CO2 and
95% O2 in standard DMEM were used as the control group.
Necrostatin-1 (Nec-1; 10 µM; Sigma-Aldrich; Merck KGaA) dissolved
in DMSO was added to the cells 0.5 h before OGD treatment.
Western blotting
Cells were lysed in RIPA buffer (Thermo Fisher
Scientific, Inc.) and the concentrations were measured by
bicinchoninic acid protein assay kit (Thermo Fisher Scientific,
Inc.). In all samples, 40 µg proteins were separated by 12%
SDS-PAGE and transferred to nitrocellulose membranes (Hybond-ECL;
GE Healthcare Life Sciences). Then, nitrocellulose membranes were
incubated with 5% skimmed milk for 1 h at room temperature. Primary
antibodies were then introduced at dilutions of 1:1,000 for
anti-RIPK1 (cat. no. ab106393; Abcam), anti-GFAP (cat. no. AB5541;
EMD Millipore), anti-NDRG2 (cat. no. 5667; Cell Signaling
Technology, Inc.) and anti-β-actin (cat. no. 4970; Cell Signaling
Technology, Inc.), and membranes were incubated at 4°C overnight.
The following secondary antibodies (1:5,000) were then added
against the primary antibodies, horseradish peroxidase
(HRP)-conjugated anti-mouse IgG (cat. no. 7076; Cell Signaling
Technology, Inc.) or HRP-conjugated anti-rabbit IgG (cat. no. 7074;
Cell Signaling Technology, Inc.), and membranes were incubated for
1 h at room temperature. The blots were detected by
chemiluminescence (Pierce; Thermo Fisher Scientific, Inc.) or
Odyssey Imaging System (LI-COR Biosciences).
Immunofluorescence assay
The brain tissue was fixed with 10% formalin
overnight at 4°C and embedded in paraffin. The sections (thickness,
6 µm) were then dewaxed in xylene three times for 5 min each at
room temperature and rehydrated in a descending series of ethanol
(100 and 95%) at room temperature for 10 min each. For the cells,
the primary cells were fixed in 4% paraformaldehyde for 10 min at
room temperature. For immunofluorescent staining, the sections were
blocked with 5% normal goat serum (cat. no. 5425; Cell Signaling
Technology, Inc.) and 0.3% Triton X-100 for 1 h at room temperature
to minimize non-specific staining. They were then incubated
overnight at 4°C with the following primary antibodies (1:200),
anti-RIPK1 (cat. no. ab106393; Abcam), anti-GFAP (cat. no. AB5541;
EMD Millipore) and anti-NeuN (cat. no. 24307; Cell Signaling
Technology, Inc.). Then, the samples were incubated with
HRP-conjugated secondary antibodies (1:500) for 1 h at room
temperature (cat. nos. A32935, A32731 and A32759; Thermo Fisher
Scientific, Inc.), and further incubated with 10 µg/ml DAPI for 5
min at room temperature for visualization of the cell nuclei. The
cells were imaged by confocal laser scanning microscopy
(magnification, ×1,000; Nikon Corporation) and the fluorescence
intensity of immunofluorescence images was measured by ImageJ v2.1
(National Institutes of Health) for quantification of five fields
per sample.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from brain tissue or primary
cells using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and then quantified. Then, 2 µg of total RNA was
reverse-transcribed using reverse transcriptase (Promega
Corporation) according to the manufacturer's instructions. qPCR was
performed with TB Green® Fast qPCR kit (Takara Bio,
Inc.) for 35 cycles of: 10 sec at 98°C, 10 sec at 55°C and 20 sec
at 72°C. The primer sequences used were as follows: GFAP, forward
5′-CGGAGACGCATCACCTCTG-3′ and reverse 5′-TGGAGGAGTCATTCGAGACAA-3′;
RIPK, forward 5′-GACAGACCTAGACAGCGGAG −3′ and reverse
5′-CCAGTAGCTTCACCACTCGAC-3′; GAPDH, forward
5′-AGGTCGGTGTGAACGGATTTG-3′ and reverse 5′-GGGGTCGTTGATGGCAACA-3′.
The mean Cq values for the target genes were normalized to the mean
Cq value for the endogenous control GAPDH as previously described
(27). The ratio of mRNA
expression of target gene to GAPDH was defined as
2−∆∆Cq.
Cell growth assay
For the crystal violet assays, 1×105
astrocytes were seeded into each well of a 12-well plate with three
replicates for each group. Cells in conditions of 5% CO2
and 95% O2 in DMEM/F12 supplemented with 10% FBS were
used as a negative control. After 12 h of OGD treatment, cell
proliferation was assessed by crystal violet staining at 37°C for 4
h, as described previously (28).
For the cell survival assays, 1×104 cells from each
treatment condition were plated in triplicate in a 96-well plate at
37°C. The survival rate was measured using a Guava Nexin assay (EMD
Millipore). Results are shown as the mean % of viable cells.
Lactate dehydrogenase (LDH) assay
A total of 1×105 astrocytes were seeded
into each well of a 6-well plate. The release of LDH was measured
using the LDH assay kit (cat. no. A020-2-2; Nanjing Jiancheng
Bioengineering Institute Co., Ltd.) according to the manufacturer's
protocols. Samples without coenzyme I treatment were used as a
negative control. LDH leakage was calculated as follows: LDH
leakage (%) = (A positive/A positive blank) / (A negative/A
negative blank) ×100%.
Statistical analysis
Data are presented as the mean ± SD. Statistical
analysis was conducted using SPSS 10.0 software (SPSS, Inc.) to
perform ANOVA followed by a Student-Newman-Keuls post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Ischemic brain injuries trigger
RIP-dependent astrocyte necroptosis
To determine the association of cerebral ischemic
injury and astrocytic features, a brain injury mouse pMCAO model
was established. The present results showed decreased grip strength
and an increased rate of right forelimb utilization in pMCAO mice
(Fig. 1A and B). In addition, a
significant infarct was found in the ipsilateral cortex of pMCAO
brains compared with sham-operated brains (Fig. 1C and D), indicating that the pMCAO
mouse model was successfully established. The present results
showed that protein and mRNA expression levels of GFAP were
decreased at 12 and 24 h after pMCAO in the ischemic cortex
(Fig. 1E, F, H and I). The protein
and mRNA expression levels of RIPK1, an initiator of necroptosis,
were increased in the pMCAO group compared with the control
(Fig. 1E, G, H and J). Therefore,
the present results suggested that ischemic brain injuries could be
associated with RIP-dependent necroptosis. Protein and mRNA
expression levels of RIPK1 were also increased in astrocytes after
OGD exposure, in addition to decreased cell survival rate and
increased LDH leakage (Fig.
1K-O).
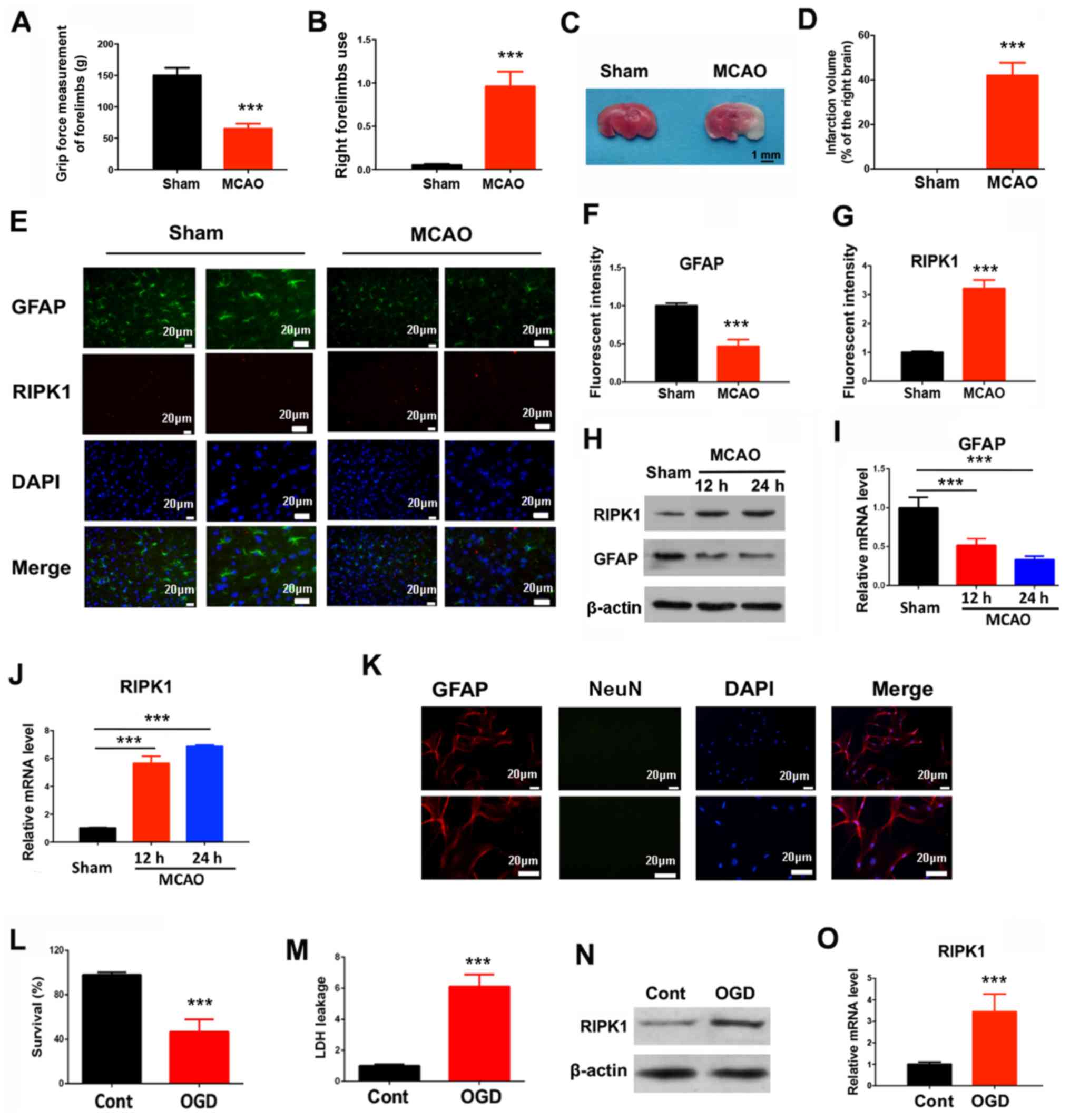 | Figure 1.Ischemic brain injuries trigger
RIP-dependent astrocyte necroptosis. In a pMCAO mouse model, 24 h
after ischemia, the (A) grip force, (B) right forelimb use and (C
and D) infarction volume were determined. Mean ± SD; n=8/group. (E)
Immunofluorescent staining in the mouse brain tissues was used to
quantify (F) GFAP and (G) RIPK1 expression levels. (H) Western
blotting results for GFAP and RIPK1 protein expression levels at
indicated times after the pMCAO operation. (I) GFAP and (J) RIPK1
mRNA expression levels were measured by reverse
transcription-quantitative PCR at indicated times after the pMCAO
operation. ***P<0.001 vs. Sham. (K) Primary cortical astrocytes
were stained with GFAP and NeuN to determine the purity. (L) Cell
survival and (M) LDH leakage of astrocytes were determined 12 h
after OGD treatment. (N) RIPK1 protein and (O) mRNA expression
levels were determined 6 h after OGD treatment. ***P<0.001 vs.
Control. RIP, receptor interacting protein; RIPK1, receptor
interacting protein kinase 1; pMCAO, permanent middle cerebral
artery occlusion; GFAP, glial fibrillary acidic protein; LDH,
lactate dehydrogenase; NeuN, neuronal nuclei; OGD, oxygen-glucose
deprivation. |
NDRG2 knockdown accelerates cerebral
ischemic injury-induced necroptosis
Previous studies have demonstrated that the tumor
suppressor NDRG2 has a neuroprotective function in various diseases
(29,30). Therefore, the present study
investigated whether NDRG2 was involved in the regulation of
cerebral ischemic injury and necroptosis. A mouse line carrying the
NDRG2 conditional knockout allele (Ndrg2-/-) with exons 2–6
flanked by locus of X-over P1 sites (Fig. 2A) was generated. Female homozygotes
(Ndrg2-/-) were crossed with CMV-Cre transgenic male mice,
which have ubiquitous Cre activity. After the pMCAO operation, an
increased infarction volume was observed in Ndrg2-/- mice
(Fig. 2B and C); thus, NDRG2 may
provide neuroprotection against pMCAO insult. Moreover, NDRG2
knockout led to a significant decrease in GFAP expression levels,
and a significant increase in the protein and mRNA expression
levels of RIPK1 (Fig. 2D-I).
Therefore, the depletion of NDRG2 could accelerate pMCAO-induced
necroptosis in brain tissue. NDRG2 may function as a
neuroprotectant during cerebral ischemic injury.
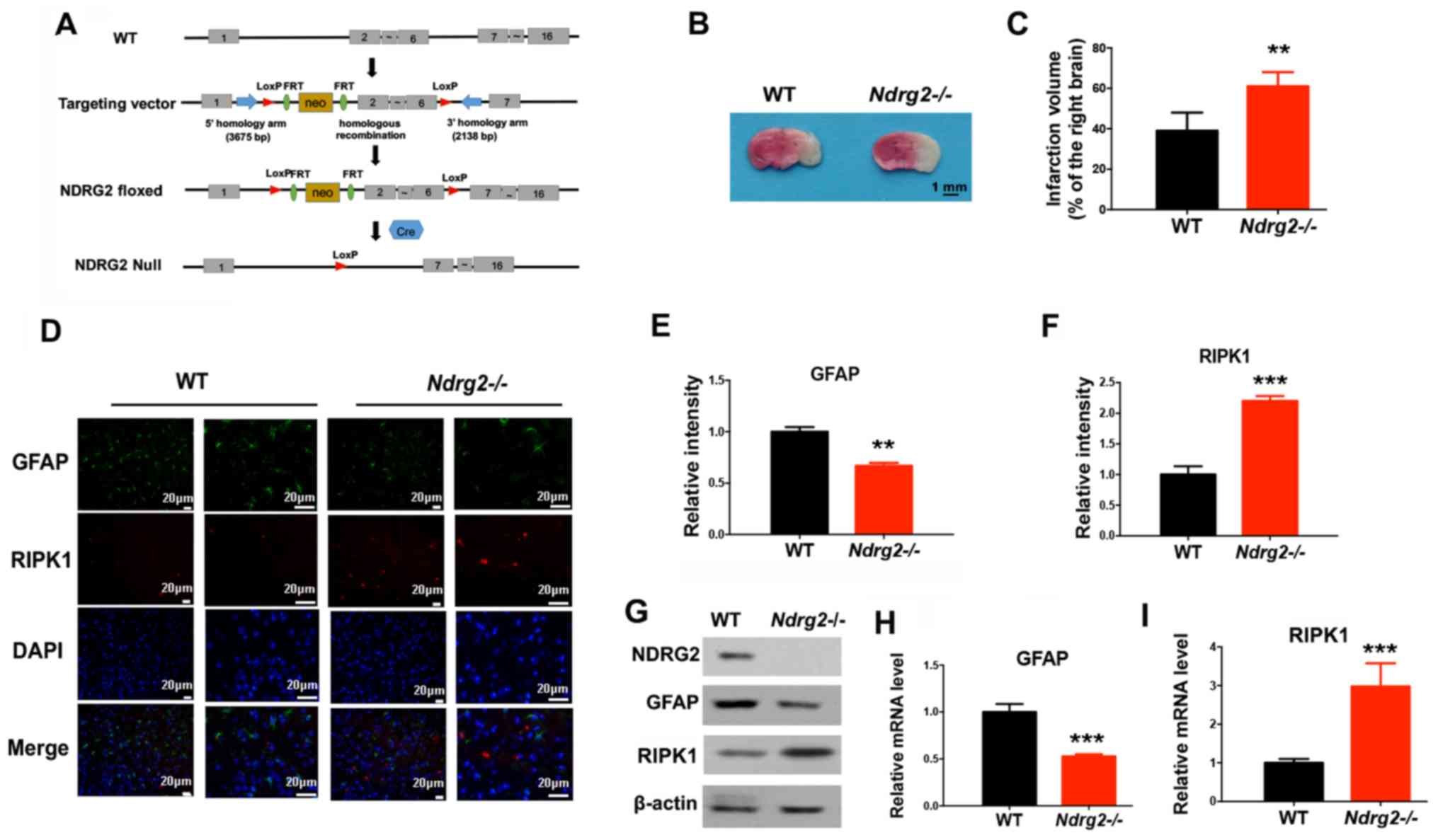 | Figure 2.NDRG2 depletion accelerates cerebral
ischemic injury-induced necroptosis. (A) Schematic representation
of the NDRG2 targeting vector: First, WT locus; second, targeted
allele; third, Ndrg2-floxed; fourth, null allele
Ndrg2-Null. Exons are represented by solid rectangles. The
targeting vector, which is flanked by 3,675 bp of 5′ homology and
2,138 bp of 3′ homology, contains a LoxP-FRT-Neo-FRT cassette and
the second LoxP sequence in introns 1 and 6, respectively. WT or
Ndrg2-/- mice received pMCAO operation. (B) At 24 h
following pMCAO surgery, the infarction volume was determined and
(C) quantified. At 12 h following pMCAO surgery, the indicated
proteins levels were determined by (D-F) immunofluorescent staining
and (G) western blotting, and mRNA expression levels of (H) GFAP
and (I) RIPK1 were determined by reverse transcription-quantitative
PCR. **P<0.01, ***P<0.001 vs. WT group. NDRG2, N-myc
downstream-regulated gene 2; WT, wild-type; RIPK1, receptor
interacting protein kinase 1; pMCAO, permanent middle cerebral
artery occlusion; GFAP, glial fibrillary acidic protein; LoxP,
locus of X-over P1. |
NDRG2 attenuates astrocytic cell death
via the suppression of RIPK1
To further determine the role of NDRG2 in regulating
astrocyte survival, astrocytes from the neonate mice were isolated
and NDRG2 was overexpressed using a lentivirus (Fig. 3A). After OGD exposure, NDRG2
overexpression could significantly block OGD-dependent decreases in
cell viability and survival, and induction of LDH leakage (Fig. 3B-D). Furthermore, NDRG2
overexpression increased GFAP expression levels and decreased RIPK1
expression levels after OGD exposure (Fig. 3E), indicating NDRG2 may attenuate
astrocytic cell death by repressing RIPK1 levels. In addition,
astrocytes were also isolated from wild-type and Ndrg2-/-
mice (Fig. 3F). NDRG2 knockout
increased OGD-induced cell death and LDH leakage (Fig. 3G-I). NDRG2 knockout also
upregulated RIPK1 and downregulated under OGD conditions GFAP
(Fig. 3J). Therefore, the present
results suggested that NDRG2 could block OGD-induced astrocytic
cell death by suppressing RIPK1.
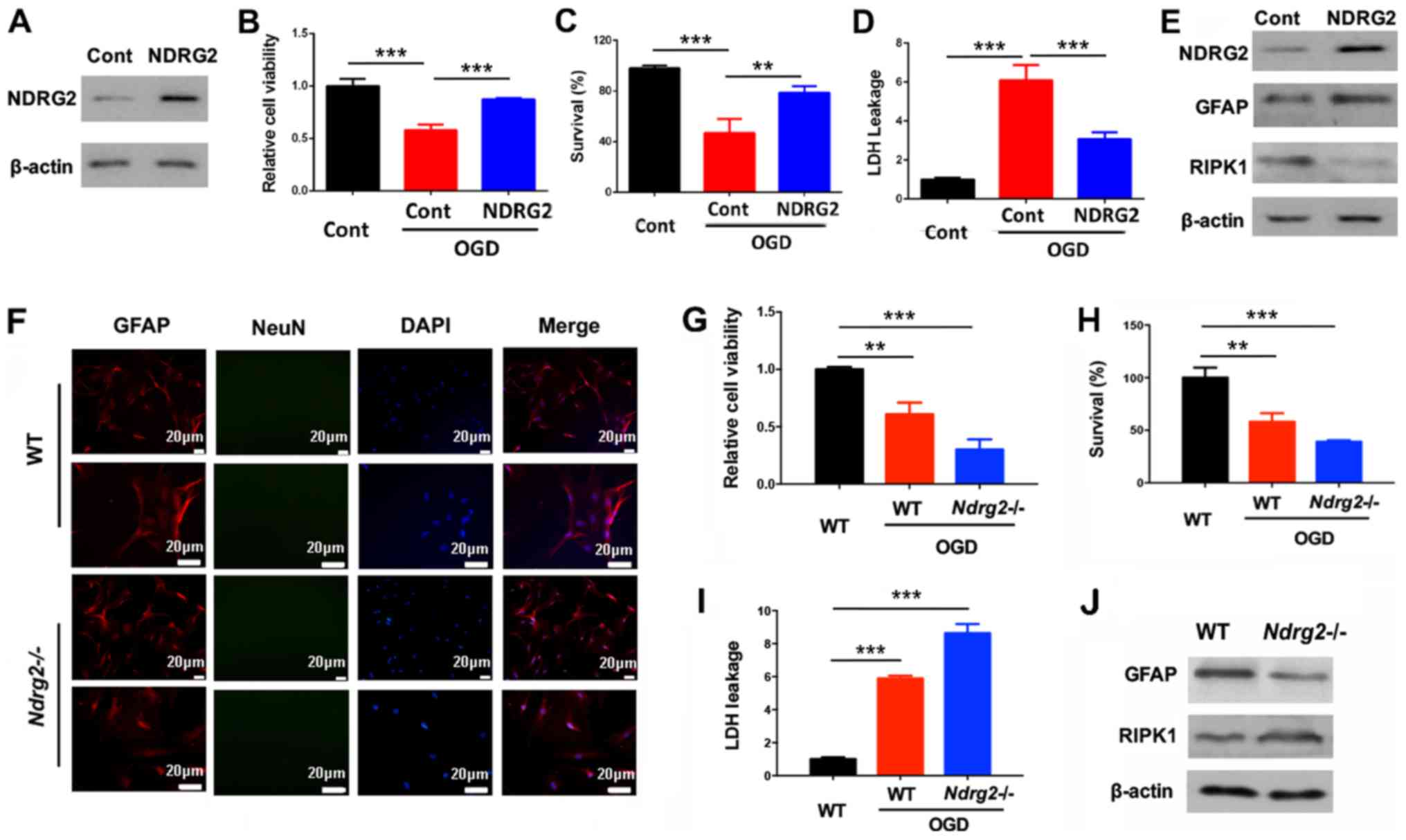 | Figure 3.NDRG2 attenuates astrocytic cell
death via the suppression of RIPK1. Astrocytes were infected with
NDRG2 overexpressing lentivirus. (A) NDRG2 expression was
determined after the lentivirus infection. (B) Cell viability, (C)
survival and (D) LDH leakage were determined 12 h after OGD
treatment. (E) Protein expression levels of GFAP and RIPK1 were
determined 6 h after OGD treatment. (F) Primary cortical astrocytes
were stained with GFAP and NeuN to determine the purity. Astrocytes
were isolated from WT or Ndrg2-/- mice. (G) Cell viability,
(H) survival and (I) LDH leakage were determined 12 h after OGD
treatment. (J) Protein levels were determined 6 h after OGD
treatment. **P<0.01, ***P<0.001. NDRG2, N-myc
downstream-regulated gene 2; WT, wild-type; RIPK1, receptor
interacting protein kinase 1; GFAP, glial fibrillary acidic
protein; LDH, lactate dehydrogenase; NeuN, neuronal nuclei; OGD,
oxygen-glucose deprivation. |
Necroptosis inhibitor necrostatin-1
blocks astrocytic cell death after NDRG2 knockdown
In order to determine whether the role of NDRG2 in
neuroprotection is the result of blocking necroptosis, the present
study used a necroptosis inhibitor Nec-1, which can inhibit
necrosome formation via the suppression of RIPK1 activity (31). Nec-1 treatment prevented the
pMCAO-induced decrease in GFAP protein and mRNA expression levels
in the ischemic cortex of Ndrg2-/- mice (Fig. 4A and B). In addition, after Nec-1
treatment, the reduced viability and survival rates of astrocytes
from Ndrg2-/- mice following OGD exposure were partly
reversed (Fig. 4C and D).
OGD-induced LDH leakage from Ndrg2-/- astrocytes was also
attenuated following Nec-1 treatment (Fig. 4E). The present results indicated
that NDRG2 could function as a neuroprotector by blocking
necroptosis, and that pharmacological inhibition of astrocyte
necroptosis promotes neuroprotection against ischemic brain
injuries after NDRG2 knockdown.
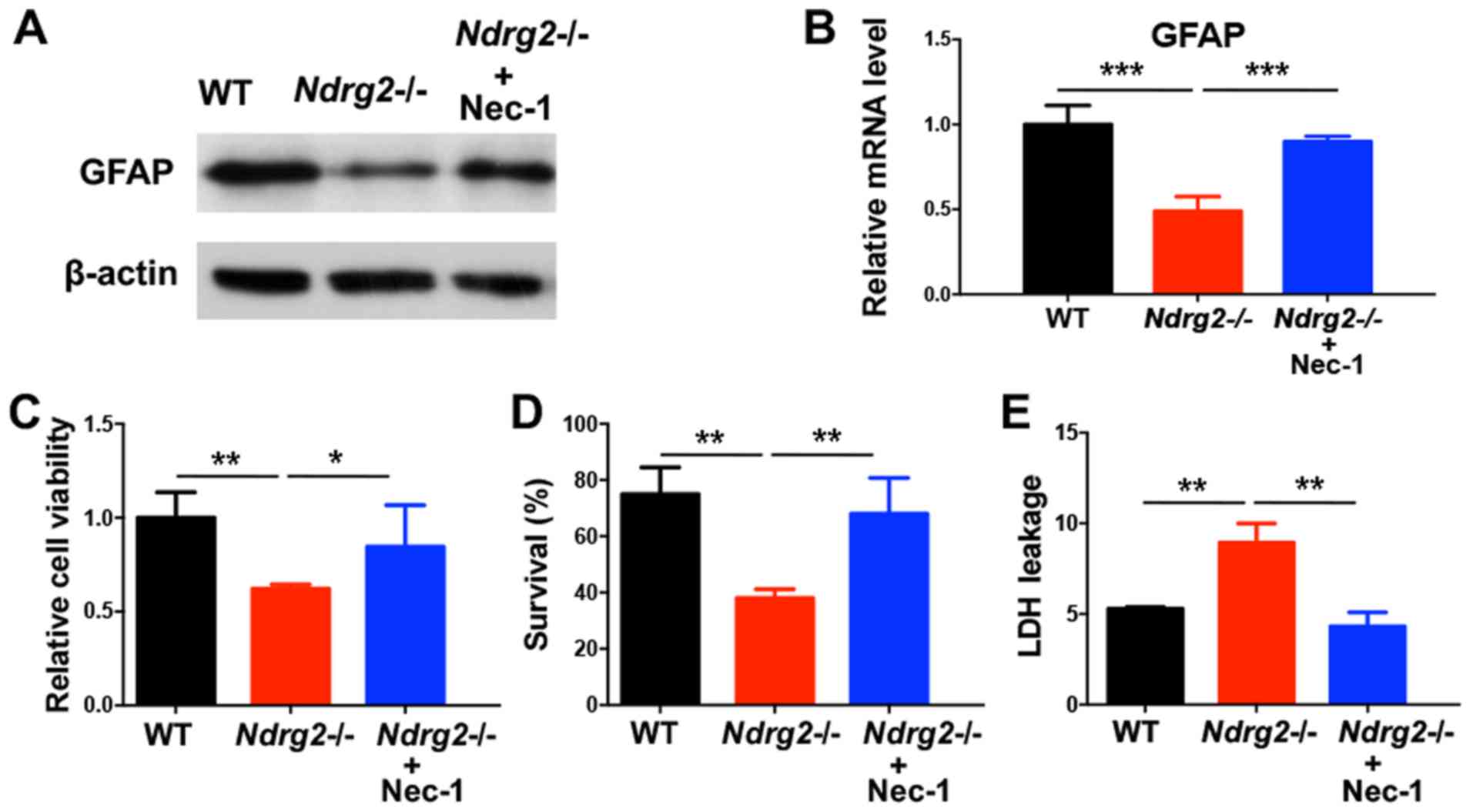 | Figure 4.Necroptosis inhibitor Nec-1 blocks
astrocytic cell death after NDRG2 knockdown. Nec-1 (10 µM) was
administrated to primary cultured astrocytes for 30 min prior to
OGD. (A) GFAP protein and (B) mRNA expression levels were
determined 6 h after OGD treatment. (C) Cell viability, (D)
survival and (E) LDH leakage were determined 12 h after OGD
treatment. *P<0.05, **P<0.01, ***P<0.001. Nec-1,
necrostatin-1; NDRG2, N-myc downstream-regulated gene 2; WT,
wild-type; RIPK1, receptor interacting protein kinase 1; GFAP,
glial fibrillary acidic protein; LDH, lactate dehydrogenase; OGD,
oxygen-glucose deprivation. |
Discussion
Cerebral ischemia is an important component of
secondary brain injury after trauma (2). Inhibiting ischemic injury has become
one of the key therapeutic methods for improving the prognosis of
brain injury (3). A highly
regulated form of necrosis, termed necroptosis, elicited
significant interest in the study of human diseases such as
ischemic stroke in order to understand its implications in
pathologies (32). The present
study investigated astrocyte activity during cerebral ischemia, and
identified that ischemic brain injuries may trigger RIP-dependent
astrocyte necroptosis. In a NDRG2 conditional knockout mouse model,
the present study found that knockdown of NDRG2 could accelerate
pMCAO-induced necroptosis in brain tissue, therefore indicating
that NDRG2 may function as a neuroprotector during cerebral
ischemic injury. The present study suggested that NDRG2 may
attenuate astrocytic cell death via the suppression of RIPK1. The
present results suggested that pharmacological inhibition of
astrocyte necroptosis by Nec-1 produced neuroprotection against
ischemic brain injuries after NDRG2 knockdown. Therefore, NDRG2
could be considered as a target for the treatment of cerebral
ischemia.
Astrocytes are the most widely distributed glial
cells in the nervous system (4,5).
Astrocytes can not only provide metabolic and nutritional support
for neurons, but also play a crucial role in promoting neuronal
survival, synaptic function, nerve regeneration and nerve repair
(4,5). It is of clinical importance to
explore the mechanism of astrocyte death to facilitate the
treatment and prognosis of post-traumatic cerebral ischemia. To
determine the association between cerebral ischemic injury and
astrocytes, the present study established a brain injury mouse
pMCAO model and identified that RIPK1, which is an initiator of
necroptosis, was upregulated after injury. OGD exposure also led to
increased RIPK1 expression levels and LDH leakage, and decreased
cell survival rate. Therefore, the present results suggested that
ischemic brain injuries may be associated with RIP-dependent
astrocyte necroptosis.
NDRG2 has been shown to function as a tumor
suppressor via the reduction of cell proliferation and metastasis,
and induction of cell differentiation in numerous cancer types
(33,34). In the central nervous system, NDRG2
is highly expressed in astrocytes, and regulates astrocytic
activity by promoting cell differentiation and stabilizing cell
morphology (21). To investigate
the role of NDRG2 in astrocytes, the present study generated
Ndrg2-/- mice. After ischemic injury, increased infarction
volume, reduction of GFAP and induction of RIPK1 were observed;
thus, NDRG2 may provide neuroprotection against cerebral ischemia.
Moreover, astrocytes from Ndrg2-/- mice showed decreased
cell viability and survival, and increased LDH leakage after OGD
exposure. The present study suggested that NDRG2 could block
OGD-induced astrocytic cell death by inhibiting RIPK1. A similar
conclusion was reached following NDRG2 overexpression in
astrocytes. Therefore, the present study suggested that NDRG2 plays
an important role in blocking cerebral ischemic injury-induced
necroptosis, potentially via the suppression of RIPK1
expression.
Necroptosis is different to traditional apoptosis.
When cells are stimulated by tumor necrosis factor-α and other
apoptotic inducers, necrotic morphological features can be induced,
and treatment with caspase inhibitors can block apoptosis pathways
(35). Nec-1 is one of the most
commonly used RIPK1 inhibitors. The effect of Nec-1 on necroptosis
is associated with allosteric inhibition of the activity of RIPK1
by interacting with the T-loop of the N-terminal kinase domain
(36). A previous study showed
that Nec-1 can decrease ischemic brain injury area
dose-dependently, and improve nerve function score, prolong nerve
protective effect and delay the onset of necroptosis by inhibiting
the activation of RIPK1/RIPK3 pathways after ischemic brain injury
(19). In addition, Nec-1 can
protect neurovasculature and improve the prognosis of animals
following neurological injury (37,38).
The present results indicated that Nec-1 treatment prevented
pMCAO-induced reductions of GFAP, ODG-induced suppression of
astrocyte viability and survival, and induction of LDH leakage in
the ischemic cortex of Ndrg2-/- mice. Therefore,
pharmacological inhibition of astrocyte necroptosis may provide
neuroprotection against ischemic brain injuries after NDRG2
knockdown. NDRG2 could be considered as a target for the treatment
of cerebral ischemia.
Acknowledgements
Not applicable.
Funding
This work was financially supported by the National
Natural Science Foundation of China (grant no. 81601719).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YHW and KX contributed to conception and design of
this study. JZ, LKY, QHW, WL and YF performed the experiments. YPX
and WLC contributed to the analysis and interpretation of data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were in accordance with the
relevant provisions of the Laboratory Animal Center and were
approved by the Experimental Animal Ethics Committee of the 101
Hospital of PLA of Anhui Medical University.
Patient consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hyder AA, Wunderlich CA, Puvanachandra P,
Gururaj G and Kobusingye OC: The impact of traumatic brain
injuries: A global perspective. NeuroRehabilitation. 22:3103–353.
2007. View Article : Google Scholar
|
|
2
|
Bouma GJ, Muizelaar JP, Choi SC, Newlon PG
and Young HF: Cerebral circulation and metabolism after severe
traumatic brain injury: The elusive role of ischemia. J Neurosurg.
75:685–693. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Piper LC, Zogg CK, Schneider EB, Orman JA,
Rasmussen TE, Blackbourne LH and Haider AH: Guidelines for the
Treatment of Severe Traumatic Brain Injury: Are They Used? JAMA
Surg. 150:1013–1015. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Davila D, Thibault K, Fiacco TA and
Agulhon C: Recent molecular approaches to understanding astrocyte
function in vivo. Front Cell Neurosci. 7:2722013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ota Y, Zanetti AT and Hallock RM: The role
of astrocytes in the regulation of synaptic plasticity and memory
formation. Neural Plast. 2013:1854632013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bambrick L, Kristian T and Fiskum G:
Astrocyte mitochondrial mechanisms of ischemic brain injury and
neuroprotection. Neurochem Res. 29:601–608. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Trendelenburg G and Dirnagl U:
Neuroprotective role of astrocytes in cerebral ischemia: Focus on
ischemic preconditioning. Glia. 50:307–320. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nakashima MN, Yamashita K, Kataoka Y,
Yamashita YS and Niwa M: Time course of nitric oxide synthase
activity in neuronal, glial, and endothelial cells of rat striatum
following focal cerebral ischemia. Cell Mol Neurobiol. 15:341–349.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Anderson MF, Blomstrand F, Blomstrand C,
Eriksson PS and Nilsson M: Astrocytes and stroke: Networking for
survival? Neurochem Res. 28:293–305. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dienel GA and Hertz L: Astrocytic
contributions to bioenergetics of cerebral ischemia. Glia.
50:362–388. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Swanson RA, Ying W and Kauppinen TM:
Astrocyte influences on ischemic neuronal death. Curr Mol Med.
4:193–205. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim SJ and Li J: Caspase blockade induces
RIP3-mediated programmed necrosis in Toll-like receptor-activated
microglia. Cell Death Dis. 4:e7162013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shindo R, Kakehashi H, Okumura K, Kumagai
Y and Nakano H: Critical contribution of oxidative stress to
TNFα-induced necroptosis downstream of RIPK1 activation. Biochem
Biophys Res Commun. 436:212–216. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
He S, Wang L, Miao L, Wang T, Du F, Zhao L
and Wang X: Receptor interacting protein kinase-3 determines
cellular necrotic response to TNF-alpha. Cell. 137:1100–1111. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ni Y, Gu WW, Liu ZH, Zhu YM, Rong JG, Kent
TA, Li M, Qiao SG, An JZ and Zhang HL: RIP1K contributes to
neuronal and astrocytic cell death in ischemic stroke via
activating autophagic-lysosomal pathway. Neuroscience. 371:60–74.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deng Y, Yao L, Chau L, Ng SS, Peng X, Liu
X, Au W-S, Wang J, Li F, Ji S, et al: N-Myc downstream-regulated
gene 2 (NDRG2) inhibits glioblastoma cell proliferation. Int J
Cancer. 106:342–347. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shen L, Qu X, Li H, Xu C, Wei M, Wang Q,
Ru Y, Liu B, Xu Y, Li K, et al: NDRG2 facilitates colorectal cancer
differentiation through the regulation of Skp2-p21/p27 axis.
Oncogene. 37:1759–1774. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mitchelmore C, Büchmann-Møller S, Rask L,
West MJ, Troncoso JC and Jensen NA: NDRG2: A novel Alzheimer's
disease associated protein. Neurobiol Dis. 16:48–58. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takahashi K and Yamada M, Ohata H, Momose
K, Higuchi T, Honda K and Yamada M: Expression of Ndrg2 in the rat
frontal cortex after antidepressant and electroconvulsive
treatment. Int J Neuropsychopharmacol. 8:381–389. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Flügge G, Araya-Callis C, Garea-Rodriguez
E, Stadelmann-Nessler C and Fuchs E: NDRG2 as a marker protein for
brain astrocytes. Cell Tissue Res. 357:31–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takeichi T, Takarada-Iemata M, Hashida K,
Sudo H, Okuda T, Kokame K, Hatano T, Takanashi M, Funabe S, Hattori
N, et al: The effect of Ndrg2 expression on astroglial activation.
Neurochem Int. 59:21–27. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu H, Zhao J, Zhou W, Li H, Zhou R, Zhang
L, Zhao H, Cao J, Zhu X, Hu H, et al: Ndrg2 regulates vertebral
specification in differentiating somites. Dev Biol. 369:308–318.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hao FL, Han XF, Wang XL, Zhao ZR, Guo AH,
Lu XJ and Zhao XF: The neurovascular protective effect of
alogliptin in murine MCAO model and brain endothelial cells. Biomed
Pharmacother. 109:181–187. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Alamri FF, Shoyaib AA, Biggers A,
Jayaraman S, Guindon J and Karamyan VT: Applicability of the grip
strength and automated von Frey tactile sensitivity tests in the
mouse photothrombotic model of stroke. Behav Brain Res.
336:250–255. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu M, Yang L, Hong LZ, Zhao XY and Zhang
HL: Direct protection of neurons and astrocytes by matrine via
inhibition of the NF-κB signaling pathway contributes to
neuroprotection against focal cerebral ischemia. Brain Res.
1454:48–64. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qin AP, Liu CF, Qin YY, Hong LZ, Xu M,
Yang L, Liu J, Qin ZH and Zhang HL: Autophagy was activated in
injured astrocytes and mildly decreased cell survival following
glucose and oxygen deprivation and focal cerebral ischemia.
Autophagy. 6:738–753. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Smith R, Owen LA, Trem DJ, Wong JS,
Whangbo JS, Golub TR and Lessnick SL: Expression profiling of
EWS/FLI identifies NKX2.2 as a critical target gene in Ewing's
sarcoma. Cancer Cell. 9:405–416. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Y, Yin A, Sun X, Zhang M, Zhang J, Wang
P, Xie R, Li W, Fan Z, Zhu Y, et al: Deficiency of tumor suppressor
NDRG2 leads to attention deficit and hyperactive behavior. J Clin
Invest. 127:4270–4284. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Takarada-Iemata M, Yoshikawa A, Ta HM,
Okitani N, Nishiuchi T, Aida Y, Kamide T, Hattori T, Ishii H,
Tamatani T, et al: N-myc downstream-regulated gene 2 protects
blood-brain barrier integrity following cerebral ischemia. Glia.
66:1432–1446. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Smith CC and Yellon DM: Necroptosis,
necrostatins and tissue injury. J Cell Mol Med. 15:1797–1806. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chu D, Zhang Z, Li Y, Wu L and Zhang J,
Wang W and Zhang J: Prediction of colorectal cancer relapse and
prognosis by tissue mRNA levels of NDRG2. Mol Cancer Ther.
10:47–56. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shi H, Jin H, Chu D, Wang W, Zhang J, Chen
C, Xu C, Fan D and Yao L: Suppression of N-myc downstream-regulated
gene 2 is associated with induction of Myc in colorectal cancer and
correlates closely with differentiation. Biol Pharm Bull.
32:968–975. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Holler N, Zaru R, Micheau O, Thome M,
Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B and Tschopp
J: Fas triggers an alternative, caspase-8-independent cell death
pathway using the kinase RIP as effector molecule. Nat Immunol.
1:489–495. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Degterev A, Hitomi J, Germscheid M, Ch'en
IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
King MD, Whitaker-Lea WA, Campbell JM,
Alleyne CH Jr and Dhandapani KM: Necrostatin-1 reduces
neurovascular injury after intracerebral hemorrhage. Int J Cell
Biol. 2014:4958172014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu X, Chua KW, Chua CC, Liu CF, Hamdy RC
and Chua BH: Synergistic protective effects of humanin and
necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain
Res. 1355:189–194. 2010. View Article : Google Scholar : PubMed/NCBI
|














