Introduction
11β-hydroxysteroid dehydrogenases (11β-HSD) are a
class of oxidoreductases that catalyze the reversible
transformation between corticosterone and dehydrocorticosterone.
11β-HSD can be divided into two isoforms in humans: 11β-HSD1 and
11β-HSD2 (1). 11β-HSD1 is a
crucial enzyme that converts inactive cortisone into active
hydrocortisone, thus regulating multiple functions of
glucocorticoids (GCs) (2).
11β-HSD1 is widely expressed in tissues (3) and is particularly abundant in the
liver, muscle and fat tissue (4).
In renal tissue, 11β-HSD1 exhibits oxidase activity, whereas in
non-renal tissues, such as liver and fat tissues, 11β-HSD1 exhibits
reductase activity. In particular, the role of 11β-HSD1 in the
liver is associated with elevated levels of active GCs, which is
consistent with increased levels of GCs in liver fibrosis (5). Therefore, it has been speculated that
11β-HSD1 is involved in the early activation and signal
transduction of hepatic stellate cells (HSCs) and may regulate the
expression of extracellular matrix (ECM) components (6–9).
Hepatic fibrosis is a common pathophysiological
feature of all chronic liver diseases (10). HSCs are the final target cell type
of various profibrotic factors (11), serving as key mediators in the
occurrence and development of hepatic fibrosis (12–15).
Once HSCs are activated by inflammatory factors, including
transforming growth factor (TGF)-β1 (16), their phenotype changes from resting
to activated and then into myofibroblasts (MFB), which secrete
abundant ECM components (17),
resulting in increased ECM deposition and reduced matrix
decomposition (18). Furthermore,
activated HSCs exhibit reduced cell cycle, accelerated
proliferation rates (19),
increased numbers (20), decreased
amounts of fat and vitamin A storage (21), and secretion of profibrotic factors
[α-smooth muscle actin (α-SMA) and connective tissue growth factor
(CTGF)], which further stimulate HSC activation (19,22),
resulting in a feedback mechanism (23). Activated HSCs promote the synthesis
and secretion of tissue inhibitor of metalloproteinase (24) and inhibit the degradation of ECM by
matrix metalloproteinases, thus promoting liver fibrogenesis
(25). ECM is primarily composed
of three types of organic substances: Collagen, glycoprotein and
proteoglycan. Among these, type IV collagen (Col IV), N-terminal
pro-peptide of collagen type III (PIIINP) and hyaluronic acid (HA)
are often used as serological indicators of the degree of hepatic
fibrosis (26,27). In addition, liver hydroxyproline
(Hyp) content, a unique amino acid component of collagen, can
reflect alterations in collagen metabolism, parallel to the degree
of hepatic fibrosis (28). As an
antigen substance, xenogeneic serum induces an immunoresponse in
the liver, leading to the formation of immune complexes. If the
excessive immune complexes are not removed in a timely manner, they
are deposited in the liver and the hepatic vascular wall, causing
cell infiltration, release of inflammatory mediators, cell
degeneration of parenchymal liver cells, fibrous hyperplasia and
hepatic fibrosis (27,29,30).
The present study developed an in vivo model
of hepatic fibrosis in rats using porcine serum. In addition, using
in vitro cell culture, LX2-HSCs were stimulated with TGF-β1.
The two experimental approaches were established to systematically
investigate alterations in 11β-HSD1 expression and activity in
liver tissue and HSCs during hepatic fibrogenesis. Furthermore, the
effects of 11β-HSD1 knockdown and overexpression on the development
and progression of hepatic fibrosis and its underlying mechanism of
action were studied in LX2-HSCs.
Materials and methods
Experimental animals
A total of 108 male Sprague Dawley (SD) rats (age,
4–6 weeks; weight, 130–150 g) in quarantine were obtained from
Hunan SJA Laboratory Animal Co. Ltd. The SD rats were housed in
barrier rooms at 22±2°C with 65–70% humidity, a 12-h light/dark
cycle and free access to water and food. Rats were randomly
assigned to the following three groups (n=36 per group): i) Normal
control group (N); ii) the model group (M); and iii) the 11β-HSD1
inhibitor group (M+I). Rats in the M and M+I groups received an
intraperitoneal injection of 0.5 ml porcine serum (Beijing Solarbio
Science & Technology Co., Ltd.) twice a week. The N group
received an equal volume of 0.9% NaCl via intraperitoneal injection
at the same frequency for the same duration. In addition to porcine
serum administration, rats in the M+I group were treated with the
11β-HSD1 inhibitor BVT.2733 (100 mg/kg), which was orally
administrated by gavage. At 5, 10 and 15 weeks following treatment,
12 rats in each group were anesthetized with an intraperitoneal
injection of 2% pentobarbital sodium (30 mg/kg body weight) and
deep anesthesia was verified by moderate respiratory and
cardiovascular depression, and good muscle relaxation. The
experimental rats were euthanized by cervical dislocation. The
abdominal aorta and right lobe of the liver were collected for
subsequent experiments. Prior to sacrifice, 2 ml blood was
collected from the abdominal aorta of the rats, which was stored at
4°C.
All animal experiments were performed in accordance
with the guidelines of The Institutional Animal Care and Use
Committee of Xiang [license no. SCXK (Xiang) 2011-0003].
Experimental reagents
The 11β-HSD1 inhibitor, BVT.2733, was purchased from
MedChem Express. Rat TGF-β1 was purchased from Abbkine Scientific
Co., Ltd. (cat. no. PRP100618). The anti-11β-HSD1 antibody was
purchased from AtaGenix (cat. no. ATA24000; 1:200). Human TGF-β1
was purchased from PeproTech, Inc. (cat. no. AF-100-21). Rat Col IV
(cat. no. MBS704982) and PIIINP ELISA kits (cat. no. MBS704287),
and the Annexin V-FITC/PI apoptosis detection kit (cat. no.
MBS355226) were purchased from MyBioSource, Inc. The HA ELISA kit
was purchased from R&D Systems (cat. no. DHYAL0). The ProCell
collagen colorimetric assay kit was purchased from Genmed
Scientifics, Inc. (cat. no. GMS10373.8). The rabbit-derived rat
α-SMA (cat. no. ab124964; 1:1,500), CTGF (cat. no. ab6992; 1:2,000)
and β-actin (cat. no. ab8227; 1:1,000) primary antibodies, and the
goat anti-rabbit secondary IgG-HRP antibody (cat. no. ab97080;
1:5,000) were all purchased from Abcam. The cortisol kit (11β-HSD1
activity detection kit) was purchased from Cisbio (cat. no.
62CRTPEG). The adenovirus and adenovirus vector were purchased from
Miao Ling Biotechnology Co., Ltd. (cat. nos. P0241 and P0653). The
Cell Counting Kit-8 (CCK-8) cell proliferation test kit (cat. no.
CK04) was purchased from Dojindo Molecular Technologies, Inc.
Pathological examination of
livers
At weeks 5, 10 and 15 of treatment, 12 rats were
randomly selected, anesthetized with 2% pentobarbital sodium and
sacrificed. The abdominal aorta and right lobe of the liver were
harvested for pathological examinations. In brief, the tissues were
fixed for 7 days in 4% neural-buffered formalin at room
temperature, dehydrated in increasing concentrations of ethanol,
embedded with paraffin wax and sliced into 4 µm-sections, and
comparative pathological alterations were observed via hematoxylin
(5–20 min) and eosin staining (10–30 min), both at room
temperature. The stages of fibrosis were examined by two
independent experienced pathologists using the following
pathological scoring criteria (31): 0, no liver fibrosis; I,
degeneration of hepatocytes with ≤0.5 of the radius of hepatic
lobules and infiltration of inflammatory cells surrounding the
portal area; II, degeneration of hepatocyte >0.5 of the radius
of hepatic lobules, with primarily balloon-like degeneration, a
large number of infiltrating inflammatory cells and obvious fibrous
tissue hyperplasia in the portal vein; and III, degeneration and
necrosis of hepatocytes extended to 0.66 of the radius of hepatic
lobules with fragmentation and bridging necrosis of hepatocytes,
and lots of fibrosis in the parenchyma.
Measurement of serum Col IV, HA and
PIIINP via ELISA
Blood samples were centrifuged at 1006.2 × g for 15
min at 4°C to obtain serum. The concentrations of Col IV, HA and
NPIIINP in the serum of the rats were detected using ELISA kits,
according to the manufacturer's protocol.
Western blotting
Total protein from the liver tissues was extracted
with ice-cold RIPA lysis buffer (Beyotime Institute of
Biotechnology), containing 50 mM Tris, 150 mM NaCl, 1% Triton
X-100, 1% sodium deoxycholate, 0.1% SDS and 1mM protease inhibitor
PMSF. Total protein was quantified using a bicinchoninic acid assay
to correct the sampling volume and 30 µg protein/lane was separated
via 10% SDS-PAGE. The separated proteins were subsequently
transferred onto nitrocellulose membranes (EMD Millipore) and
blocked with 5% BSA (Beyotime Institute of Biotechnology) for 1 h
at room temperature. The membranes were then incubated with the
primary antibodies for 1 h at room temperature. Following the
primary antibody incubation, the membranes were washed with 1X
TBS-1% Tween-20 (TBST) three times for 15 min each and incubated
with the secondary antibodies (1:2,000; Boster Biological
Technology; cat. nos. BA1050 and BA1054) 1 h at room temperature.
The membrane was subsequently washed with 1X TBST twice for 15 min
and 1X TBS once for 15 min. Protein bands were visualized using a
SuperSignal West Pico Chemiluminescent substrate (Thermo Fisher
Scientific, Inc.) and protein expression was quantified using
ImageJ v1.8.0 software (National Institutes of Health).
Measurement of Hyp in liver
tissues
Hyp content in the liver tissue was detected using
an A030-2 kit (Nanjing Jiancheng Bioengineering Institute)
according to the manufacturer's instructions. In brief, liver
tissues (~200 mg) were homogenized and incubated overnight at
110°C. After filtering acid hydrolysates with a 0.45 µm filter,
samples or Hyp standards (50 µl) were dried at 60°C, dissolved with
methanol (50 µl) and treated with 1.2 ml 50% isopropanol and 200 µl
chloramine-T solution at room temperature for 10 min. Ehrlich's
solution (1.3 ml) was added and the samples were further incubated
at 50°C for 90 min. The optical density was read at a wavelength of
558 nm using a spectrophotometer. A standard curve was constructed
using serial two-fold dilutions of 1 mg hydroxyproline
solution.
Detection of 11β-HSD1 activity in
liver tissue and HSCs
Liver tissues (1.5-2.0 g) were homogenized in PBS,
and the resulting homogenate was centrifuged at 1,000 × g at 4°C
for 5 min. The supernatant was collected and centrifuged at 42,000
× g for 3 min at 4°C. The protein concentration of the supernatant
was determined using the bicinchoninic acid method. The supernatant
was placed in an ice bath for subsequent analysis. HSCs were lyzed
and total protein was quantified using the bicinchoninic acid
assay. 11β-HSD1 activity in the supernatant of rat liver tissue and
total protein of HSCs was detected using the cortisol kit according
to the manufacturer's instructions.
Preparation, culture and verification
of rat primary HSCs
Primary HSCs were obtained from 20 additional SD
rats (all details on rats and housing the same as above) by
perfusion with Pronase E/Collagenase IV. Briefly, the rats were
euthanized with 30 mg/kg body weight sodium pentobarbital and
CO2 (20% container volume/min). After determining that
the rats were immobile, not breathing and the appearance of pupil
dilation, the abdomen of the rats was disinfected with 75% ethanol.
The abdominal cavity was opened to expose the inferior vena cava
and liver. The inferior vena cava was clipped, and the portal vein
was cut, and a syringe pump was used to infuse EGTA solution (20
ml/ml at 40°C) from the inferior vena cava into the liver at a
constant rate until the liver turned white. Subsequently, the liver
was infused with 15 ml Pronase E solution and 20 ml Collagenase IV
solution for 20 min each at 37°C. The liver was separated from the
abdominal cavity and plated in a 100-mm Petri dish, then 5 ml
Pronase E (0.2 g/l)/Collagenase IV (0.2 g/l) digestion solution was
added to digest the liver tissue at 37°C for 15 min. The liver was
crushed with ophthalmic scissors and a pipette was used to disperse
the hepatocytes. The digested hepatocyte suspension was transferred
to a 50 ml centrifuge tube and shook for 25 min on a plate shaker
at 37°C. Subsequently, 10 ml pre-chilled rat HSC complete medium
(4°C; cat. no. CM-R041; Procell Life Science & Technology Co.,
Ltd.) was added to stop the digestion. After filtering through a
200-mesh screen, the suspension was placed in a 50 ml centrifuge
tube to undergo one-step density gradient centrifugation with 18%
Nycodenz (W/V) at 1,450 × g for 22 min at room temperature. Trypan
blue staining was conducted to evaluate the relative population of
living HSCs at 37°C for 3 min. Quiescent HSCs were examined under a
fluorescence microscope and appeared blue under UV light
irradiation (328 nm wavelength). Desmin immunocytochemistry was
conducted to determine the purity of the isolated primary HSCs.
After culture in DMEM (Sigma-Aldrich; Merck KGaA) supplemented with
20% FBS (Biological Industries) at 37°C for 24 h, the majority of
HSCs had attached to the dishes. Cell culture medium was replaced
with DMEM containing 0.5% FBS and cultured at 37°C for another 24
h. Subsequently, 5×106 HSCs were treated with or without
5 ng/ml TGF-β1 at 37°C for another 48 h.
Immunocytochemistry
Rat HSCs were inoculated into a petri dish
containing coverslips and treated with or without TGF-β1. After
cells were grown to a monolayer, the coverslips were removed and
washed twice with 1X PBS. The cells were fixed with 4%
paraformaldehyde for 10 min at room temperature, after which the
slides were washed with 1X PBS for 5 min and the cells were
permeabilized with 0.5% Triton for 15 min at room temperature. The
slides were then washed twice with 1X PBS for 5 min each, blocked
with 1% BSA (Beyotime Institute of Biotechnology) solution for 30
min at room temperature and incubated with the primary antibody
diluted with 1% BSA solution at 37°C for 2 h. Subsequently, the
slides were incubated with Alexa Fluor 488-labeled goat anti-rabbit
secondary antibody (cat. no. bs-0295G-AF488; BIOSS) diluted with 1%
BSA solution at 37°C for 1 h, washed twice with 1X PBS for 5 min
each time and stained with a 5% DAPI solution at room temperature
for 2 min. Subsequently, the slides were mounted with an
anti-quenching sealer and visualized under a fluorescence
microscope (magnification, ×200).
11β -HSD1 gene knockdown and
overexpression in LX2-HSCs
To overexpress 11β-HSD1, the expression plasmid
pSuper-11β-HSD1 was constructed. Briefly, the full-length 11β-HSD1
cDNA was synthesized by Miao Ling Biotechnology Co., Ltd. and
inserted into the parental vector pSuper (Addgene, Inc.). The
pSuper-11β-HSD1 expression vector was confirmed by double enzymatic
digestion with BamH1/XhoI and sequencing. The custom
interference plasmids, including small interfering (si)RNA-11β-HSD1
and non-specific control (NC) siRNA-11β-HSD1-NC, were purchased
from Miao Ling Biotechnology Co., Ltd. The sequences of the siRNAs
were as follows: siRNA-11β-HSD1 sense,
5′-CAGAGAUGCUCCAAGGAAAGATT-3′ and antisense,
5′-UUUCCUUGGAGCAUCUCUGGUTT-3′; and siRNA-11β-HSD1-NC sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. A total of 5×106 LX2-HSCs
cells were divided into the following groups: i) Normal control
group (N); ii) the siRNA group, in which LX2-HSCs were transfected
with 2 µg interference plasmid siRNA-11β-HSD1 and expression of
11β-HSD1 was silenced; iii) the negative control group (NC), in
which LX2-HSCs were transfected with 50 nM plasmid
siRNA-11β-HSD1-NC; iv) the pSuper group, in which LX2-HSCs were
transfected with 1 µg pSuper-11β-HSD1 plasmid to overexpress
11β-HSD1; and v) the Vec group, in which LX2-HSCs were transfected
with 1 µg pSuper-11β-HSD1 plasmid, which was used as a control. All
cells were transfected using Lipofectamine® 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). At 72 h
post-transfection, total protein was extracted and the expression
of 11β-HSD1 was measured in each experimental group by western
blotting.
Cell cycle distribution and HSC
proliferation
HSC proliferation was measured after 12, 24, 48, 72
and 96 h of culture using the CCK-8 cell proliferation kit,
according to the manufacturer's instructions. HSC cell cycle
distribution was detected using propidium iodide (PI) staining.
Briefly, HSC cells were trypsinized, collected and then fixed and
permeabilized using 70% (v/v) cold ethanol for 2 h at room
temperature. Cells were subsequently incubated with 10 µg/ml
PI/0.1% Triton X-100/0.1% RNase in PBS solution at 37°C for 30 min
in the dark. Stained cells were analyzed on a BD FACS Calibur (BD
Biosciences) using a fluorescence emission wavelength of 585 nm
after an excitation wavelength of 488 nm. For each analysis, 10,000
events were evaluated, and DNA content was determined using ModFit
LT v3.0 software (Verity Software House, Inc.).
Effect of changes in 11β-HSD1
expression on the release of collagen in HSCs
Cells from each group were collected and the
concentration of collagen was measured using the ProCell collagen
colorimetric assay kit, according to the manufacturer's protocol.
Briefly, Sircol dye reagent was added to the cell culture
supernatants (1,000 × g; 10 min; room temperature), stirred for 20
min at room temperature and centrifuged at 15,000 × g for 10 min at
room temperature. The absorbance of the bound dye was measured at a
wavelength of 555 nm on a spectrophotometer.
Statistical analysis
Statistical analyses were conducted using SPSS
statistical software (version 17.0; SPSS, Inc.). At least three
independent experiments were performed for each analysis. Data are
expressed as the mean ± standard deviation. The normality of the
data was analyzed using the Kolmogorov-Smirnov test. The
homoscedasticity of data was analyzed using the Levene test. For
normally distributed data with homogeneity, differences among
groups were analyzed using one-way ANOVA followed by Fisher's LSD
post hoc test. Data containing >3 groups were compared with the
control group using one-way ANOVA followed by Dunnett's post hoc
test. For comparisons within groups (TGF-β1− and
TGF-β1+) and among groups analyzed using two-way ANOVA
followed by the Bonferroni post hoc test. For non-normally
distributed data, the Kruskal-Wallis test followed by the
Bonferroni post hoc test was used. P<0.05 was considered to
indicate a statistically significant difference.
Results
Role of 11β-HSD1 inhibition in liver
fibrosis
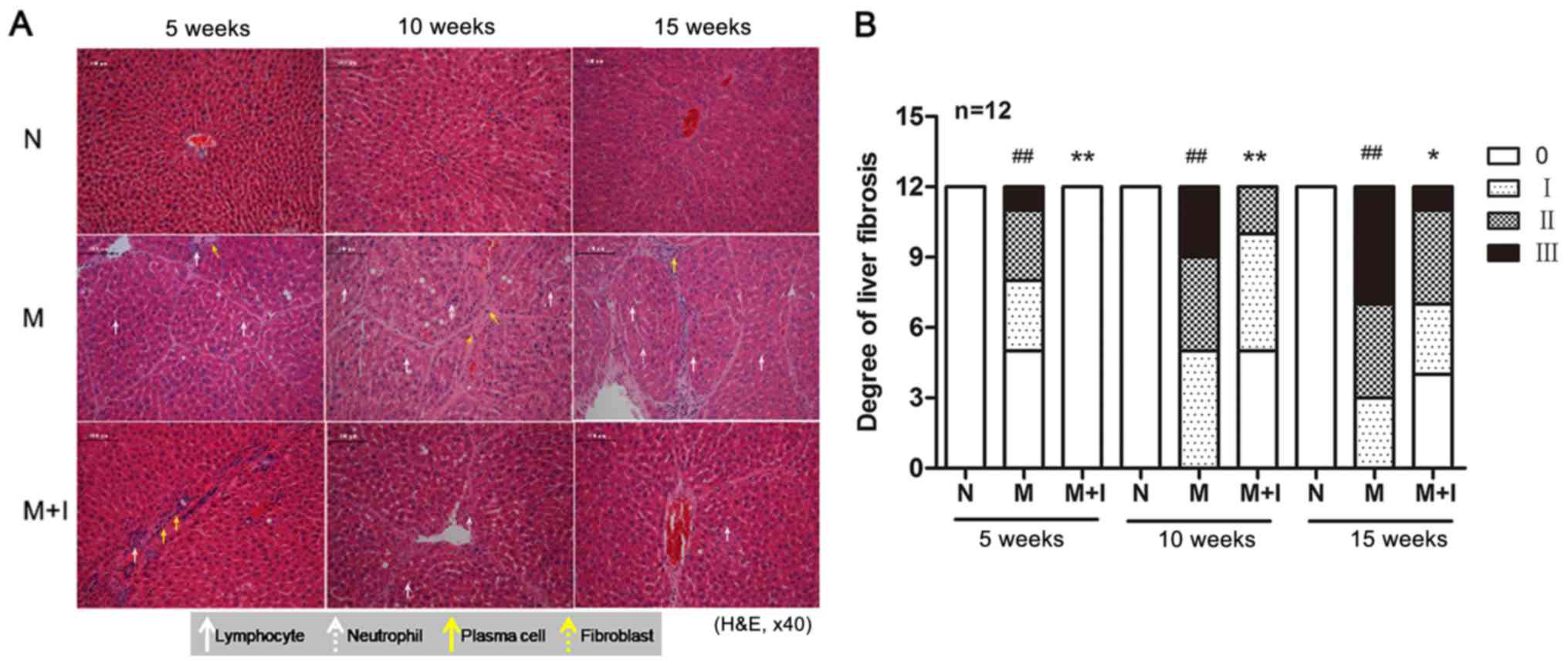
As shown in Fig. 1A and
B, excessive formation of fibrotic tissues was observed in the
livers of rats treated with porcine serum (M group), whereas slight
periportal inflammatory infiltration was observed in fibrotic rats
treated with the 11β-HSD1 inhibitor (M+I group). Histopathological
analysis demonstrated that the degree of liver fibrosis was
significantly higher in the M group compared with the N group
(P<0.01) and the M+I group (P<0.05). Together, the results
suggested that 11β-HSD1 inhibition was effective against
experimental liver fibrosis.
Serum markers of fibrosis are
ameliorated after treatment with an 11β-HSD1 inhibitor
As hepatic fibrosis developed in the experimental
model, the levels of Col IV, PIIINP and HA significantly increased
in a time-dependent manner in the M group (P<0.01), which was
not observed in the N group (Fig.
2A-C). Although the markers of liver fibrosis increased as
hepatic fibrosis developed in the presence of the 11β-HSD1
inhibitor (M+I group), the increase was significantly decreased
compared with the M group (P<0.01).
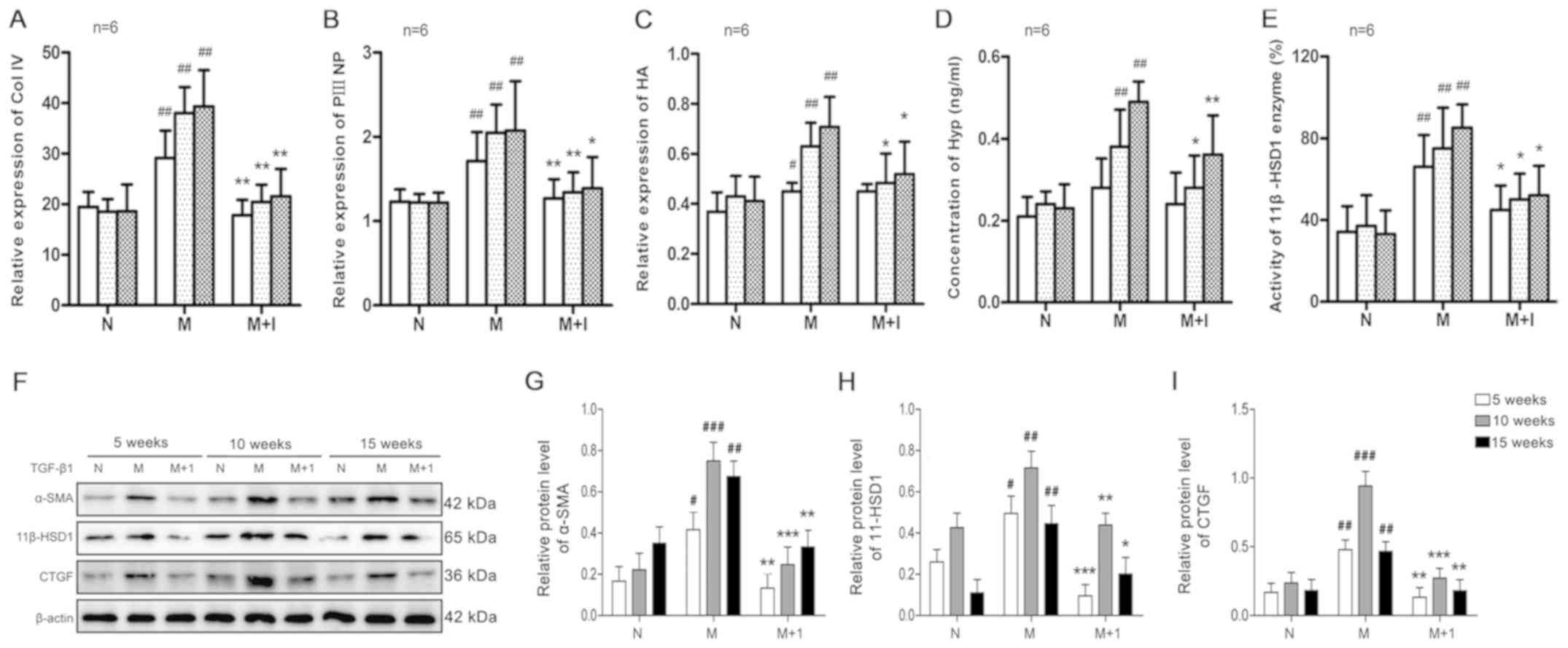 | Figure 2.Effect of porcine serum on liver
fibrosis. As liver fibrosis progressed, the expression of (A) Col
IV, (B) PIIINP and (C) HA in the serum was measured.
#P<0.05, ##P<0.01 vs N; *P<0.05,
**P<0.01 vs. M. The liver (D) Hyp content and (E) 11β-HSD1
enzyme activity. ##P<0.01 vs. N; **P<0.01 and
*P<0.05 vs. M. Protein expression levels were (F) determined by
western blotting and semi-quantified for (G) α-SMA, (H) 11β-HSD1
and (I) CTGF. #P<0.05, ##P<0.01 and
###P<0.001 vs. N; *P<0.05, **P<0.01 and
***P<0.001 vs. M. Col IV, type IV collagen; PIIINP, N-terminal
pro-peptide of collagen type III; HA, hyaluronic acid; N, normal
control group; M, model group; M+I, 11β-HSD1 inhibitor (BVT.2733)
group; Hyp, hydroxyproline; 11β-HSD1, 11β-hydroxysteroid
dehydrogenase 1; α-SMA, α-smooth muscle actin; CTGF, connective
tissue growth factor. |
11β-HSD1 inhibition decreases liver
Hyp content and enzyme activity
Subsequently, whether the Hyp content and 11β-HSD1
activity in the liver tissues of fibrotic rats were altered in
response to fibrosis induction was investigated. Hyp content and
11β-HSD1 enzyme activity were significantly increased in the M
group compared with the N group (P<0.01; Fig. 2D and E). By contrast, Hyp content
and 11β-HSD1 enzyme activity were significantly decreased in the
M+I group compared with the M group (P<0.01).
HSC activation decreases following
11β-HSD1 inhibition
Concomitant with serum markers of liver fibrosis,
the levels of α-SMA, 11β-HSD1 and CTGF gradually increased in the M
group as hepatic fibrogenesis progressed compared with the N group
(P<0.01; Fig. 2F-I). By
contrast, protein expression levels of α-SMA, 11β-HSD1 and CTGF in
the liver tissues of the M+I group were significantly decreased
compared with the M group (P<0.01), indicating that the 11β-HSD1
inhibitor prevented HSC activation.
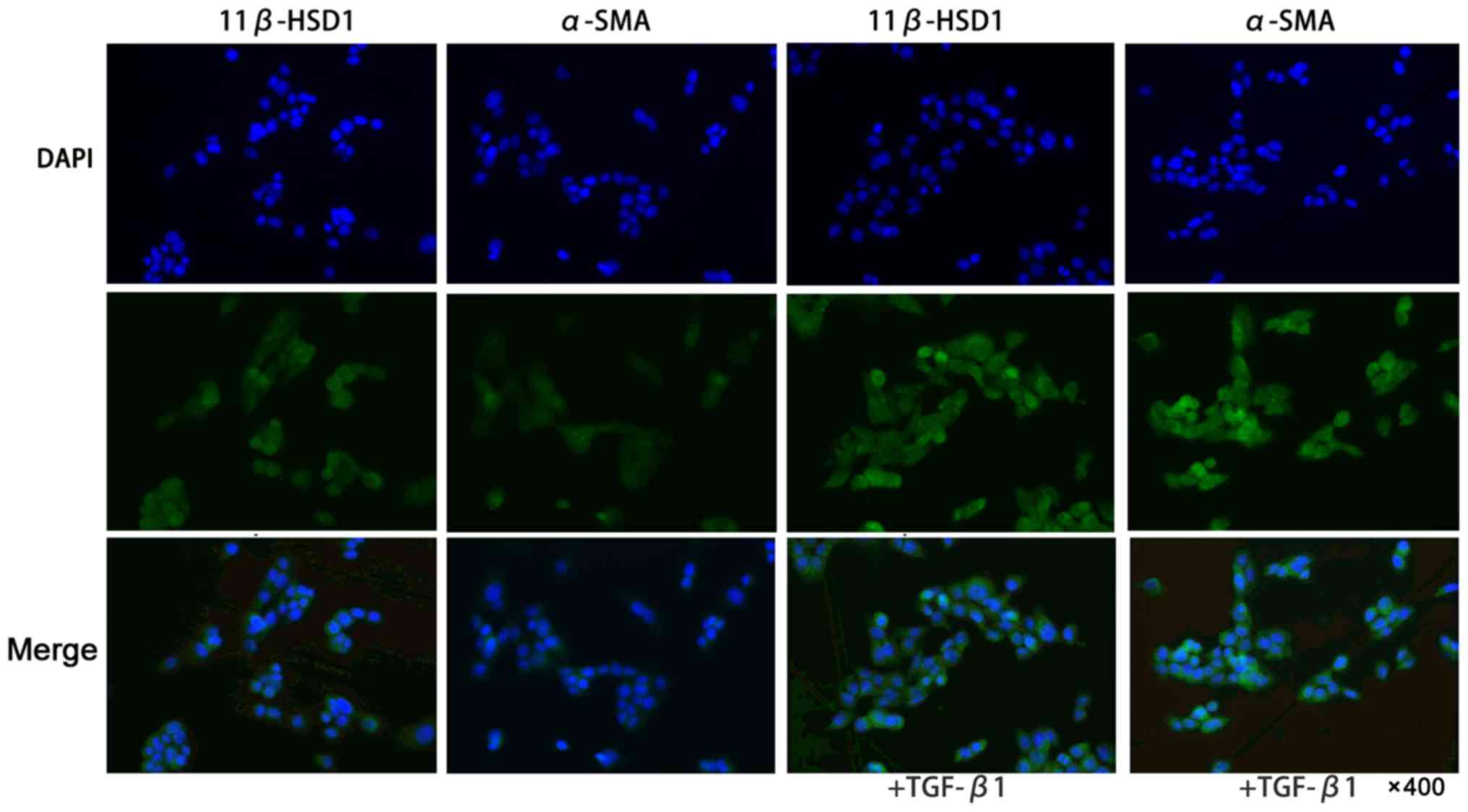
Alteration of 11β-HSD1 during primary
HSC activation
To determine whether 11β-HSD1 expression could be
altered during HSC activation, primary HSCs were freshly prepared
and treated with TGF-β1 in vitro. As shown in Fig. 3, the 11β-HSD1 and α-SMA expression
levels were higher after the TGF-β1-mediated activation of primary
HSCs compared with the controls.
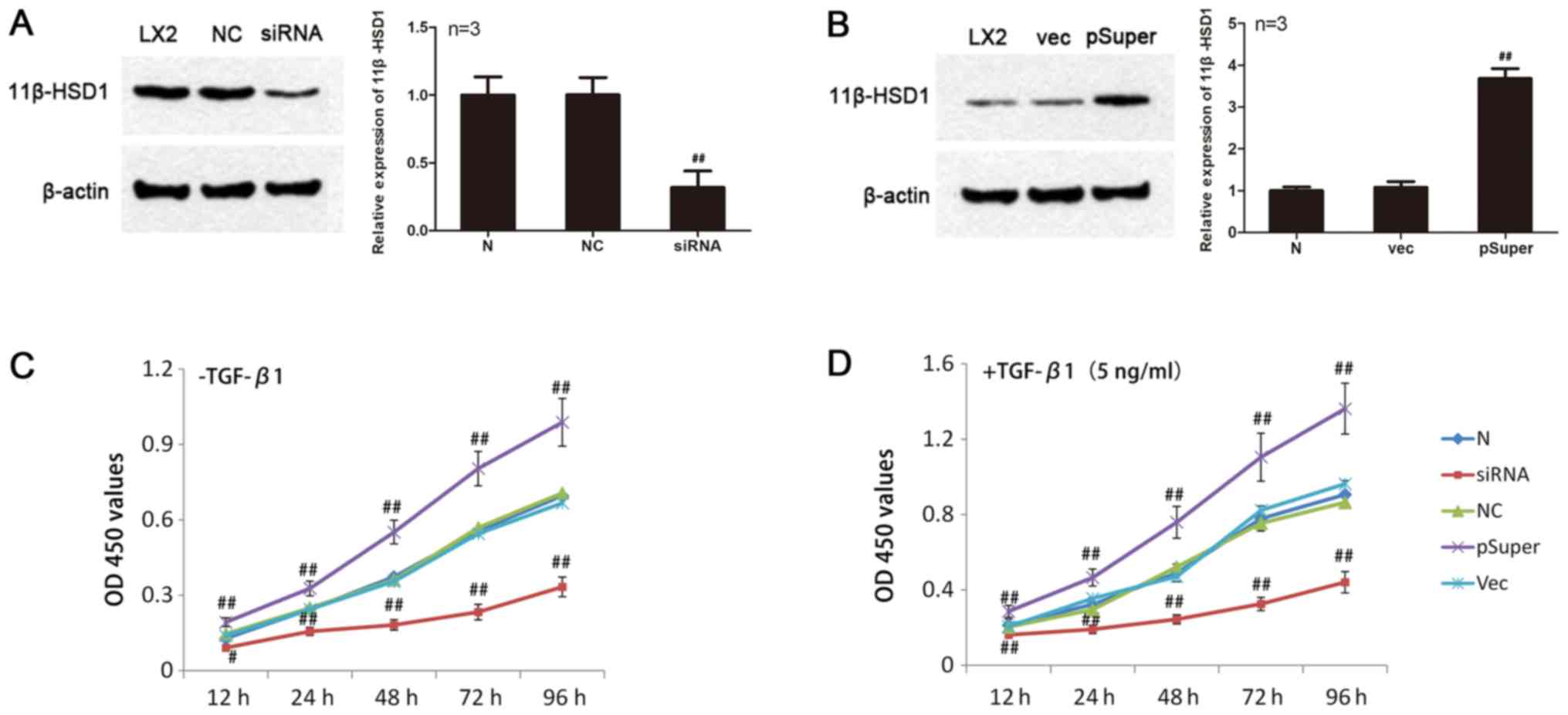
Silencing of the 11β-HSD1 gene in HSCs
via siRNA transfection
As shown in Fig.
4A, the protein expression levels of 11β-HSD1 in LX2-HSCs
transfected with siRNA-11β-HSD1 were significantly lower compared
with the NC group (P<0.01). Furthermore, siRNA-mediated
knockdown of 11β-HSD1 resulted in an 85% decreased in 11β-HSD1
expression levels. No significant difference in the expression
levels of 11β-HSD1 protein were observed between the NC and the N
groups.
Overexpression of 11β-HSD1 in HSCs
following transfection of the pSuper-11β-HSD1 expression
vector
Eukaryotic expression of pSuper-11β-HSD1 plasmids
was identified and the sequencing results demonstrated that the
recombinant plasmid had no deletions, mutations or other
alterations (data not shown). As shown in Fig. 4B, 11β-HSD1 protein expression in
the pSuper group was significantly higher (~x3.4) compared with the
Vec group (P<0.01). No significant difference in 11β-HSD1
protein expression was observed between the Vec and N groups.
Effect of alterations in 11β-HSD1
expression on HSC proliferation
HSCs with 3 different expression levels of 11β-HSD1
were challenged with TGF-β1 (5 ng/ml). Proliferation was measured
at different time points (12, 24, 48, 72 and 96 h) using the CCK-8
assay.
As observed in Fig.
4C, under normal culture conditions, LX2-HSC proliferation was
higher in the pSuper group compared with the N group (P<0.01),
whereas LX2-HSC proliferation was decreased in the siRNA group
compared with the N group (12 h, P<0.05; 24, 48, 72 and 96 h,
P<0.01). However, no significant differences were observed
between the NC and Vec groups. Following TGF-β1 induction, LX2-HSC
proliferation was enhanced compared with untreated LX2-HSCs. Cell
proliferation was significantly increased in the pSuper group
compared with the N group (P<0.01). Although LX2-HSC
proliferation in the siRNA group was higher following TGF-β1
treatment compared with untreated cells, the effect of TGF-β1 on
the LX2-HSC proliferation was significantly reduced compared with
the N group (P<0.01). No significant differences were observed
between the NC and Vec groups.
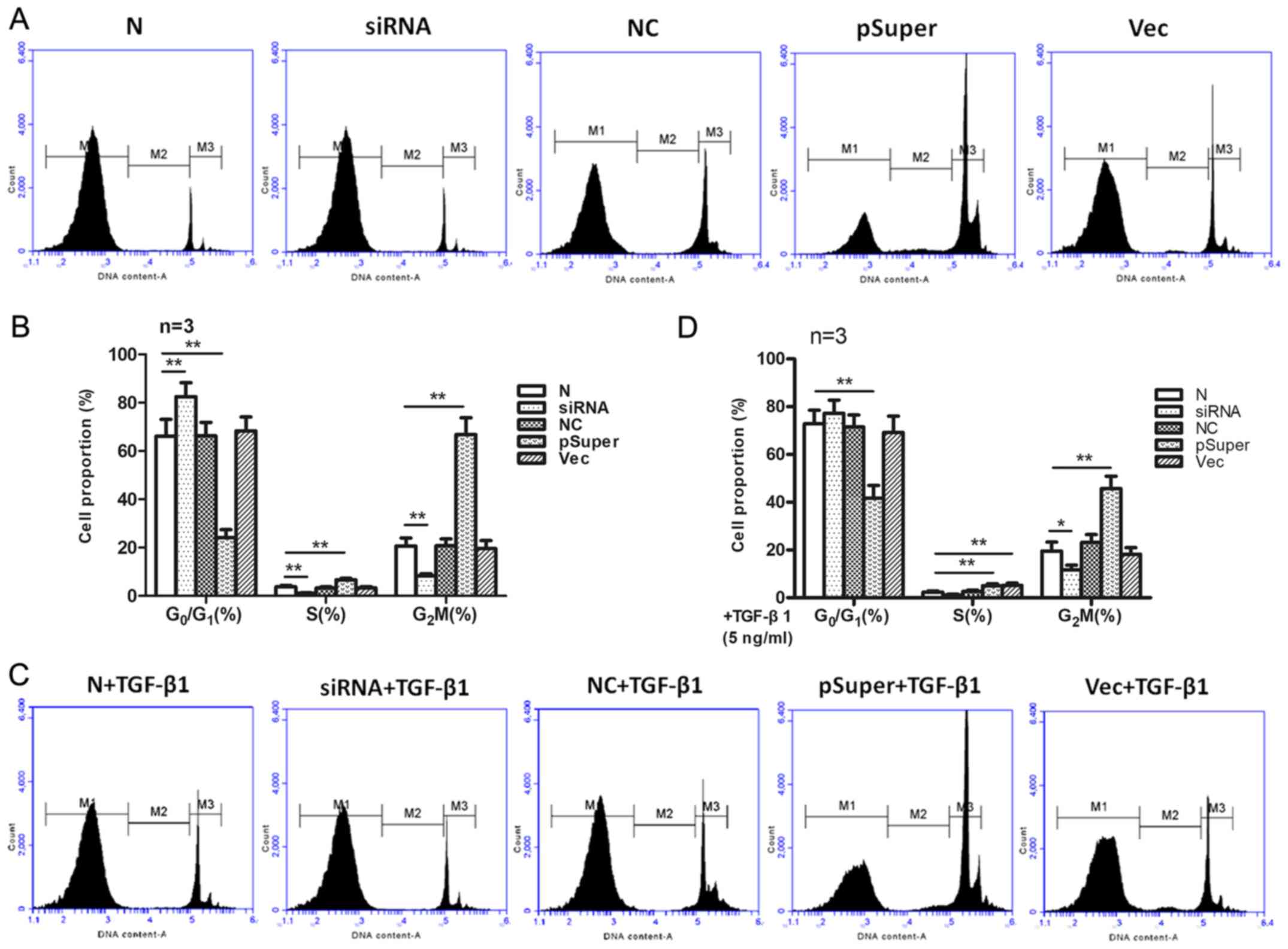
Effect of TGF-β1 induction on cell
cycle distribution in LX2-HSCs
HSC cycle distribution was detected via flow
cytometry (Fig. 5). In the siRNA
group (11β-HSD1 knockdown group), the cells displayed increased
G0/G1 phase arrest compared with the NC
group. No significant differences were observed in the cell cycle
distribution between the NC and N groups. In the pSuper group
(11β-HSD1 overexpression group), a decreased number of cells were
in the G0/G1 phase and an increased number of
cells were in the S and G2/M phases compared with the N
group. However, no significant differences were observed in cell
cycle distribution between the Vec and N groups.
Influence of TGF-β1 on the expression
of α-SMA, 11β-HSD1 and CTGF in vitro
TGF-β1 treatment significantly increased the protein
expression levels of α-SMA, 11β-HSD1 and CTGF in LX2-HSCs cells
compared with untreated cells in all experimental groups (Fig. 6A-H). The pSuper group (11β-HSD1
overexpression group) displayed altered the protein expression
levels of α-SMA, 11β-HSD1 and CTGF (Fig. 6E-H) to a greater extent compared
with the siRNA group (11β-HSD1 knockdown group; Fig. 6A-D), whereas no statistical
differences were observed between the NC and Vec groups (Fig. 6A-H).
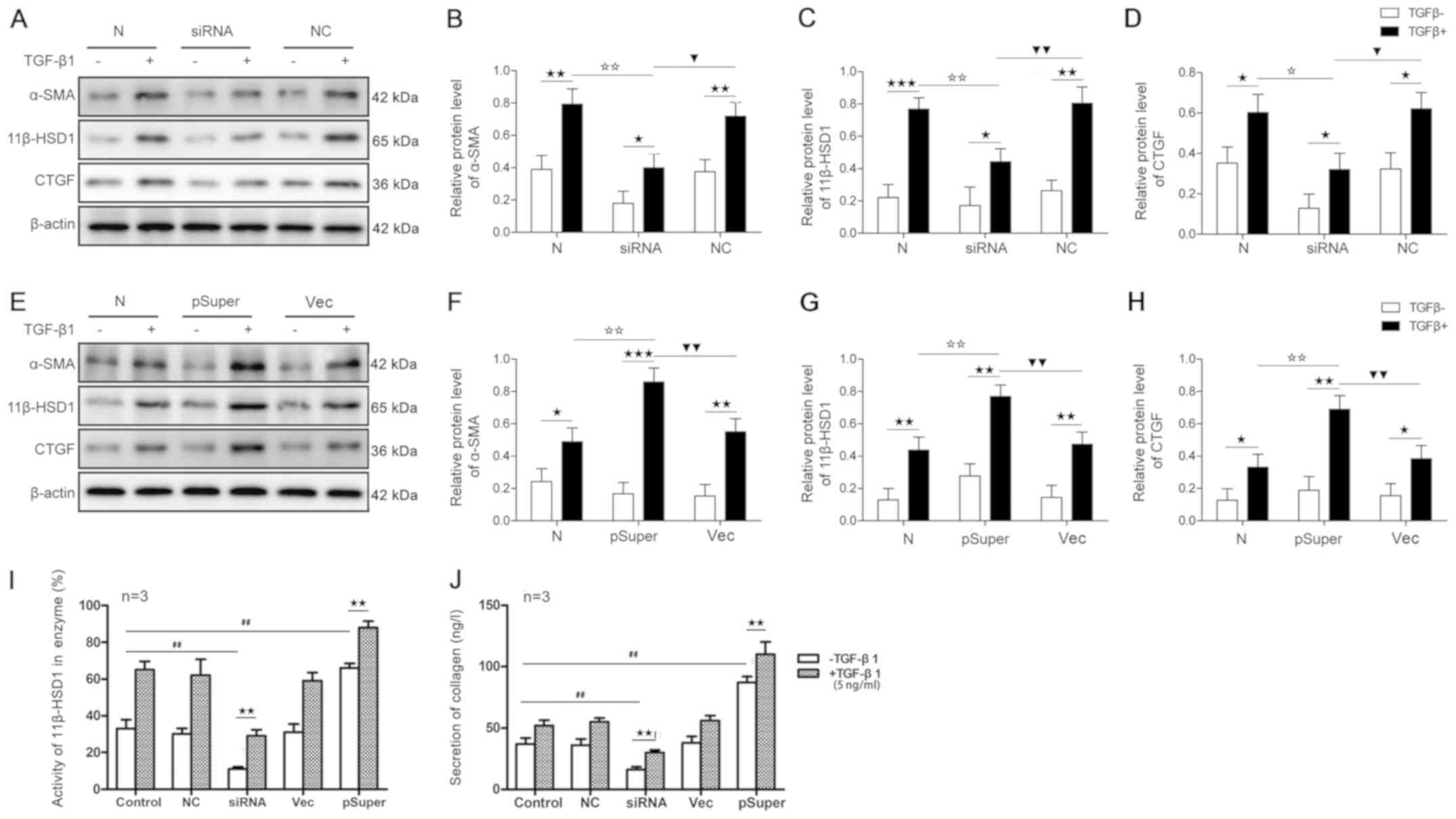 | Figure 6.Effects of siRNA-11β-HSD1 or
pSuper-11β-HSD1 on protein levels of α-SMA, 11β-HSD1 and CTGF,
enzyme activity of 11β-HSD1 and collagen secretion in LX2-HSCs.
Protein expression levels were (A) determined by western blotting
and semi-quantified for (B) α-SMA, (C) 11β-HSD1 (D) CTGF following
siRNA transfection. Protein expression levels were (E) determined
by western blotting and semi-quantified for (F) α-SMA, (G) 11β-HSD1
and (H) CTGF following pSuper transfection. Effects of
siRNA-11β-HSD1 or pSuper-11β-HSD1on (I) 11β-HSD1enzyme activity and
(J) collagen secretion in LX2-HSCs. ★P<0.05,
★★P<0.01 and ★★★P<0.001;
✩P<0.05 and ✩✩P<0.01;
▼P<0.05 and ▼▼P<0.01;
##P<0.01. siRNA; small interfering RNA; 11β-HSD1,
11β-hydroxysteroid dehydrogenase 1; α-SMA, α-smooth muscle actin;
CTGF, connective tissue growth factor; HSCs, hepatic stellate
cells; N, normal control group; NC, non-specific control; Vec,
group in which LX2-HSCs were transfected with the
pSuper-11β-HSD1-vector plasmid; TGF-β1, transforming growth factor
β1. |
Effect of TGF-β1 on collagen release
and 11β-HSD1 enzyme activity in HSCs in culture
As shown in Fig. 6I and
J, LX2-HSCs in the pSuper group displayed increased collagen
secretion, which was associated with enhanced 11β-HSD1 enzyme
activity, compared with the control group. By contrast, LX2-HSC
collagen secretion was significantly reduced in the siRNA group,
which was accompanied by a decrease in 11β-HSD1 enzyme activity
compared with the control group. No significant differences were
observed between the NC and Vec groups. Following TGF-β1 induction,
collagen secretion and 11β-HSD1 enzyme activity was increased in
all experimental groups. Significant increases in these parameters
were observed in the pSuper group.
Discussion
Consistent with previous studies (27,32,33),
the present study indicated that there was an increase in collagen
fibers in the liver, which was associated with significantly
elevated levels of Hyp, and serum Col IV, HA and PIIINP in an
experimental rat model of hepatic fibrosis compared with control
rats.
11β-HSD1 is a key enzyme that catalyzes GC synthesis
and promotes the conversion of GCs to active forms (34). It is a bidirectional catalytic
enzyme with redox capacity, which catalyzes the conversion between
the biologically inactive forms of cortisol (cortisone) and the
biologically active form (hydrocortisone) (35). 11β-HSD1 is distributed in the
liver, fat and kidney, as well as other organs, and serves various
physiological roles, such as the intracellular metabolism of GCs
and oxysterol metabolism, while its aberrant expression is
associated with insulin resistance, visceral obesity and
hypotension (36). In the present
study, a hepatic fibrosis rat model was established by
intraperitoneal injection of porcine serum. Alterations in 11β-HSD1
expression and biochemical parameters associated with the
development of hepatic fibrosis were systematically evaluated. The
results demonstrated that, in addition to the progression of liver
fibrosis, 11β-HSD1 expression levels increased in the liver, which
may be attributed to enhanced 11β-HSD1 activity and increased
levels of markers of liver fibrosis, including Col IV, HA, PIIINP,
α-SMA and CTGF. The use of BVT.233, an inhibitor of 11β-HSD1,
markedly reduced levels of the biochemical markers of liver
fibrosis and the severity of hepatic fibrosis. Subsequently, the
results suggested that 11β-HSD1 overexpression promoted cell cycle
progression, enhanced LX2-HSC proliferation, increased α-SMA, CTGF
and collagen secretion, and induced TGF-β1 in LX2-HSCs. By
contrast, 11β-HSD1 knockdown displayed the opposite effects,
including diminishing TGF-β1-mediated stimulation of LX2-HSCs. The
results suggested that 11β-HSD1 may participate in the development
and progression of hepatic fibrosis by activating HSCs and
regulating the expression of profibrotic markers.
Several studies on hepatic 11β-HSD1 have generated
conflicting results. For example, Konopelska et al (37) reported that there was no
association between hepatic 11β-HSD1 expression and the pathology
of fatty liver or non-alcoholic steatohepatitis (NASH) in humans,
whereas Ahmed et al (38)
suggested that in the early stages of nonalcoholic fatty liver
disease (NAFLD), with steatosis alone, hepatic 11β-HSD1 activity
decreases with the progression to NASH, which is associated with
increased 11β-HSD1 levels. Notably, a more recent study conducted
by Zou et al (39)
demonstrated that specific deletion or inhibition of 11β-HSD1
enhances the activation of HSCs in a murine model of liver fibrosis
induced by carbon tetrachloride (CCl4), which
contradicts the results of the present study that used a rat model
of liver fibrosis induced by persistent immune injury. In the face
of this disagreement, independent experiments were carefully
performed, and the findings were confirmed in the present study.
There is a possibility that the measured outcomes varied due to the
differences in animal models of liver fibrosis induced by chronic
and acute liver injury. Chemical liver injury induced by
CCl4 is the usual fibrosis model of acute liver injury;
however, in the present study, porcine serum was used to induce
liver fibrosis. Chronic viral hepatitis, especially chronic
hepatitis B, is the major cause of liver fibrosis in China, and
liver fibrosis caused by viral hepatitis is a process of chronic
immune injury rather than a simple, acute chemical injury (40). Therefore, heterogeneous serum
samples were selected to induce hepatic fibrosis in rats in the
present study. Notably, Chen et al (41) reported that gossypol, an inhibitor
of 11β-HSD1, ameliorates liver fibrosis in a rat model of type 2
diabetes-related liver fibrosis, which was consistent with the
observations of the present study, suggesting the antifibrotic
effects of the 11β-HSD1 specific siRNA and inhibitor in rats. In
addition, another study demonstrated that gossypol had an
antifibrotic effect on lung fibrosis (42). Chen et al (41) demonstrated that the number of
activated HSCs is significantly reduced following treatment with
the 11β-HSD1 inhibitor. Indeed, the present study demonstrated that
a decrease in 11β-HSD1 expression significantly inhibited LX2 cell
proliferation, with cell cycle arrested in the
G0/G1 phase. In support of the role of
11β-HSD1 in cell cycle, 11β-HSD1 overexpression significantly
promoted LX2 cell proliferation with increased numbers of cells in
the S and G2/M phases. Given the key events in liver
fibrosis, including HSC activation and proliferation, the present
study hypothesized that there may be a mechanism that partially
explains why the inhibition of 11β-HSD1 led to an antifibrotic
effect. The results of the present study and the aforementioned
studies have provided scientific evidence that inhibiting 11β-HSD1
holds therapeutic potential for chronic inflammatory liver
fibrosis.
As 11β-HSD1 blocks hydroxycortisone, the active form
that can activate HSCs, it is worth determining whether the
hydroxycortisone could rescue 11β-HSD1 inhibition. Collectively,
the study of 11β-HSD1 in liver fibrosis requires further
investigation to improve the understanding of the role of 11β-HSD1
and its underlying mechanisms in liver fibrosis.
In conclusion, the results suggested that
upregulation of 11β-HSD1 in the liver may be associated with the
progression and development of liver fibrogenesis, and its
mechanism may be related to HSC activation and proliferation, as
well as increased CTGF and α-SMA production.
Acknowledgements
Not applicable.
Funding
The present study was financially supported by the
National Natural Science Foundation of China (grant no.
81771827).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WX and ZGL designed the study. WX, MHL, PFR, HYZ, JG
and YQP performed the experiments. WX collected the data. WX, MHL,
HYG and ZGL analyzed the data. WX, PFR, HYZ, JG and YQP interpreted
the data. WX and ZGL drafted the manuscript, which was revised for
content by MHL and HYG. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Animal Care and Use Committee [Xiang license no. SCXK (Xiang)
2011-0003]. All procedures involving animals were in accordance
with the ethical standards of the institution or practice at which
the studies were conducted.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Eijken M, Hewison M, Cooper MS, de Jong
FH, Chiba H, Stewart PM, Uitterlinden AG, Pols HA and van Leeuwen
JP: 11beta-Hydroxysteroid dehydrogenase expression and
glucocorticoid synthesis are directed by a molecular switch during
osteoblast differentiation. Mol Endocrinol. 19:3191–631. 2005.
View Article : Google Scholar
|
|
2
|
Gyllenhammer LE, Alderete TL, Mahurka S,
Allayee H and Goran MI: Adipose tissue 11βHSD1 gene expression,
βcell function and ectopic fat in obese African Americans versus
Hispanics. Obesity (Silver Spring). 22:14–18. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tomlinson JW, Walker EA, Bujalska IJ,
Draper N, Lavery GG, Cooper MS, Hewison M and Stewart PM:
11beta-hydroxysteroid dehydrogenase type 1: A tissue-specific
regulator of glucocorticoid response. Endocr Rev. 25:831–866. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wake DJ and Walker BR: 11
beta-hydroxysteroid dehydrogenase type 1 in obesity and the
metabolic syndrome. Mol Cell Endocrinol. 215:45–54. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chapman K, Holmes M and Seckl J:
11β-hydroxysteroid dehydrogenases: Intracellular gate-keepers of
tissue glucocorticoid action. Physiol Rev. 93:1139–1206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Necela BM and Cidlowski JA: Mechanisms of
glucocorticoid receptor action in noninflammatory and inflammatory
cells. Proc Am Thorac Soc. 1:239–246. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rhen T and Cidlowski JA: Antiinflammatory
action of glucocorticoids - new mechanisms for old drugs. N Engl J
Med. 353:1711–1723. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang TY and Daynes RA: Macrophages from
11beta-hydroxysteroid dehydrogenase type 1-deficient mice exhibit
an increased sensitivity to lipopolysaccharide stimulation due to
TGF-beta-mediated up-regulation of SHIP1 expression. J Immunol.
179:6325–6335. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gilmour JS, Coutinho AE, Cailhier JF, Man
TY, Clay M, Thomas G, Harris HJ, Mullins JJ, Seckl JR, Savill JS,
et al: Local amplification of glucocorticoids by 11
beta-hydroxysteroid dehydrogenase type 1 promotes macrophage
phagocytosis of apoptotic leukocytes. J Immunol. 176:7605–7611.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ezhilarasan D, Sokal E and Najimi M:
Hepatic fibrosis: It is time to go with hepatic stellate
cell-specific therapeutic targets. Hepatobiliary Pancreat Dis Int.
17:192–197. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Koyama Y, Xu J, Liu X and Brenner DA: New
Developments on the Treatment of Liver Fibrosis. Dig Dis.
34:589–596. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schnabl B, Kweon YO, Frederick JP, Wang
XF, Rippe RA and Brenner DA: The role of Smad3 in mediating mouse
hepatic stellate cell activation. Hepatology. 34:89–100. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pellicoro A, Ramachandran P, Iredale JP
and Fallowfield JA: Liver fibrosis and repair: Immune regulation of
wound healing in a solid organ. Nat Rev Immunol. 14:181–194. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fabris L, Strazzabosco M, Crosby HA,
Ballardini G, Hubscher SG, Kelly DA, Neuberger JM, Strain AJ and
Joplin R: Characterization and isolation of ductular cells
coexpressing neural cell adhesion molecule and Bcl-2 from primary
cholangiopathies and ductal plate malformations. Am J Pathol.
156:1599–1612. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang YH, Chen YX, Zhang LJ, Chen ZX and
Wang XZ: Hydrodynamics-based transfection of rat interleukin-10
gene attenuates porcine serum-induced liver fibrosis in rats by
inhibiting the activation of hepatic stellate cells. Int J Mol Med.
34:677–686. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pereira RM, dos Santos RA, da Costa Dias
FL, Teixeira MM and Simões e Silva AC: Renin-angiotensin system in
the pathogenesis of liver fibrosis. World J Gastroenterol.
15:2579–2586. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lam BP, Jeffers T, Younoszai Z, Fazel Y
and Younossi ZM: The changing landscape of hepatitis C virus
therapy: Focus on interferon-free treatment. Therap Adv
Gastroenterol. 8:298–312. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Friedman SL: Liver fibrosis - from bench
to bedside. J Hepatol. 38 (Suppl 1):S38–S53. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Inagaki Y and Okazaki I: Emerging insights
into Transforming growth factor beta Smad signal in hepatic
fibrogenesis. Gut. 56:284–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kluwe J, Mencin A and Schwabe RF:
Toll-like receptors, wound healing, and carcinogenesis. J Mol Med
(Berl). 87:125–138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Görbig MN, Ginès P, Bataller R, Nicolás
JM, Garcia-Ramallo E, Tobías E, Titos E, Rey MJ, Clària J, Arroyo
V, et al: Atrial natriuretic peptide antagonizes endothelin-induced
calcium increase and cell contraction in cultured human hepatic
stellate cells. Hepatology. 30:501–509. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Grieb G, Steffens G, Pallua N, Bernhagen J
and Bucala R: Circulating fibrocytes - biology and mechanisms in
wound healing and scar formation. Int Rev Cell Mol Biol. 291:1–19.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schlernitzauer A, Oiry C, Hamad R, Galas
S, Cortade F, Chabi B, Casas F, Pessemesse L, Fouret G,
Feillet-Coudray C, et al: Chicoric acid is an antioxidant molecule
that stimulates AMP kinase pathway in L6 myotubes and extends
lifespan in Caenorhabditis elegans. PLoS One. 8:e787882013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Di Sario A, Bendia E, Svegliati Baroni G,
Ridolfi F, Casini A, Ceni E, Saccomanno S, Marzioni M, Trozzi L,
Sterpetti P, et al: Effect of pirfenidone on rat hepatic stellate
cell proliferation and collagen production. J Hepatol. 37:584–591.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Iredale JP: A cut above the rest? MMP-8
and liver fibrosis gene therapy. Gastroenterology. 126:1199–1201.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cremer MA, Rosloniec EF and Kang AH: The
cartilage collagens: A review of their structure, organization, and
role in the pathogenesis of experimental arthritis in animals and
in human rheumatic disease. J Mol Med (Berl). 76:275–288. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bhunchet E, Eishi Y and Wake K:
Contribution of immune response to the hepatic fibrosis induced by
porcine serum. Hepatology. 23:811–817. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim JH, Lee S, Lee MY and Shin HK:
Therapeutic effect of Soshiho-tang, a traditional herbal formula,
on liver fibrosis or cirrhosis in animal models: A systematic
review and meta-analysis. J Ethnopharmacol. 154:1–16. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Andrade RG, Gotardo BM, Assis BC, Mengel J
and Andrade ZA: Immunological tolerance to pig-serum partially
inhibits the formation of septal fibrosis of the liver in
Capillaria hepatica-infected rats. Mem Inst Oswaldo Cruz.
99:703–707. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cai DY, Zhao G, Chen JC, Ye GM, Bing FH
and Fan BW: Therapeutic effect of Zijin capsule in liver fibrosis
in rats. World J Gastroenterol. 4:260–263. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Goodman ZD: Grading and staging systems
for inflammation and fibrosis in chronic liver diseases. J Hepatol.
47:598–607. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee JH, Jang EJ, Seo HL, Ku SK, Lee JR,
Shin SS, Park SD, Kim SC and Kim YW: Sauchinone attenuates liver
fibrosis and hepatic stellate cell activation through TGF-β/Smad
signaling pathway. Chem Biol Interact. 224:58–67. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang Y, Wang H, Lv X, Wang Q, Zhao H, Yang
F, Yang Y and Li J: Involvement of cAMP-PKA pathway in adenosine A1
and A2A receptor-mediated regulation of acetaldehyde-induced
activation of HSCs. Biochimie. 115:59–70. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Legeza B, Marcolongo P, Gamberucci A,
Varga V, Bánhegyi G, Benedetti A and Odermatt A: Fructose,
Glucocorticoids and Adipose Tissue: Implications for the Metabolic
Syndrome. Nutrients. 9:4262017. View Article : Google Scholar
|
|
35
|
Morton NM, Holmes MC, Fiévet C, Staels B,
Tailleux A, Mullins JJ and Seckl JR: Improved lipid and lipoprotein
profile, hepatic insulin sensitivity, and glucose tolerance in
11beta-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem.
276:41293–41300. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Seckl JR, Morton NM, Chapman KE and Walker
BR: Glucocorticoids and 11beta-hydroxysteroid dehydrogenase in
adipose tissue. Recent Prog Horm Res. 59:359–393. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Konopelska S, Kienitz T, Hughes B, Pirlich
M, Bauditz J, Lochs H, Strasburger CJ, Stewart PM and Quinkler M:
Hepatic 11beta-HSD1 mRNA expression in fatty liver and nonalcoholic
steatohepatitis. Clin Endocrinol (Oxf). 70:554–560. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ahmed A, Rabbitt E, Brady T, Brown C,
Guest P, Bujalska IJ, Doig C, Newsome PN, Hubscher S, Elias E, et
al: A switch in hepatic cortisol metabolism across the spectrum of
non alcoholic fatty liver disease. PLoS One. 7:e295312012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zou X, Ramachandran P, Kendall TJ,
Pellicoro A, Dora E, Aucott RL, Manwani K, Man TY, Chapman KE,
Henderson NC, et al: 11Beta-hydroxysteroid dehydrogenase-1
deficiency or inhibition enhances hepatic myofibroblast activation
in murine liver fibrosis. Hepatology. 67:2167–2181. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang R, Xu Y, Dai Z, Lin X and Wang H: The
Immunologic Role of Gut Microbiota in Patients with Chronic HBV
Infection. J Immunol Res. 2018:23619632018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen G, Wang R, Chen H, Wu L, Ge RS and
Wang Y: Gossypol ameliorates liver fibrosis in diabetic rats
induced by high-fat diet and streptozocin. Life Sci. 149:58–64.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kottmann RM, Trawick E, Judge JL, Wahl LA,
Epa AP, Owens KM, Thatcher TH, Phipps RP and Sime PJ: Pharmacologic
inhibition of lactate production prevents myofibroblast
differentiation. Am J Physiol Lung Cell Mol Physiol.
309:L1305–L1312. 2015. View Article : Google Scholar : PubMed/NCBI
|