Introduction
Cerebral ischemia-reperfusion (I/R) is a devastating
cerebrovascular disease, accounting for significant morbidity and
mortality rates worldwide, with an estimated 41.6% death rate every
year (1). The prevention and
treatment of I/R injuries remains an important challenge in
clinical practice (1). Various
pathophysiological processes have been discovered to be tightly
involved with cerebral I/R injury, with excessive inflammation and
neuronal apoptosis among the most critical responses (2,3).
However, the specific mechanisms triggered by I/R injury remain
unclear, although numerous mechanisms are known to contribute to
neuronal death.
There is increasing evidence to suggest that
endoplasmic reticulum (ER) stress is tightly involved with the
pathophysiological processes of numerous types of central nervous
system disease, including Alzheimer's disease, Parkinson's disease,
amyotrophic lateral sclerosis and Huntington's disease (4,5).
Moreover, ER stress has been noted to serve an essential role in
cerebral I/R injury (6). The ER is
the primary site for the synthesis, processing and transportation
of functional proteins, and has a vital role in the maintenance of
cellular homeostasis (7). ER
function has been identified to be inhibited by several factors,
including unfolded protein accumulation and an imbalance of
Ca2+ homeostasis, both of which contribute to ER stress
(7). The accumulation of misfolded
proteins within the ER initiates the unfolded protein response
(UPR) (8), which is regulated by
several signaling pathways, including activating transcription
factor 6 (ATF6) and protein kinase RNA-like endoplasmic reticulum
kinase (PERK), contributing to apoptotic activity and neuronal
apoptosis (9).
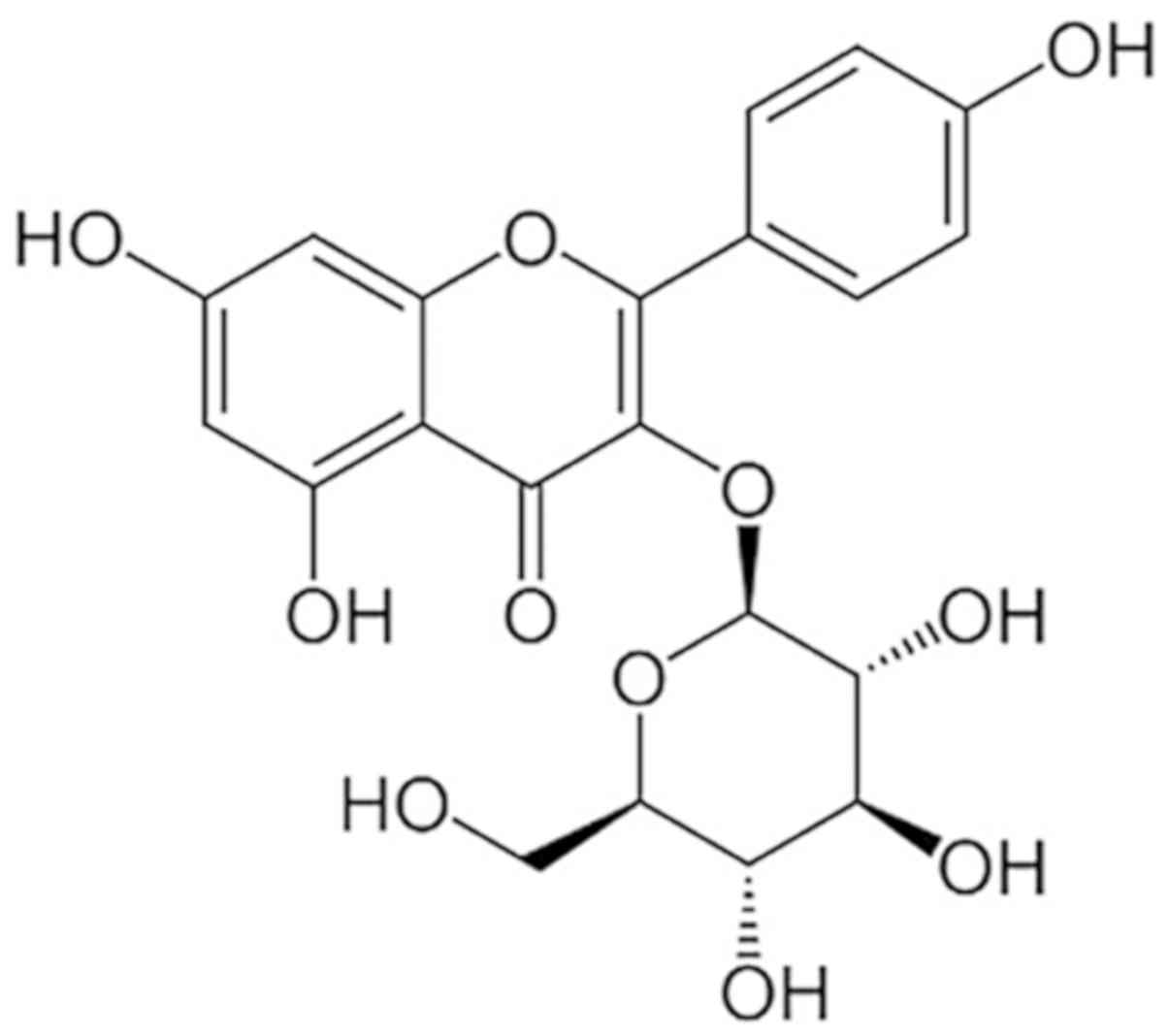
Astragalin (AST), also known as
kaempferol-3-O-glucoside, is a flavonoid compound that is isolated
from various traditional herbs, such as leaves of persimmon or Rosa
agrestis (10). AST has been
discovered to exert inhibitory effects on apoptosis and
inflammation in the treatment of various types of disease; for
example, AST was reported to improve antioxidant activity and
inhibit the inflammatory responses during spermatogenesis in a
streptozotocin-induced diabetes model (11), while another study revealed that
AST regulated the activity of ER stress in varicocelized rats
(12). However, to the best of our
knowledge, it remains unclear whether AST serves a role in ischemic
stroke. The present study aimed to determine the effect of AST and
its potential mechanism of action in cerebral I/R injury in
vivo and in vitro. The effects of AST on cerebral I/R
injury were hypothesized to be mediated through the regulation of
inflammation and apoptosis.
Materials and methods
Animal model
Male Sprague-Dawley rats (300–320 g; 8-weeks-old;
n=30) were purchased from the Animal Center of the Chinese Academy
of Sciences (Shanghai, China) Animals were housed at 23±2°C and
40–70% humidity with a 12-h light/dark cycle and ad libitum
access to food and water. Rats (n=10) were assigned into three
groups: i) Sham; ii) I/R; and iii) I/R + AST. The cerebral I/R
injury model was developed according to the intraluminal occlusion
method previously described (13).
Briefly, animals were anesthetized using 50 mg/kg pentobarbital. A
nylon thread (0.3 mm) was then embedded into the external carotid
artery and extended to the middle cerebral artery. The thread was
removed to induce I/R injury after 2 h of ischemia. In the sham
group, the thread was extended to the middle cerebral artery and
immediately removed. Animals in the I/R + AST group received 50
mg/kg/day of AST orally, beginning immediately post operation
(14). Rats in the other groups
received equal volumes of saline. The animals were sacrificed 3
days after brain I/R injury for further research (western blotting
and PCR) (n=5 in each group). All animal research was approved by
the Animal Care and Use Committee of Hainan Medical University
(Haikou, China).
Neurological score
Neurological outcomes were measured using a foot
fault test, as previously described (15). The animals were trained for 3 days
pre-operation; an initial neurological outcome was then recorded
for use as a baseline at day 0. The neurological scores (15) (left forelimb score + foot fault
score) of each group (Sham, I/R, I/R + AST; n=15) were subsequently
recorded at 1, 3, 5, 7, 14 and 28 days post operation.
Chemicals and reagents
AST (purity >98%) was acquired from Chengdu Must
Bio-Technology Co., Ltd. (Fig. 1).
Thapsigargin (TG), a commonly used inducer of ER stress (6), was acquired from Sigma-Aldrich; Merck
KGaA. Primary antibodies against CHOP (cat. no. 2895),
glucose-regulated protein 78 kDa (GRP78; cat. no. 3183), Bax (cat.
no. 2772), Bcl-2 (cat. no. 3498), cleaved (c)-caspase-3 (cat. no.
9661) and GADPH, and a caspase-3 activity kit and anti-mouse (cat.
no. 7076) and anti-rabbit (cat. no. 7074) horseradish peroxide
(HRP)-conjugated secondary antibodies (1:10,000) were acquired from
Cell Signaling Technology, Inc. An in situ cell death
detection kit (cat. no. PH0534) was acquired from Roche
Diagnostics. Other reagents were acquired from Sigma-Aldrich; Merck
KGaA, unless otherwise specified.
Cell culture, drug treatment and Cell
Counting Kit-8 (CCK-8) assay
The human-derived neuroblastoma SH-SY5Y cells were
acquired from the American Type Culture Collection. SH-SY5Y cells
were passaged every 3–5 days in DMEM (Gibco; Thermo Fisher
Scientific, Inc.) with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) in a cell incubator (5% CO2,
37°C).
To determine the effects of AST against TG-induced
apoptotic activity, cells were seeded into a 96-well plate
(8×103 cells/well). Cells were subsequently incubated
with a range of AST concentrations (0–200 µM) with or without TG
(10 µM) for 8 h at 37°C. Following the incubation, 10 µl CCK-8 dye
was added into every well for 1 h at 37°C, and the optical density
(450 nm) was detected on a microplate spectrophotometer according
to the manufacturer's protocol.
Western blotting
SH-SY5Y cells were subsequently incubated with AST
concentrations (50 µM) with or without TG (10 µM) for 24 h at 37°C.
Ischemic cortex and neuronal cell culture samples were homogenized
in RIPA lysis buffer (Sigma-Aldrich; Merck KGaA) supplemented with
PMSF (1:100) and quantified using a BCA protein assay kit to assess
protein concentration. Proteins (50 µg) were resolved on an 8 and
10% SDS-PAGE gel and transferred onto a nitrocellulose membrane.
Samples were then incubated with 5% skimmed milk for 90 min at room
temperature. Subsequently, the membranes were incubated with the
following primary antibodies at 4°C overnight: Anti-Bax (1:1,000),
anti-Bcl-2 (1:1,000), anti-c-caspase-3 (1:1,000), anti-GRP78
(1:1,000), anti-CHOP (1:1,000) and anti-GAPDH (1:5,000). Following
the primary antibody incubation, the membranes were incubated with
a HRP-conjugated secondary antibody (1:10,000) for 60 min at room
temperature. Finally, protein bands were detected using western
blotting detection reagents (MAC Gene Technology Ltd.) by an
imaging system (Bio-Rad Laboratories, Inc.). Data was analyzed
using the Image Lab 3.0 software (Bio-Rad Laboratories, Inc.).
Apoptosis analysis
SH-SY5Y cells were planted onto cover slips in
6-well plates at a density of 5×105 cells/ml. Cells were
subsequently incubated with AST concentrations (50 µM) with or
without TG (10 µM) for 24 h at 37°C. Apoptotic activity was
detected by both a TUNEL kit (and caspase-3 activity kit. Briefly,
SH-SY5Y cells (6×104/ml) were incubated with precooled
4% paraformaldehyde for 1 h, followed by 3% (v/v)
H2O2 for 15 min and then 0.1% Triton X-100
for 8 min at room temperature. Specimens were analyzed using an
TUNEL kit solution at 37°C for 2 h and nuclei were visualized
following incubation with DAPI for 8 min at room temperature. TUNEL
positive cells were analyzed using a Nikon Eclipse Ti confocal
microscope (magnification, ×10; Nikon Corporation), and the
percentage of apoptotic cells was counted in three random
high-power fields of three different slides and analyzed using
GraphPad Prism (Version 8.0, GraphPad Software, Inc.). Caspase-3
activity was detected as a marker of apoptosis using a Caspase-3
Activity assay kit according the manufacturer's protocol.
Immunofluorescence assay
SH-SY5Y cells were planted onto cover slips in
6-well plates at a density of 5×105 cells/ml. Cells were
subsequently incubated with AST concentrations (50 µM) with or
without TG (10 µM) for 24 h at 37°C. Following drug treatment,
cells were incubated with 4% paraformaldehyde for 1 h, 0.5% Triton
X-100 for 8 min, then 5% BSA (Sigma-Aldrich; Merck KGaA) for 45
min. Samples were subsequently incubated with an anti-caspase 12
primary antibody (1:200; cat. no. 62484; Abcam) overnight at 4°C.
Following the primary antibody incubation, the sections were
incubated with Donkey Anti-Rabbit IgG-TRITC secondary antibody
(1:500; cat. no. 7980; Abcam) for 1 h at room temperature. Nuclei
were visualized by incubation with DAPI for 8 min at room
temperature. Samples were then visualized using a Nikon Eclipse Ti
microscope (magnification, ×40; Nikon Corporation).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the ischemic cortex of
rats using TRIzol® reagent (Sigma-Aldrich; Merck KGaA).
RNA was then quantified by spectrophotometry by calucluating the
optical density (OD)260 and OD280 ratio. Total RNA(1 µg) was
reverse transcribed into cDNA using a PrimeScript RT reagent kit
(Takara Bio, Inc.) as follows: 2 µl 5X PrimeScript buffer, 0.5 µl
PrimeScript RT Enzyme Mix, 2 µl total RNA, 5 µl RNase Free
distilled H2O. The reaction was carried out in water
bath at 37°C for 15 min, and then at 85°C for 15 sec. qPCR was
subsequently performed using a SYBR Premix Ex Taq mixture (Takara
Bio, Inc.). The expression levels of target genes in the ischemic
cortex were quantified using the 2−ΔΔCq method (16). PCR primers were as follows: IL-1β
forward, 5′-AATGACCTGTTCTTTGAGGCTGAC-3′ and reverse,
5′-CGAGATGCTGCTGTGAGATTTGAAG-3′; IL-6 forward
5′-AGGAACGAAAGTCAACTCCATCTG-3′ and reverse,
5′-GGCAGTGGCTGTCAACAACATC-3′; TNF-α forward,
5′-AGTCCGGGCAGGTCTACTTT-3′ and reverse, 5′-TTCAGCGTCTCGTGTGTTTC-3′;
IL-8 forward, 5′-CAGAGACTTGGGAGCCACTC-3′ and reverse
5′-GCTGAAATTATCCACCCTGATT-3′; cyclooxygenase (COX)-2 forward,
5′-TGAGTACCGCAAACGCTTCTC-3′ and reverse,
5′-TGGACGAGGTTTTTCCACCAG-3′; inducible nitric oxide synthase (iNOS)
forward, 5′-ATGGAACAGTATAAGGCAAACACC-3′ and reverse,
5′-GTTTCTGGTCGATGTCATGAGCAAAGG-3′; β-actin forward,
5′-GAGAGGGAAATCGTGCGT-3′ and reverse, 5′-GGAGGAAGAGGATGCGG-3′.
Statistical analysis
Each experiment was performed at ≥3 times and data
are represented as the mean ± SD. All statistical significances
between groups were calculated using a one-way ANOVA and the
Tukey's post hoc test using Prism (Version 8.0, GraphPad Software,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
AST improves long-term neurological
outcomes in rats with cerebral I/R injury
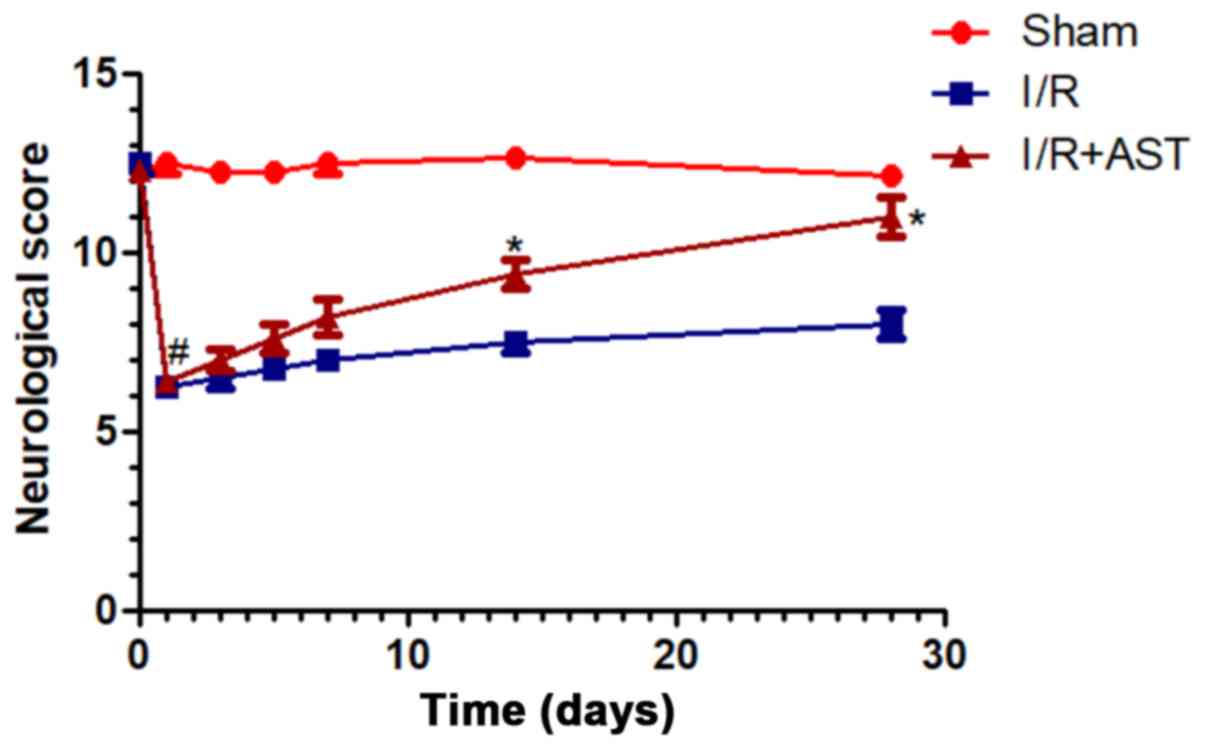
To investigate the effects of AST on cerebral I/R
injury, animals were examined following I/R injury with and without
AST treatment. Compared with sham group, neurological scores
markedly decreased in I/R model rats at 1-day post operation, and
then recovered slowly during one-month post operation (Fig. 2). Notably, AST administration
significantly improved neurological scores by day-14 and day-28
post operation compared with the I/R group, which indicated that
AST administration may improve the long-term neurological outcomes
in rats with cerebral I/R injury.
AST suppresses mRNA expression levels
of inflammatory cytokines induced by cerebral I/R injury in
vivo
The mRNA expression levels of the inflammatory
factors, IL-1β, IL-6, IL-8, TNF-α, COX-2 and iNOS, were analyzed by
RT-qPCR. Significant inflammatory-related changes were evident in
the rats with I/R injury; the expression levels of the inflammatory
factors were significantly upregulated in the I/R group compared
with the sham group (Fig. 3). In
contrast, the I/R + AST group demonstrated significantly
downregulated mRNA expression levels of inflammatory cytokines
compared with the I/R group, which indicated that AST may regulate
inflammatory gene responses in the cerebral I/R injury model.
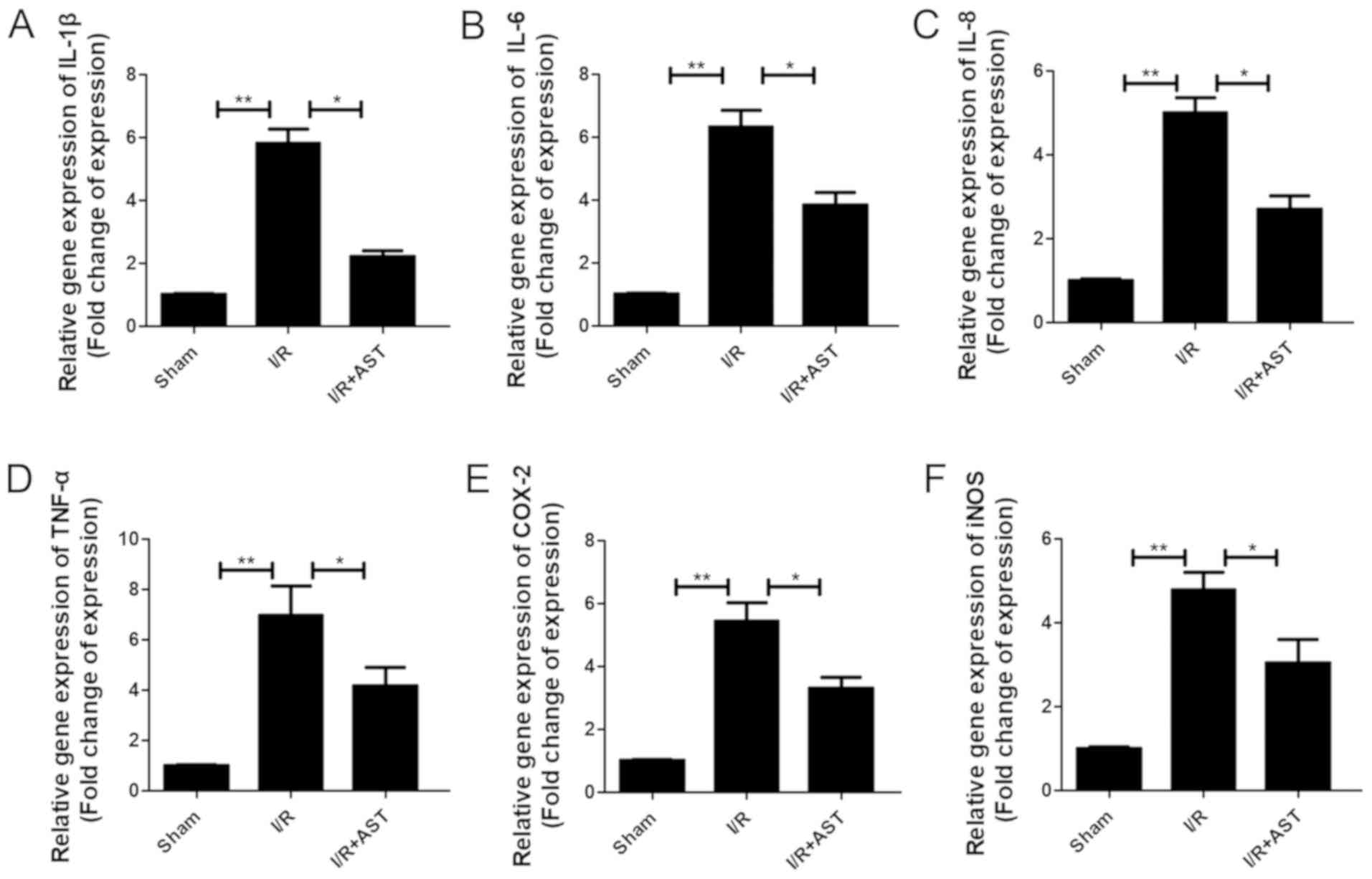 | Figure 3.AST suppresses the release of
inflammatory cytokines in cerebral I/R injury model rats. The
relative mRNA expression levels of (A) IL-1β, (B) IL-6, (C) IL-8,
(D) TNF-α, (E) COX-2 and (F) iNOS in cerebral I/R injury model rats
with or without AST treatment (50 µM) were analyzed using reverse
transcription-quantitative PCR. Data are presented as the mean ±
SD, n=5/group. *P<0.05, **P<0.01. AST, astragalin; COX-2,
cyclooxygenase-2; I/R, ischemia/reperfusion; iNOS, inducible nitric
oxide synthase. |
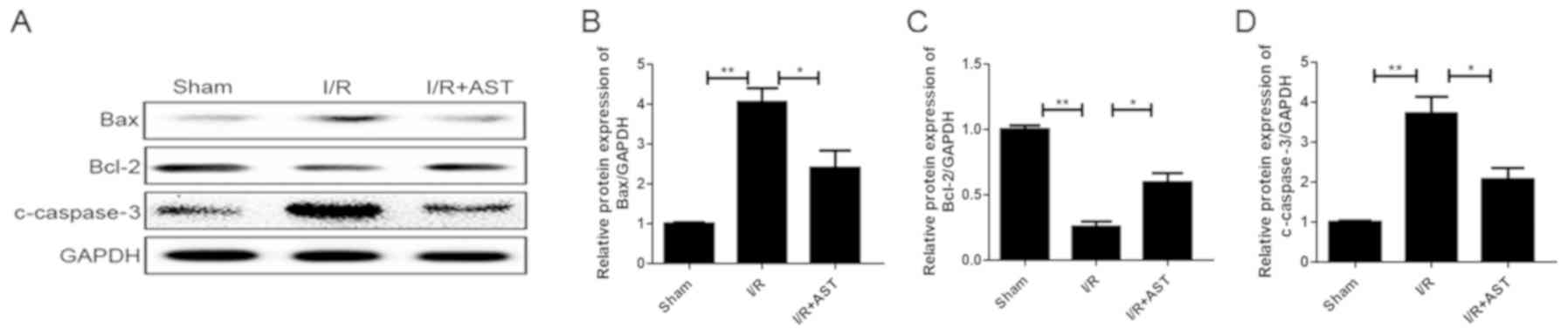
AST administration downregulates the
expression levels of apoptotic-related proteins in vivo
To determine whether AST could affect the protein
expression levels of apoptosis-related proteins following I/R
injury, the expression levels of apoptosis-related proteins
[apoptotic protein (Bax and c-caspase3) and anti-apoptotic protein
Bcl-2] were analyzed using western blotting analysis. The I/R group
exhibited significantly (~4 fold) upregulated Bax expression levels
compared with the sham group, which were subsequently significantly
downregulated in the I/R + AST treatment group (Fig. 4A and B). Furthermore, the I/R + AST
group exhibited a significant upregulation in the expression levels
of the anti-apoptotic protein Bcl-2 compared with the I/R model
group (Fig. 4A and C). Finally,
the expression levels of c-caspase-3 were significantly upregulated
in the I/R group compared with the sham group, but subsequently
significantly downregulated in the I/R + AST group compared with
the I/R group (Fig. 4A and D).
Together, these findings suggested that AST may downregulate the
apoptotic rate in a cerebral I/R injury model.
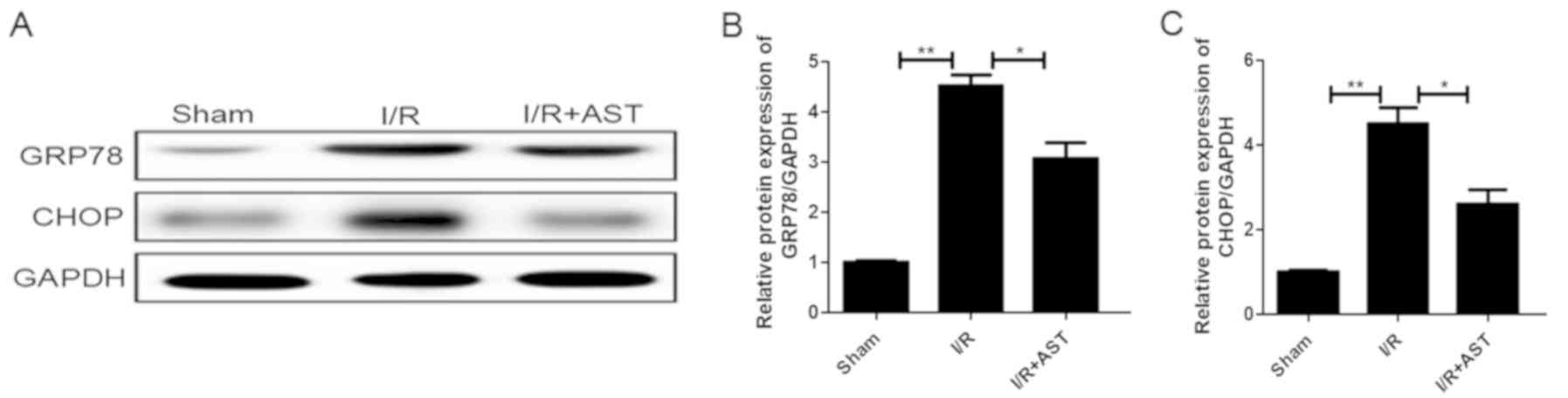
AST attenuates ER stress and related
apoptotic protein expression levels induced by cerebral I/R injury
in vivo
To determine whether the protective effects of AST
were tightly associated with its regulation of ER stress, the
expression levels of the ER stress-related proteins GRP78 and CHOP
in the rats following AST treatment were compared with the rats
that did not receive AST treatment. The I/R group exhibited a
significant upregulation of GRP78 (Fig. 5A and B) and CHOP (Fig. 5A and C) expression levels compared
with the sham group, which were subsequently significantly
downregulated following the administration of AST in both cases.
These findings indicated that ER stress may be associated with the
protective effects of AST in rats with cerebral I/R injury.
AST improves survival in TG-treated
SH-SY5Y cells
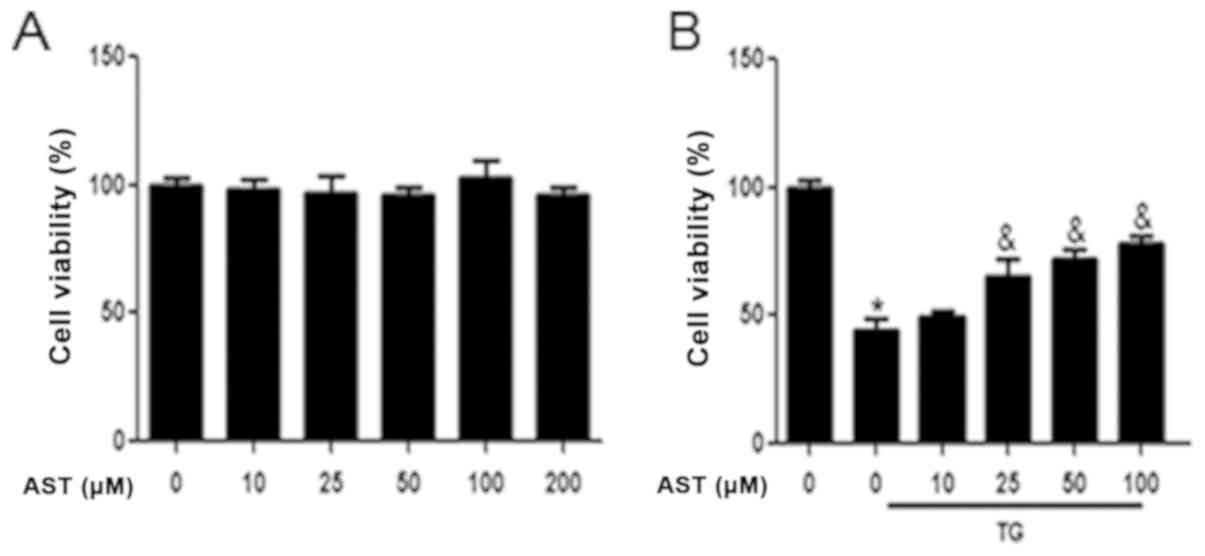
CCK-8 assays were used to analyze the cytoprotective
role of AST on ER stress-stimulated SH-SY5Y cells. Preliminary
analyses revealed that AST did not exhibit any significant cellular
toxicity at doses of up to 200 µM (Fig. 6A). Thus, to exclude the potential
toxic effects of AST on cells, 0–100 µM AST was used in all
subsequent experiments. To further investigate whether the
cytoprotective effects of AST were related to its regulation of ER
stress, SH-SY5Y cells were treated with TG, a commonly used inducer
of ER stress. The viability of TG-treated SH-SY5Y cells was
significantly decreased compared with the control group; this
viability was significantly rescued by the administration of AST
(25–100 µM; Fig. 6B). Together,
these data indicated that AST may exert a cytoprotective effect on
TG-treated SH-SY5Y cells.
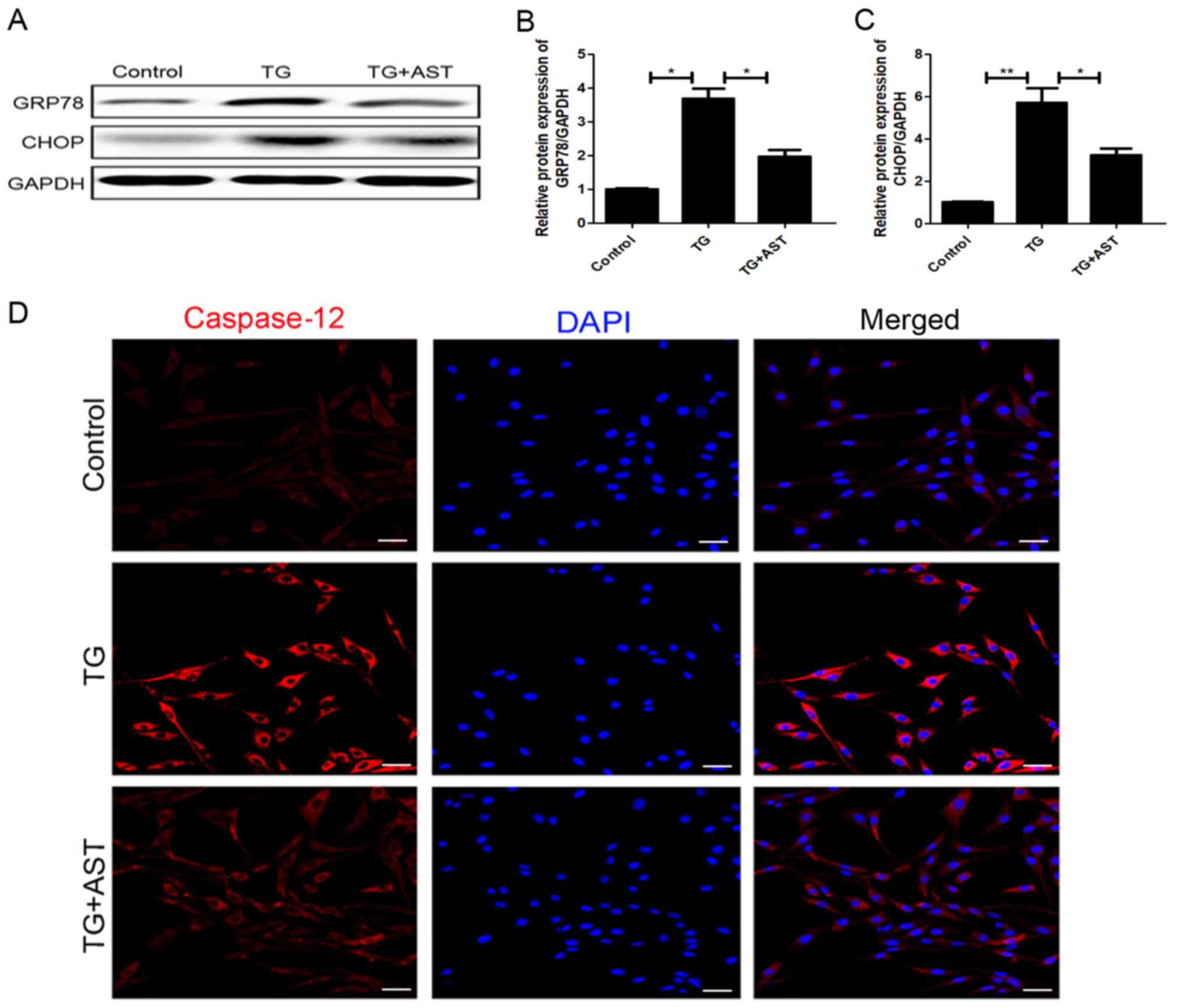
AST inhibits TG-induced ER stress and
downregulates apoptotic protein expression levels in SH-SY5Y
cells
Western blotting analysis discovered that the
TG-treated cells exhibited significantly upregulated expression
levels of GRP78 and the downstream apoptotic protein CHOP compared
with the control cells. Notably, the upregulation of both GRP78 and
CHOP expression levels was significantly reversed following the
administration of AST (Fig. 7A-C).
Immunofluorescence analysis of caspase-12 expression levels
suggested there was higher level of caspase12-positive puncta in
TG-induced apoptotic cells, which was reversed by the treatment of
AST (Fig. 7D). Together, these
data provided strong evidence to suggest that AST treatment may
attenuate ER stress.
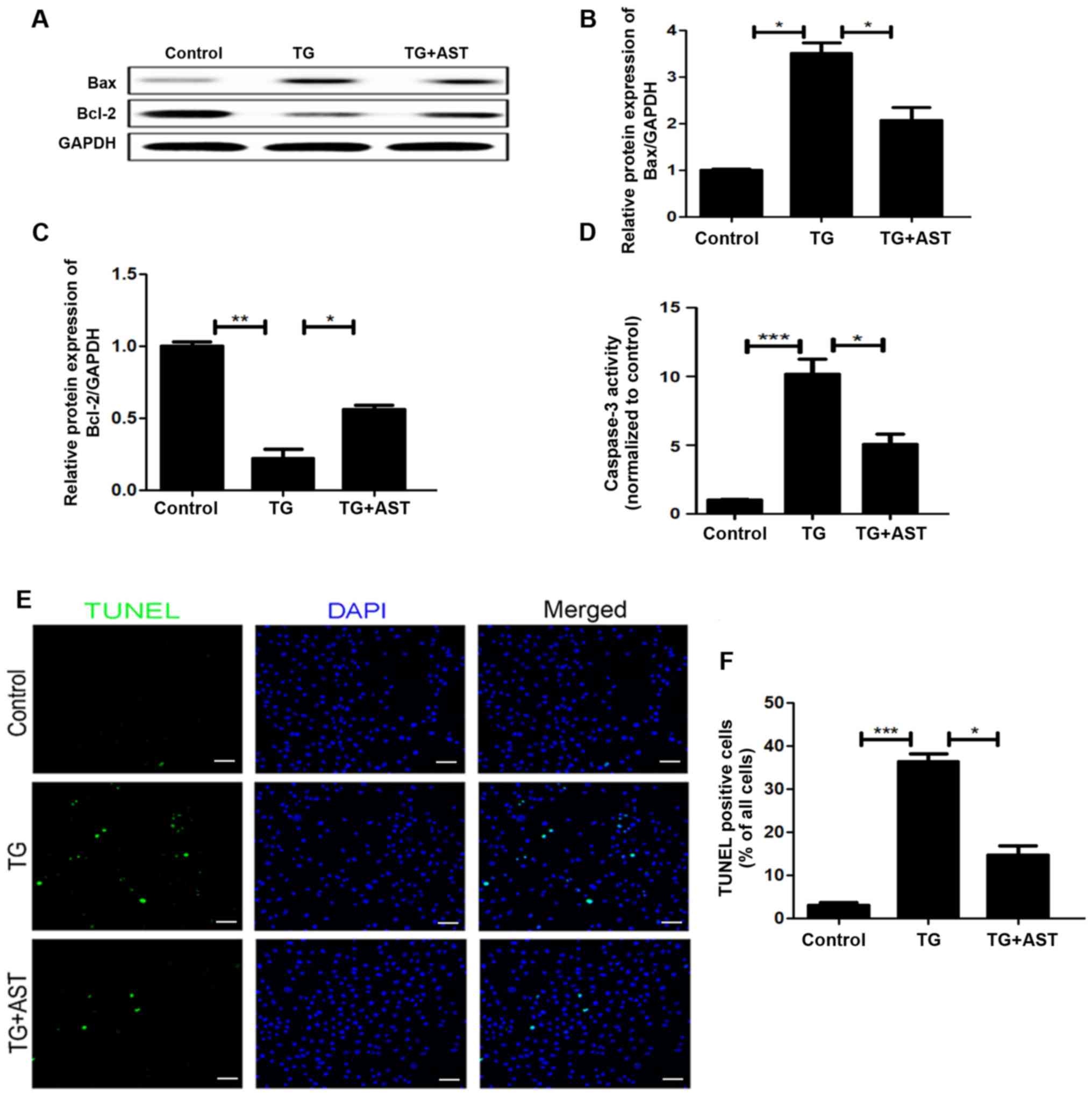
AST inhibits TG-induced apoptosis in
vitro
To further investigate the association between ER
stress and the anti-apoptotic effects of AST in the cerebral I/R
injury model, the expression levels of apoptotic proteins were
analyzed in SH-SY5Y cells. Western blotting analysis revealed that
AST administration partially reversed the significant upregulation
of Bax and downregulation of Bcl-2 expression levels observed in
TG-treated SH-SY5Y cells compared with the control cells (Fig. 8A-C). Analysis of caspase-3 activity
suggested there was higher apoptotic activity in TG-induced
apoptotic cells compared with that of the control group, which was
reversed by the treatment of AST (Fig.
8D). TUNEL staining and subsequent semi-quantification
demonstrated that AST treatment significantly reduced the numbers
of apoptotic cells compared with TG-treated SH-SY5Y cells, which
were significantly increased compared with the control cells
(Fig. 8E and F). Together, these
results suggested that the anti-apoptotic roles of AST may be
related to the regulation of ER stress.
Discussion
Cerebral I/R remains one of the most devastating
cerebrovascular events, which results in significant mortality and
long-term disability, with an estimated 41.6% death rate every year
(1), thereby exerting a heavy
social and economic burden. It has been hypothesized that the only
effective approach for the treatment of cerebral I/R injury is
restoration of blood perfusion within an effective time window
(17). Various studies have
reported that the restoration of blood flow initiated a complex
series of molecular events, including oxidative stress,
inflammation, mitochondrial dysfunction and ER dysfunction,
ultimately contributing to cerebral I/R injury (18,19).
Cerebral I/R injury may also occur in the perioperative period,
especially in elderly or critically ill patients (1). Thus, the development of an effective
strategy to both treat and prevent cerebral I/R injury has become
the primary focus of I/R research (2,3).
Numerous studies have demonstrated that the
neurological damage that occurs during cerebral I/R injury may be
enhanced under various complex pathophysiological conditions such
as inflammation and apoptosis, which subsequently regulates the
cell death-related signaling pathway (2,20).
Previous studies have revealed that excessive inflammation and
neuronal apoptosis were critical factors for cerebral I/R injury
(2,3,21).
In fact, several inflammatory mediators (TNF-α, IL-1β and iNOS)
were discovered to damage microvessels, further enhancing the
expression of inflammatory factors through the activation of
leukocytes and causing secondary neuronal damage (22,23).
It has been demonstrated that inflammation has an essential role in
cerebral I/R injury (24,25). In an analysis of 37 patients with
ischemic stroke, peak IL-6 plasma concentrations were identified to
be closely related to brain infarct volume and neurological
outcomes (26). Treatment with
recombinant IL-33 reduced the infarct area, attenuated microglia
activation and decreased proinflammatory macrophage infiltration in
a mouse model of middle cerebral artery occlusion (MCAO) (27). Infarct size and neurological
outcomes were also improved in Toll-like receptor 4-deficient mice
of cerebral I/R injury (28).
The enhancement of neuronal apoptotic activity was
discovered to be a main feature of cerebral I/R injury (29). The suppression of growth arrest
through RNA interference was demonstrated to markedly upregulate
apoptotic protein expression levels and enlarge the infarct size in
rats with MCAO (29). Previous
studies have also noted the roles of AST in the regulation of
inflammation and apoptosis in various types of disease; for
example, AST improved diabetes-induced spermatogenic dysfunction in
male mice through the regulation of antioxidant activities and
inflammation (11). Similar
effects were also observed in varicocelized Sprague-Dawley rats, in
which AST promoted spermatogenesis by regulating inflammation,
oxidative stress, ER stress and apoptosis (12). From a mechanistic standpoint, AST
has been illustrated to confer various cardioprotective effects
through a combination of anti-oxidative, anti-apoptotic and
anti-inflammatory activities (30). Among IL-1β-stimulated human
chondrocytes, these effects were mediated by the suppression of
various inflammatory factors through the regulation of NF-κB and
MAPK signaling (31). The present
study observed significant cell death following cerebral I/R
injury, which was significantly attenuated by treatment with AST.
AST also significantly downregulated the expression levels of the
pro-apoptotic proteins Bax and c-caspase-3, inhibited caspase-3
activity and upregulated the expression levels of the
anti-apoptotic protein Bcl-2 following I/R. Moreover, AST
suppressed the release of inflammatory cytokines, resulting in
improved long-term neurological outcomes of cerebral I/R injury
in vivo. Together, these results demonstrated the
neuroprotective effects of AST against cerebral I/R injury in
vivo and in vitro.
Numerous studies have proved that ER stress
contributed to apoptotic activity following cerebral I/R injury
(6,32–35).
These effects appeared to be partially mediated by the upregulation
of ER stress markers, including CHOP, eukaryotic initiation factor
2α and GRP78 (6,32). Previous work has demonstrated that
ER stress exerted a positive effect on neuronal apoptosis and the
detection of cellular damage in ischemic stroke (33,34).
GRP78 is an ER resident protein triggered by microenvironmental
damage that disturbs ER function (35). There is increasing evidence to
suggest that GRP78 may regulate the UPR in the ER, and that it is a
critical regulator of cellular apoptosis under stress conditions
(36). There is also accumulating
evidence to support the essential role of CHOP during apoptosis
which is triggered by ER stress (37). Under conditions of ER stress,
caspase-12 becomes dissociated from the membrane, leading to the
elevated production of c-caspase-12, which then activates various
apoptotic signaling mechanisms such as PERK/ATF6 (38–40).
The UPR activates the PERK/ATF6 signaling pathway and enhances
chaperone expression 9GRP78 and protein disulfide isomerase (PDI)],
which results in the elevated production of CHOP and caspase-12, as
well as increased apoptosis (39,40).
In the present study, the neuroprotective effects of AST were
analyzed along with its potential association with ER stress and
apoptotic signaling. The present findings demonstrated that the
administration of AST downregulated the expression levels of GRP78,
CHOP and caspase-12, indicating a potential mechanism for AST. To
further investigate the effect of ER stress on cerebral I/R injury,
TG, a well-established inducer of ER stress, was used in an in
vitro neuronal cell culture model. AST treatment significantly
reduced TG-stimulated ER stress related-protein (GRP78, CHOP and
caspase-12) in SH-SY5Y cells. Furthermore, AST treatment partially
reversed the upregulation in Bax and downregulation in Bcl-2
expression levels, contributing to the significant attenuation of
neuronal apoptosis. These data suggested that ER stress may be
tightly associated with the protective effects of AST.
However, some limitations should be noted in the
current study. AST has been demonstrated to have a
concentration-dependent protective effect on other types of
disease, such as spermatogenesis and osteoarthritis (11,30).
However, only one drug dose was used in the present animal study
and the mechanism was not clarified, thus other concentrations and
in-depth investigations into the mechanisms need to be further
studied.
In conclusion, the findings of the present study
indicated that the administration of AST may improve long-term
neurological outcomes in cerebral I/R injury model rats. These
protective effects were regulated through the inhibition of
neuronal apoptosis, the inflammatory response and ER stress
following injury. In vitro analyses further supported these
observations: AST significantly attenuated ER stress and apoptosis
in a neuronal cell culture model. Thus, these results suggested
that the regulation of ER stress by AST may be associated with
neuronal apoptosis and neurological recovery, highlighting the
potential use of AST as a therapeutic candidate in the treatment of
cerebral I/R injury.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Research and
Cultivation Fund Project of Hainan Medical University (grant no.
HYPY201925).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DL and WC designed the study and supervised the
experiments. DL and WW contributed to the statistical analysis,
data interpretation and manuscript preparation. DL and YG performed
the experiments and data interpretation. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal Care
and Use Committee of Hainan Medical University (approval no.
201819; Haikou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Moskowitz MA, Lo EH and Iadecola C: The
science of stroke: Mechanisms in search of treatments. Neuron.
67:181–198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Radak D, Katsiki N, Resanovic I, Jovanovic
A, Sudar-Milovanovic E, Zafirovic S, Mousad SA and Isenovic ER:
Apoptosis and acute brain ischemia in ischemic stroke. Curr Vasc
Pharmacol. 15:115–122. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dziedzic T: Systemic inflammation as a
therapeutic target in acute ischemic stroke. Expert Rev Neurother.
15:523–531. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xiang C, Wang Y, Zhang H and Han F: The
role of endoplasmic reticulum stress in neurodegenerative disease.
Apoptosis. 22:1–26. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sprenkle NT, Sims SG, Sanchez CL and
Meares GP: Endoplasmic reticulum stress and inflammation in the
central nervous system. Mol Neurodegener. 12:422017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nakka VP, Gusain A and Raghubir R:
Endoplasmic reticulum stress plays critical role in brain damage
after cerebral ischemia/reperfusion in rats. Neurotox Res.
17:189–202. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Secondo A, Petrozziello T, Tedeschi V,
Boscia F, Vinciguerra A, Ciccone R, Pannaccione A, Molinaro P,
Pignataro G and Annunziato L: ORAI1/STIM1 interaction intervenes in
stroke and in neuroprotection induced by ischemic preconditioning
through store-operated calcium entry. Stroke. 50:1240–1249. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hetz C: The unfolded protein response:
Controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee JY, Maeng S, Kang SR, Choi HY, Oh TH,
Ju BG and Yune TY: Valproic acid protects motor neuron death by
inhibiting oxidative stress and endoplasmic reticulum
stress-mediated cytochrome C release after spinal cord injury. J
Neurotrauma. 31:582–594. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim MS and Kim SH: Inhibitory effect of
astragalin on expression of lipopolysaccharide-induced inflammatory
mediators through NF-κB in macrophages. Arch Pharm Res.
34:2101–2107. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Han XX, Jiang YP, Liu N, Wu J, Yang JM, Li
YX, Sun M, Sun T, Zheng P and Yu JQ: Protective effects of
Astragalin on spermatogenesis in streptozotocin-induced diabetes in
male mice by improving antioxidant activity and inhibiting
inflammation. Biomed Pharmacother. 110:561–570. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Karna KK, Choi BR, You JH, Shin YS, Cui
WS, Lee SW, Kim JH, Kim CY, Kim HK and Park JK: The ameliorative
effect of monotropein, astragalin, and spiraeoside on oxidative
stress, endoplasmic reticulum stress, and mitochondrial signaling
pathway in varicocelized rats. BMC Complement Altern Med.
19:3332019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nan D, Jin H, Deng J, Yu W, Liu R, Sun W
and Huang Y: Cilostazol ameliorates ischemia/reperfusion-induced
tight junction disruption in brain endothelial cells by inhibiting
endoplasmic reticulum stress. FASEB J. 33:10152–10164. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Soromou LW, Chen N, Jiang L, Huo M, Wei M,
Chu X, Millimouno FM, Feng H, Sidime Y and Deng X: Astragalin
attenuates lipopolysaccharide-induced inflammatory responses by
down-regulating NF-κB signaling pathway. Biochem Biophys Res
Commun. 419:256–261. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Metz GA and Whishaw IQ: Cortical and
subcortical lesions impair skilled walking in the ladder rung
walking test: A new task to evaluate fore- and hindlimb stepping,
placing, and co-ordination. J Neurosci Methods. 115:169–179. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rabinstein AA: Treatment of acute ischemic
stroke. Continuum (Minneap Minn). 23:62–81. 2017.PubMed/NCBI
|
|
18
|
Bakthavachalam P and Shanmugam PST:
Mitochondrial dysfunction-silent killer in cerebral ischemia. J
Neurol Sci. 375:417–423. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma XD, Song JN, Zhang M, An JY, Zhao YL
and Zhang BF: Advances in research of the neuroprotective
mechanisms of cerebral ischemic postconditioning. Int J Neurosci.
125:161–169. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dong J, Yuan X and Xie W: Pentoxifylline
exerts anti-inflammatory effects on cerebral ischemia
reperfusioninduced injury in a rat model via the p38
mitogen-activated protein kinase signaling pathway. Mol Med Rep.
17:1141–1147. 2018.PubMed/NCBI
|
|
21
|
Cheng CY, Kao ST and Lee YC: Ferulic acid
ameliorates cerebral infarction by activating Akt/mTOR/4EBP1/Bcl2
antiapoptotic signaling in the penumbral cortex following permanent
cerebral ischemia in rats. Mol Med Rep. 19:792–804. 2019.PubMed/NCBI
|
|
22
|
Ji K, Xue L, Cheng J and Bai Y:
Preconditioning of H2S inhalation protects against cerebral
ischemia/reperfusion injury by induction of HSP70 through
PI3K/Akt/Nrf2 pathway. Brain Res Bull. 121:68–74. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moritz M, Pfeifer S, Balmayor ER,
Mittermayr R, Wolbank S, Redl H and van Griensven M: VEGF released
from a fibrin biomatrix increases VEGFR-2 expression and improves
early outcome after ischaemia-reperfusion injury. J Tissue Eng
Regen Med. 11:2153–2163. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jin R, Liu L, Zhang S, Nanda A and Li G:
Role of inflammation and its mediators in acute ischemic stroke. J
Cardiovasc Transl Res. 6:834–851. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He F, Zhang N, Lv Y, Sun W and Chen H:
Lowdose lipopolysaccharide inhibits neuronal apoptosis induced by
cerebral ischemia/reperfusion injury via the PI3K/Akt/FoxO1
signaling pathway in rats. Mol Med Rep. 19:1443–1452.
2019.PubMed/NCBI
|
|
26
|
Smith CJ, Emsley HC, Gavin CM, Georgiou
RF, Vail A, Barberan EM, del Zoppo GJ, Hallenbeck JM, Rothwell NJ,
Hopkins SJ and Tyrrell PJ: Peak plasma interleukin-6 and other
peripheral markers of inflammation in the first week of ischaemic
stroke correlate with brain infarct volume, stroke severity and
long-term outcome. BMC Neurol. 4:22004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang SR, Piepke M, Chu HX, Broughton BR,
Shim R, Wong CH, Lee S, Evans MA, Vinh A, Sakkal S, et al: IL-33
modulates inflammatory brain injury but exacerbates systemic
immunosuppression following ischemic stroke. JCI Insight.
3:e1215602018. View Article : Google Scholar
|
|
28
|
Caso JR, Pradillo JM, Hurtado O, Lorenzo
P, Moro MA and Lizasoain I: Toll-like receptor 4 is involved in
brain damage and inflammation after experimental stroke.
Circulation. 115:1599–1608. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu B, Zhang YH, Jiang Y, Li L, Chen Q, He
GQ, Tan XD and Li CQ: Gadd45b is a novel mediator of neuronal
apoptosis in ischemic stroke. Int J Biol Sci. 11:353–360. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qu D, Han J, Ren H, Yang W, Zhang X, Zheng
Q and Wang D: Cardioprotective effects of astragalin against
myocardial ischemia/reperfusion injury in isolated rat heart. Oxid
Med Cell Longev. 2016:81946902016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma Z, Piao T, Wang Y and Liu J: Astragalin
inhibits IL-1β-induced inflammatory mediators production in human
osteoarthritis chondrocyte by inhibiting NF-κB and MAPK activation.
Int Immunopharmacol. 25:83–87. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Noh MR, Kim JI, Han SJ, Lee TJ and Park
KM: C/EBP homologous protein (CHOP) gene deficiency attenuates
renal ischemia/reperfusion injury in mice. Biochim Biophys Acta.
1852:1895–1901. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qi W, Cao D, Li Y, Peng A, Wang Y, Gao K,
Tao C and Wu Y: Atorvastatin ameliorates early brain injury through
inhibition of apoptosis and ER stress in a rat model of
subarachnoid hemorrhage. Biosci Rep. 38:BSR201710352018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu Y, Zhou H, Xiong Y and Liu J: Exosomal
miR-199a-5p derived from endothelial cells attenuates apoptosis and
inflammation in neural cells by inhibiting endoplasmic reticulum
stress. Brain Res. 1726:1465152020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Suyama K, Watanabe M, Sakabe K, Okada Y,
Matsuyama D, Kuroiwa M and Mochida J: Overexpression of GRP78
protects glial cells from endoplasmic reticulum stress. Neurosci
Lett. 504:271–276. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang C, Jiang K, Gao D, Kang X, Sun C,
Zhang Q, Li Y, Sun L, Zhang S, Guo K and Liu Y: Clusterin protects
hepatocellular carcinoma cells from endoplasmic reticulum stress
induced apoptosis through GRP78. PLoS One. 8:e559812013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cai XH, Li XC, Jin SW, Liang DS, Wen ZW,
Cao HC, Mei HF, Wu Y, Lin ZD and Wang LX: Endoplasmic reticulum
stress plays critical role in brain damage after chronic
intermittent hypoxia in growing rats. Exp Neurol. 257:148–156.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Voccoli V, Mazzoni F, Garcia-Gil M and
Colombaioni L: Serum-withdrawal-dependent apoptosis of hippocampal
neuroblasts involves Ca++ release by endoplasmic
reticulum and caspase-12 activation. Brain Res. 1147:1–11. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li YH, Tardif G, Hum D, Kapoor M, Fahmi H,
Pelletier JP and Martel-Pelletier J: The unfolded protein response
genes in human osteoarthritic chondrocytes: PERK emerges as a
potential therapeutic target. Arthritis Res Ther. 18:1722016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|