Introduction
Congenital bilateral absence of the vas deferens
(CBAVD) accounts for 2–6% of male infertility cases and up to 25%
of cases of obstructive azoospermia (1,2). In
addition to CBAVD, the clinical symptoms and characteristics of
this condition generally include normal or slightly small
testicles, atrophy or absence of seminal vesicle and cauda
epididymis and normal serum follicle-stimulating hormone (3). The hallmarks of CBAVD include
azoospermia, reduced semen volume (<1 ml), pH value (≤7.0) and
seminal fructose, as well as decreased production of spermatozoa in
the testicles (4).
CBAVD may occur as an isolated manifestation or as
an atypical symptom of cystic fibrosis (CF), one of the most common
autosomal recessive genetic disorders in the Caucasian population
(5). CF has an incidence of
~1:2,500 and is usually caused by mutations in the CF transmembrane
conductance regulator (CFTR) gene (5). Since almost 97% of male patients with
CF are infertile due to CBAVD with resulting obstructive
azoospermia, it may be hypothesized that isolated CBAVD and CF have
a common genetic origin (1). The
anatomical defects associated with CBAVD arise at the embryonic
stage, and normal development of the male reproductive tract may
require CFTR or CFTR-mediated anion secretion (3,6). In
addition, 60–70% of patients with CBAVD carry pathogenic CFTR
mutations (Online Mendelian Inheritance in Man no. 602421), usually
one severe and one mild, in compound heterozygosity (7,8).
However, the exact role of CFTR in male reproductive physiology
remains to be fully elucidated. The present review summarized
recent findings linking CFTR gene mutations to CBAVD and
highlighted the assisted reproductive techniques (ART) and genetic
counseling provided clear diagnosis and treatment options for such
patients. Literature databases, such as Pubmed (pubmed.ncbi.nlm.nih.gov/) and Medline (de.medline.eu/), and Shanxi Medical University library
(library.sxmu.edu.cn/) were searched for
articles on CFTR and male fertility with relevant keywords such as
cystic fibrosis transmembrane conductance regulator gene (CFTR),
Congenital bilateral absence of the vas deferens (CBAVD),
variation, pre-implantation genetic diagnosis (PGD), and assisted
reproductive techniques (ART).
Overview of CFTR structure and function
The CFTR gene was first cloned and identified by
Riordan et al (9) in 1989.
In humans, it is located on chromosome 7q31.2 and spans a total
length of 230 kb comprising 27 exons; the composition of the gene
is outlined in Table I (10). CFTR encodes a protein containing
1,480 amino acid residues, with a molecular weight of 168,138 Da
(11). The CFTR protein is an
ATP-bound glycosylated transmembrane protein located in the apical
membrane of several types of exocrine epithelial cells, which
provides a selective channel for chloride ions across the
epithelium (12). The structure of
CFTR consists of five domains, including two membrane-spanning
domains (MSD-1 and −2) together forming a selective channel, two
nucleotide-binding domains (NBD-1 and −2) and a regulatory domain
(R domain) (Fig. 1). Each MSD is
composed of six transmembrane helices, of whichTM1-6 constitute
MSD-1 and TM7-12 constitute MSD-2, followed by a cytoplasmic NBD
(13). The MSD-2 and NBD-1 modules
are connected by the R domain, a regulatory region with disordered
structure that is unique to the CFTR gene and not found in other
transporters (14). The two MSD
domains form a selective chloride channel and the two NBDs regulate
the gating of this channel (15).
Furthermore, phosphorylation of the R domain controls channel
activity (15). The R domain
contains several phosphorylation sites for protein kinase A, a
cyclic AMP-dependent kinase that is highly conserved between
species (16).
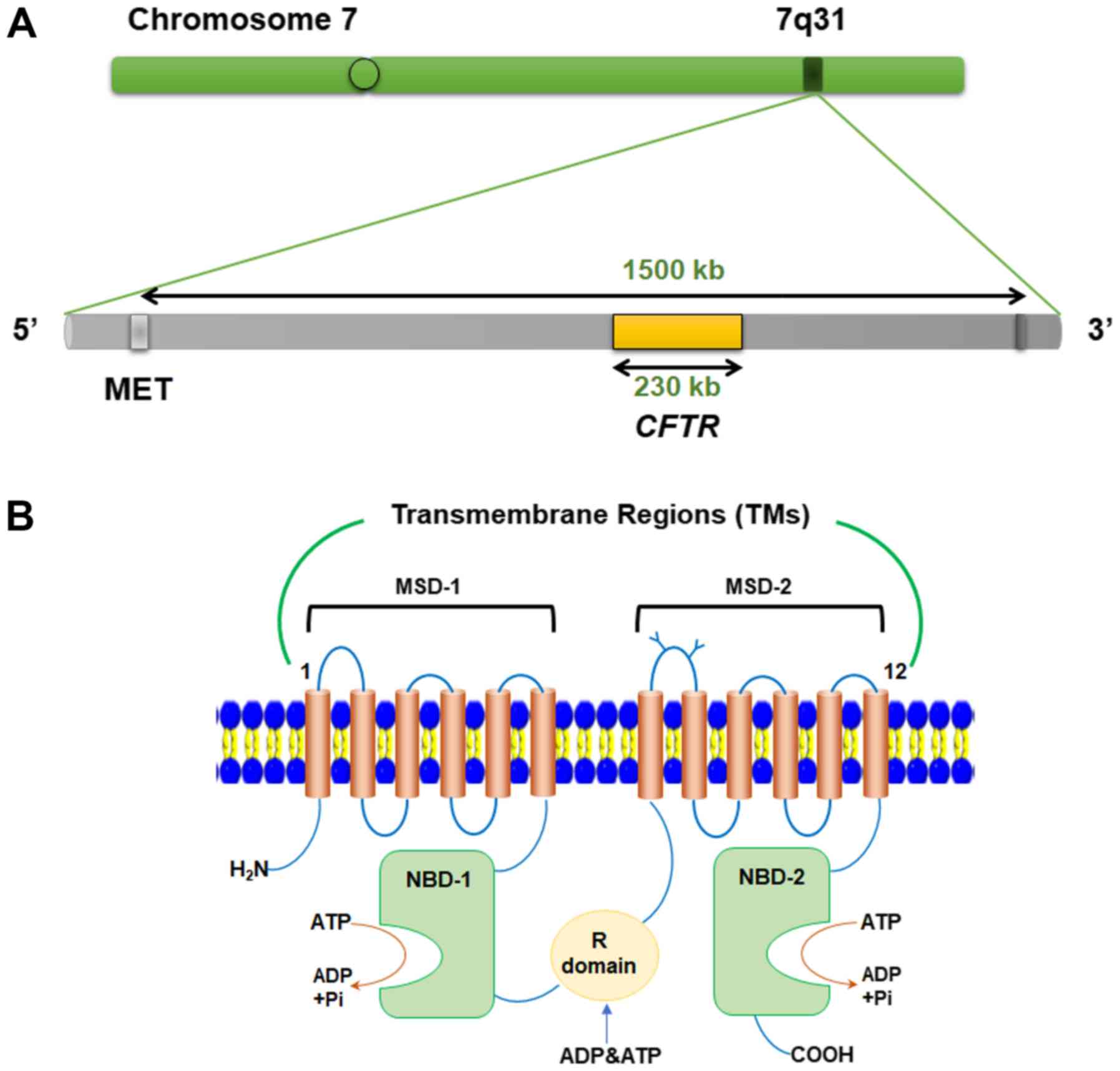 | Figure 1.Schematic representation of the CFTR
gene and protein. (A) Chromosomal localization of the CFTR gene.
(B) CFTR protein structure and functional domain composition. CFTR,
cystic fibrosis transmembrane conductance regulator; kb, kilobase;
Met, methionine; TM, transmembrane region; MSD, membrane-spanning
domain; NBD, nucleotide-binding domain; R domain, regulatory
domain; Pi, pyrophosphate; H2N, N-terminus; COOH,
C-terminus. |
 | Table I.Description of the structure of the
cystic fibrosis transmembrane conductance regulator gene. |
Table I.
Description of the structure of the
cystic fibrosis transmembrane conductance regulator gene.
| A, Exons |
|---|
|
|---|
| Name | Historical
name | Start, base
position | End, base
position | Length,
basepairs |
|---|
| Exon 1 | Exon 1 | 117,120,017 | 117,120,201 | 185 |
| Exon 2 | Exon 2 | 117,144,307 | 117,144,417 | 111 |
| Exon 3 | Exon 3 | 117,149,088 | 117,149,196 | 109 |
| Exon 3 | Exon 3 | 117,170,953 | 117,171,168 | 216 |
| Exon 5 | Exon 5 | 117,174,330 | 117,174,419 | 90 |
| Exon 6 | Exon 6a | 117,175,302 | 117,175,465 | 164 |
| Exon 7 | Exon 6b | 117,176,602 | 117,176,727 | 126 |
| Exon 8 | Exon 7 | 117,180,154 | 117,180,400 | 247 |
| Exon 9 | Exon 8 | 117,182,070 | 117,182,162 | 93 |
| Exon 10 | Exon 9 | 117,188,695 | 117,188,877 | 183 |
| Exon 11 | Exon 10 | 117,199,518 | 117,199,709 | 192 |
| Exon 12 | Exon 11 | 117,227,793 | 117,227,887 | 95 |
| Exon 13 | Exon 12 | 117,230,407 | 117,230,493 | 87 |
| Exon 14 | Exon 13 | 117,231,988 | 117,232,711 | 724 |
| Exon 15 | Exon 14a | 117,234,984 | 117,235,112 | 129 |
| Exon 16 | Exon 14b | 117,242,880 | 117,242,917 | 38 |
| Exon 17 | Exon 15 | 117,243,586 | 117,243,836 | 251 |
| Exon 18 | Exon 16 | 117,246,728 | 117,246,807 | 80 |
| Exon 19 | Exon 17a | 117,250,573 | 117,250,723 | 151 |
| Exon 20 | Exon 17b | 117,251,635 | 117,251,862 | 228 |
| Exon 21 | Exon 18 | 117,254,667 | 117,254,767 | 101 |
| Exon 22 | Exon 19 | 117,267,576 | 117,267,824 | 249 |
| Exon 23 | Exon 20 | 117,282,492 | 117,282,647 | 156 |
| Exon 24 | Exon 21 | 117,292,896 | 117,292,985 | 90 |
| Exon 25 | Exon 22 | 117,304,742 | 117,304,914 | 173 |
| Exon 26 | Exon 23 | 117,305,513 | 117,305,618 | 106 |
| Exon 27 | Exon 24 | 117,306,962 | 117,308,715 | 1,754 |
|
| B,
Introns |
|
| Name | Historical
name | Start | End | Length,
basepairs |
|
| Intron 1–2 | − | 117,120,202 | 117,144,306 | 24,105 |
| Intron 2–3 | – | 117,144,418 | 117,149,087 | 4,670 |
| Intron 3–4 | – | 117,149,197 | 117,170,952 | 21,756 |
| Intron 4–5 | – | 117,171,169 | 117,174,329 | 3,161 |
| Intron 5–6 | – | 117,174,420 | 117,175,301 | 882 |
| Intron 6–7 | – | 117,175,466 | 117,176,601 | 1,136 |
| Intron 7–8 | – | 117,176,728 | 117,180,153 | 3,426 |
| Intron 8–9 | – | 117,180,401 | 117,182,069 | 1,669 |
| Intron 9–10 | – | 117,182,163 | 117,188,694 | 6,532 |
| Intron 10–11 | – | 117,188,878 | 117,199,517 | 1,064 |
| Intron 11–12 | – | 117,199,710 | 117,227,792 | 28,083 |
| Intron 12–13 | – | 117,227,888 | 117,230,406 | 2,519 |
| Intron 13–14 | – | 117,230,494 | 117,231,987 | 1,494 |
| Intron 14–15 | – | 117,232,712 | 117,234,983 | 2,272 |
| Intron 15–16 | – | 117,235,113 | 117,242,879 | 7,767 |
| Intron 16–17 | – | 117,242,918 | 117,243,585 | 668 |
| Intron 17–18 | – | 117,243,837 | 117,246,727 | 2,891 |
| Intron 18–19 | – | 117,246,808 | 117,250,572 | 3,765 |
| Intron 19–20 | – | 117,250,724 | 117,251,634 | 911 |
| Intron 20–21 | – | 117,251,863 | 117,254,666 | 2,804 |
| Intron 21–22 | – | 117,254,768 | 117,267,575 | 12,808 |
| Intron 22–23 | – | 117,267,825 | 117,282,491 | 14,667 |
| Intron 23–24 | – | 117,282,648 | 117,292,895 | 10,248 |
| Intron 24–25 | – | 117,292,986 | 117,304,741 | 11,756 |
| Intron 25–26 | – | 117,304,915 | 117,305,512 | 598 |
| Intron 26–27 | – | 117,305,619 | 117,306,961 | 1,343 |
CFTR is expressed in epithelial tissues of various
organs, including the pancreas, intestine, sweat glands and vas
deferens, and participates in the secretion of substances and
fluids and maintains the balance of electrolyte and homeostasis in
the lumen (17). In the steady
state, the chloride channel is blocked by anions or negative
charges in the regulatory R domain. Phosphorylation of the R domain
is required for channel activity (18). As the concentration of
intracellular chloride ion increases, Protein Kinase A (PKA) is
activated, causing the channel to open (19). Similarly, the extracellular
chloride concentration also regulates channel gating, and increased
concentrations of extracellular chloride promote the opening of
channels (20). Thus, CFTR
facilitates two-way permeability of chloride ions (20). Previous studies have suggested
that, in addition to regulating chloride ion transport through
epithelial cells, CFTR is involved in the regulation of various ion
transporters, including epithelial sodium channels,
chloride/bicarbonate exchangers, sodium/proton exchangers and water
channels (20–22). Therefore, CFTR-dependent
physiological processes have a key role in maintaining homeostasis
of pH, ions and water levels in the fluids of secretory epithelium
(21).
The CFTR gene and development of the vas
deferens
The majority of male patients with CF and CBAVD have
primary infertility (22),
suggesting that the CFTR protein has an important role in the
development of the male reproductive tract (23). CFTR mutations among patients with
CBAVD (58.1%) are common but not as frequent as in patients with
unilateral absence of vas deferens (75%), and a normal amount of
functional CFTR protein may be required to ensure the normal
development of the vas deferens (24). Mutations in the CFTR gene impair
the normal function of chloride channels and prevent them from
regulating the flow of chloride ions and water across the cell
membrane, resulting in the production of highly viscous mucus by
epithelial cells of the male genital tract (25). This mucus, in turn, causes blockage
of the vas deferens during embryonic development, causing it to
denature or even deteriorate.
In 12–18 week-old abortive fetuses with CFTR gene
mutations, the vas deferens is obstructed and denatured by mucus,
and the vas deferens may be further aggravated in embryonic
development (26). Furthermore,
Gaillard et al (26)
demonstrated that the expression of the CFTR in epididymal
epithelium in human fetuses carrying CFTR mutations at 10–33 weeks
of gestation was low, resulting in the production of highly viscous
mucus by epithelial cells of the male genital tract. In addition,
CFTR mutations are absent in cases of CBAVD with renal
abnormalities, suggesting that the mesonephric duct has an
important role in the development of the vas deferens (26). It has also been demonstrated that
fluid secretion is necessary for the normal development of the
mesonephric duct (25). Abnormal
fluid secretion impairs normal development, leading to hypoplasia
of the mesonephric duct and degeneration during early embryonic
development (25,26). Therefore, CFTR may have an
important role in the development of the vas deferens and mutations
of CFTR may be associated with the occurrence of CBAVD.
The CFTR gene in patients with CBAVD
Numerous studies have investigated the genetic link
between CFTR mutations and the risk of CBAVD (27–30).
A large number of CFTR mutant alleles have been found in patients
with CBAVD. CFTR mutations display high heterogeneity in terms of
spectrum and frequency (27,28).
A total of 50,022 CFTR gene variants have been identified to date
(National Center for Biotechnology Information; ncbi.nlm.nih.gov/variation/view/?q=1080%5Bgeneid%5D&assm=GCF_000001405.25),
most minor allele frequency (MAF) of which are <0.01%. Only 605
variants are >1% and 347 variants are >5%. A total of 1.68%
of CFTR gene variants are considered to be potentially pathogenic,
while 1.16% are not known to cause functional changes and another
97.16% have not been characterized (27). In addition, 90.43% of the mutations
involve sequences that are only a few bases long. Of these,
missense mutations were the most common, accounting for 2.97% of
all mutations, while gene rearrangement was rare, accounting for
only 1.13% of mutations (27).
To date, >2,000 CF-causing CFTR mutations have
been identified (Table II;
genet.sickkids.on.ca/StatisticsPage.html). CFTR gene
mutations are divided into six classes, according to the extent of
CFTR protein downregulation (28–30).
Class I mutations refer to the production of non-functional protein
or non-functional mRNA products that are degraded by
nonsense-mediated mRNA decay (31). Class I mutations usually result
from splice site mutations, nonsense mutations, frameshift
mutations or large fragment insertion-deletion, the most common
ones being these three types (p.G542X, p.R533X and p.W1282X) of
missense mutations (31). Class II
mutations result in defects of protein maturity (32). A common example is the p.F508del
mutation, which leads to abnormal folding of the CFTR protein,
resulting in partial function and low CFTR protein concentration
(32). Class III mutations, such
as p.G551D, refer to defects in chloride channel gating function
despite normal protein structure, which are usually caused by
impaired binding of the NBD domain with ATP and hydrolysis
(33). Class IV mutations in the
CFTR gene, such as p.R117, p.HR334W and p.R347P, lead to abnormal
regulation of chloride channel function, usually due to abnormal
structure of MSDs (34). With
class V mutations, protein function is normal, but CFTR protein
synthesis is reduced (35). For
instance, the c.3717+12191 C>T mutation in intron 19 causes
leakage in transcription of exon 19, resulting in a decrease in
mRNA to only 5–10% of the normal mRNA levels (35).
| Table II.Mutations in the cystic fibrosis
transmembrane conductance regulator. |
Table II.
Mutations in the cystic fibrosis
transmembrane conductance regulator.
| Mutation type | Number of known
mutations | Frequency |
|---|
| Missense | 810 | 38.87 |
| Frameshift | 336 | 16.12 |
| Splicing | 229 | 10.99 |
| Nonsense | 175 | 8.40 |
| In-frame indel | 43 | 2.06 |
| Large indel | 59 | 2.83 |
| Promoter | 17 | 0.82 |
| Sequence
variation | 269 | 12.91 |
| Unknown | 146 | 7.01 |
A novel class of mutations, Class VI, has recently
been identified, which may be further divided into subclasses A and
B (36). Class VI-A mutations lead
to instability of CFTR protein, with a half-life reduced to only
about one-fifth of normal proteins. Examples of Class VI-A mutation
include p.Q1412X, c.4323delTC and c.4279insA, which result in the
loss of 70–100 bp of mRNA and abnormal folding of the C-terminal
end of the CFTR protein (36,37).
Class VI-B mutations, such as the p.G551D missense mutation,
usually affect the interaction between MSDs of the CFTR protein
(38).
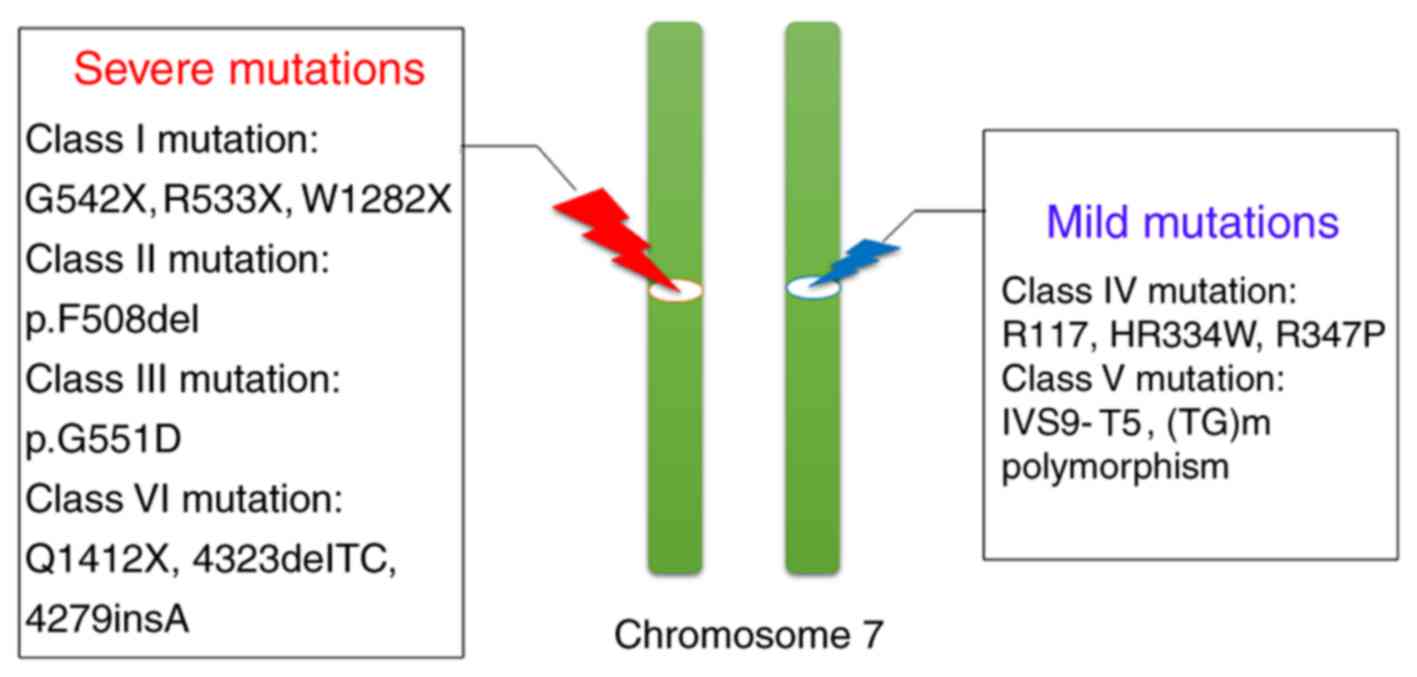
Class I, II and VI mutations are generally
considered severe, with near-complete loss of gene function. The
transcription level of the CFTR gene is <3%, which is easy to
cause CF and high clinical mortality (31,32,36).
By contrast, class IV and class V mutations are mild mutations with
slightly higher residual function, accounting for 3–10% of all CFTR
mutations, and lead to CF-associated diseases, such as p.R117H-T7,
lead to CBAVD (36).
A meta-analysis by Yu et al (8) suggested that 78% of patients with
CBAVD had at least one type of CFTR gene mutation, among which 48%
had two simultaneous mutations. Most of these mutations in patients
with CBAVD were point mutations (class IV or V) with a mild
phenotype, with few (<1%) gene deletions or rearrangements. In
addition, 15–25% of patients with CF displayed gene rearrangements
(Fig. 2). The mutation frequency
and genomic region of CFTR in which mutations occur vary by region
and ethnicity. For instance, the CFTR mutation rate in Caucasian
populations is significantly higher than in Asians. Furthermore, CF
is the most common lethal autosomal recessive genetic disease in
Caucasians (8,39). The most common mutant alleles in
Caucasians with CBAVD are p.F508del, c.IVS9-10 T5 and p.R117H, with
frequencies of 13–21, 22–29 and 2–4%, respectively (8,39).
A severe heterozygous mutation on one allele
combined with a heterozygous mutation on the other (88%), or two
mild heterozygous mutations (12%) usually lead to the occurrence of
CBAVD. However, a severe homozygous mutation (88%) or two severe
heterozygous mutations (12%) may lead to the occurrence of CF
(40). This suggests that CFTR
gene mutations are ethnically specific and their non-coding regions
may lead to mutations in CBAVD disease, while these types of
mutations do not lead to CF.
Hotspot mutation types in patients with
CBAVD
M470V
The p.M470V mutation is an amino acid substitution
caused by A to G polymorphism of the nucleotide at position 1,540
in exon 11 of the CFTR gene. It is a missense mutation and affects
the chloride channel activity of CFTR (41). Residue 470 codes for part of the
first NBD domain, and both M470 and V470 may lead to the synthesis
of a completely glycosylated CFTR protein. However, the chloride
channel activity of the M470 variant is twice as high as that of
V470 (41,42). Residue 470 also interacts with
other sites on the CFTR gene, thereby influencing the expression
levels of the CFTR protein (43).
The M470V mutation is frequently combined with the (TGm) and Tn
polymorphisms. The expression of the V470 CFTR variant decreases
with increasing numbers of TG repeats and shorter T repeats,
leading to an increased risk of CBAVD (43).
The ratio of M470 to V470 varies among different
populations. This ratio is normally ~1:1 in the healthy Caucasian
population. However, the ratio in the normal and infertile
population remains to be determined. Furthermore, in Chinese
patients with CBAVD, the ratio of M470 to V470 is 0.7:1, in line
with the Hardy-Weinberg equilibrium, and it is thought to be
unrelated to the pathogenesis of CBAVD (44).
p.F508del
p.F508del refers to the deletion of three bases
(CTT) at positions 1,653-1,655 in exon 11. The resulting deletion
of residue F508 of the CFTR protein leads to abnormal protein
folding and a significant reduction in CFTR protein levels on the
cell membrane of exocrine gland epithelial cells. Failure to form
effective chloride channels on the epithelium, in turn, results in
severe impairment of small molecule transport (45). p.F508del is the most common
mutation in Caucasian patients with CBAVD, with a mutation
frequency ranging from 35 to 74.6% (45). However, the incidence of this
mutation is low in Asians (46,47).
p.D1152H
p.D1152H mutation is an amino acid substitution
caused by G to C polymorphism at position 3,586 in exon 21 of the
CFTR gene. This mutation is a class IV mild mutation, resulting in
partial CFTR protein function and mild clinical manifestations of
CF disease. In males, the major clinical manifestation is CBAVD
(48). The incidence of the
p.D1152H mutation is 12% in Ashkenazi Jewish (49) and 6.27% in Hispanic and African
American individuals (50), which
included pre-marital and pre-natal screening in certain areas with
a high incidence of CF (48).
p.R117H
p.R117H results from a G to A substitution at
position 4,482 of exon 4; the resulting change from arginine to
histidine is considered a mild mutation. However, pR117H combined
with the intervening sequence (IVS) 9–10 T5 variant leads to severe
clinical manifestations of CF (51). In addition, p.R117H combined with
IVS9-10 T9 does not cause disease. Furthermore, when combined with
IVS9-10 T7, this mutation frequently leads to CBAVD and results in
CF when combined with IVS9-10 T5 (52). p.R117H has a high incidence (~3%)
in Caucasian patients with CF.
Mutations in the promoter region
Promoters regulate gene transcription levels by
interacting with trans-acting factors (17,53).
The CFTR gene promoter is a housekeeping-type, GC-rich (<65% GC
content) promoter lacking the typical TATA box, and its specific
cis-acting element location and regulatory mechanism are
poorly characterized (53).
Yoshimura et al (54)
suggested that the core sequence of the CFTR gene promoter may be
located ~3.8 kb upstream of the ATG transcription initiation site
and contains conserved sequences acting as binding sites for
several important transcription proteins, such as CTCC-binding
factor (CTCF), activator protein-1 (AP-1), specificity protein 1
(SP1), greater curvature (GRE), carbapenem-resistant (CRE),
CCAAT/enhancer-binding protein (C/EBP) and Y-box proteins (55). Giordano et al (56) examined the CFTR gene promoter in
patients with CF and identified 23 mutations. All of these were
point mutations that affected transcription, resulting in lack of
regulation, upregulation and downregulation of the CFTR gene
(56). Lopez et al
(67) demonstrated that the
forkhead box I1 (FOXI1) transcription factor was able to
downregulate CFTR gene transcription. Indeed, the c.-165G>A
promoter mutation enhanced FOXI1 binding, thereby inhibiting
transcription of the CFTR gene. Feng et al (53) recently demonstrated that the
c.-195C>A promoter mutation significantly enhanced the
inhibitory effect of specificity protein (SP) on CFTR
expression.
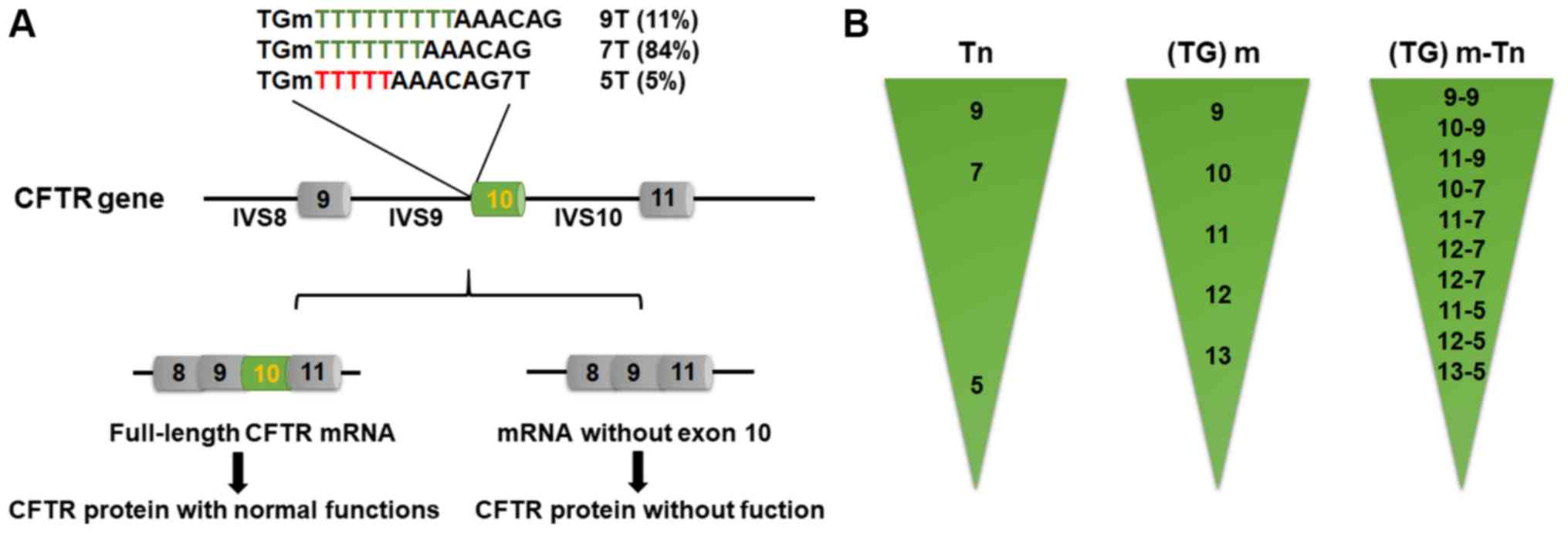
Intron 9–10 (TG)m and Tn
polymorphism
(TG)m and Tn polymorphism inIVS 9–10 of the CFTR
gene are associated with the occurrence of CBAVD. Tn refers to the
length of a noncoding thymidine sequence. The Tn sequence is
usually 7 or 9 bases long (T7 and T9, respectively) in the normal
population, while the incidence of 5T is 5.2% (57,58).
The shorter the Tn sequence, the lower the transcription level of
the CFTR gene.
In patients who are homozygous for the IVS9-10 5T
(c.1210-7_1210-6delTT variant, formerly known as IVS8 5T), a
sequence of 5 consecutive thymidines in intron 9 may cause
alternative splicing of exon 10 mRNA, resulting in missing
transcription of the CFTR gene. This, in turn, causes a reduction
in the synthesis of CFTR protein with normal functions (only 10% of
the normal protein); these patients with CBAVD do not exhibit other
symptoms of CF (Fig. 3A) (30). Exon 10 encodes 60 amino acids of
the CFTR protein NBD-1. Thus, a mutant CFTR protein lacking this
amino acid sequence loses chloride channel activity, thus reducing
the permeability of chloride to the epithelial cell membrane of the
exocrine gland and increasing the reabsorption of sodium ions in
the exocrine fluid, eventually leading to CF and associated
diseases including chronic obstructive pulmonary disease,
pancreatic exocrine dysfunction and meconium ileus (59).
Radpour et al (60) examined the distribution of Tn
alleles in 106 patients with CBAVD and demonstrated that the
incidence of T5 was 25.94%, although T5 distribution was not
investigated among males with normal fertility. However, the
incidence of Tn polymorphism in patients with CBAVD varies with
ethnicity, ranging from 13 (Asian) to 43.7% (Caucasian) (61). Disset et al (62) identified a rare IVS9 T3 mutation in
one case. Functional experiments demonstrated that this mutation
led to severe cystic fibrosis (62), consistent with the hypothesis that
leakage in exon 10 transcription becomes more serious with the
decrease in length of the Tn sequence (59). The T5 variant is currently
considered to be a mild CFTR gene mutation rather than a
polymorphism.
Furthermore, the (TG)m (c.1210-34) polymorphism also
affects the transcription of exon 10 (Fig. 3B). In association with the T7
variant, loss of exon 10 transcription caused by TG11 and TG12 is
2.8 and 6.0 times more likely than with TG10, respectively
(63). Thus, (TG)m and Tn
synergistically affect the transcription of exon 10. Indeed, the
greater the number of TG repeats, and the shorter the number of T
repeats and the higher the probability of transcription loss of
exon 10 is (63). IVS9-10 (TG)m Tn
is a common mutation type of the CFTR gene. Furthermore, the
frequency of this mutation in Chinese patients with CBAVD is
slightly higher than that in Caucasian patients (44).
Intron 10–11 (TAAA)n short-tandem
repeats
Short-tandem repeats are widely distributed in the
genome and the length of repeats is closely associated with the
occurrence of diseases, such as neuromuscular disorders (64) and gynecomastia (65). Previous studies have also
demonstrated that short-tandem repeats in promoters and introns may
affect gene transcription (64–66).
A large number of dimer and tetramer short-tandem repeats have been
identified in the noncoding region of the CFTR gene that affect its
expression. (TAAA)n is a short-tandem repeat composed of four bases
in IVS10-11 that may impair the transcription of exon 10. However,
it does not affect the transcription of exon 10 as Tn in intron
9–10 does. Lopez et al (67) suggested that FOXI1 transcription
factor binds to IVS10-11 (TAAA)n short-tandem repeats to negatively
regulate the expression of the CFTR gene. With decreasing numbers
of repeats, binding of FOXI1 transcription factor and promoter was
enhanced, thus reducing the transcriptional levels of the CFTR
gene. Furthermore, combination with the c.-165G>A mild promoter
mutation or IVS9-10 (TG)mTn may lead to the occurrence of CBAVD
(67).
Although the CFTR gene was identified >20 years
ago, the relationship between its genotype and clinical phenotype
remains controversial, mostly due to the wide variety of CFTR gene
mutations and differences across ethnicities. Furthermore, other
factors may also be involved, including environmental factors,
viruses or bacteria (27,50,61).
In addition, it has been observed that 6–15% of
patients with CBAVD did not carry any CFTR gene mutations (1), further highlighting possible limits
of detection during diagnosis or the possibility that other genetic
mutations are present in certain patients.
Assisted reproductive techniques and genetic
counselling
Compared with patients with CF, patients CBAVD are
more likely to have mild CFTR gene mutations (class IV and V)
associated with CFTR protein dysfunction. Most patients with CBAVD
have normal spermatogenic function, and their spouses may be
impregnated via testicular tissue sperm extraction (TESE) or
intracytoplasmic sperm injection (ICSI), a procedure, through which
a single sperm is injected into the cytoplasm of a mature oocyte to
obtain a viable embryo (68). The
first pregnancy for a couple in whom the male had CBAVD was
reported in 1987 (68). A recent
meta-analysis suggested that the results of ICSI were not affected
by the source of sperm (fresh, frozen, epididymal or testicular),
but a lower rate of fertilization and a higher rate of miscarriage
were observed in patients with CBAVD as compared with those with
acquired aspermia (69).
Due to possible inheritance of the CFTR mutation and
risk of CF occurrence, genetic counseling prior to the use of
assisted reproductive technology (ART) is particularly important,
especially for patients with a CBAVD familial history. To date,
CFTR gene mutation detection has been applied in pre-implantation
genetic diagnosis (PGD) in the USA and certain European countries
such as the Netherlands (70),
Russia (71) and France (72). CFTR mutation screening as part of a
PGD screening program may select embryos carrying a healthy CFTR
gene, which has certain clinical significance for couples affected
by CBAVD and their potential children. Liu et al (70) reported the first successful PGD in
a couple affected by CBAVD, in which both partners were
heterozygotes with p.F508del mutations. In this instance, three
carrier embryos were implanted and a healthy male infant was born.
The presence of CF or CBAVD-associated mutations in the male
partner does not currently appear to significantly affect in
vitro oocyte fertilization, embryo implantation rates or
successful delivery of asymptomatic offspring after PGD (73,74).
For a couple affected by CBAVD with CFTR deficiency
planning to have their own children, the risk for both male and
female offspring to have CF or associated diseases, and for male
offspring to have CBAVD depends on whether the female partner is a
carrier, as one mutant allele is always inherited from the male. If
the female does not carry the CFTR gene mutation, the probability
of CBAVD in the male offspring is low, estimated to be <1%. The
inheritance of CBAVD is more complex than that of CF, as genetic
analysis may include but not exclude the diagnosis of reproductive
CF. Furthermore, when rare mutations are detected in males or
females, the risk of CF or CBAVD in the offspring may be
unpredictable. The couple should be informed that tests cannot
detect all mutations in the gene; therefore, negative mutation
screening may reduce but not eliminate the risk of being a
carrier.
Clinical examination and follow-up of children born
to couples affected by CBAVD are of great significance, as it may
provide information regarding the variability in phenotypes
associated with CFTR mutations. Identifying a CFTR mutation in a
male affected by CBAVD is important not only for the patient
himself, but also for his family. Healthy siblings of a male child
born to a parent with CBAVD have a 50% chance of being carriers of
CF. Therefore, both the patient and their partner should be
screened for CFTR gene mutations. If no mutation is detected in the
male partner, it is not certain that the unborn child will be free
of CF, and the risk may be <1:1,000 (27). Among male infertile heterosexual
couples not affected by CF, the risk of CF in the offspring was no
different from that of couples in the general population. The risk
of infertility in their male offspring remains elusive. In
addition, for all couples with positive results, PGD and prenatal
diagnosis (PND) are recommended, together with genetic counseling
(Fig. 4).
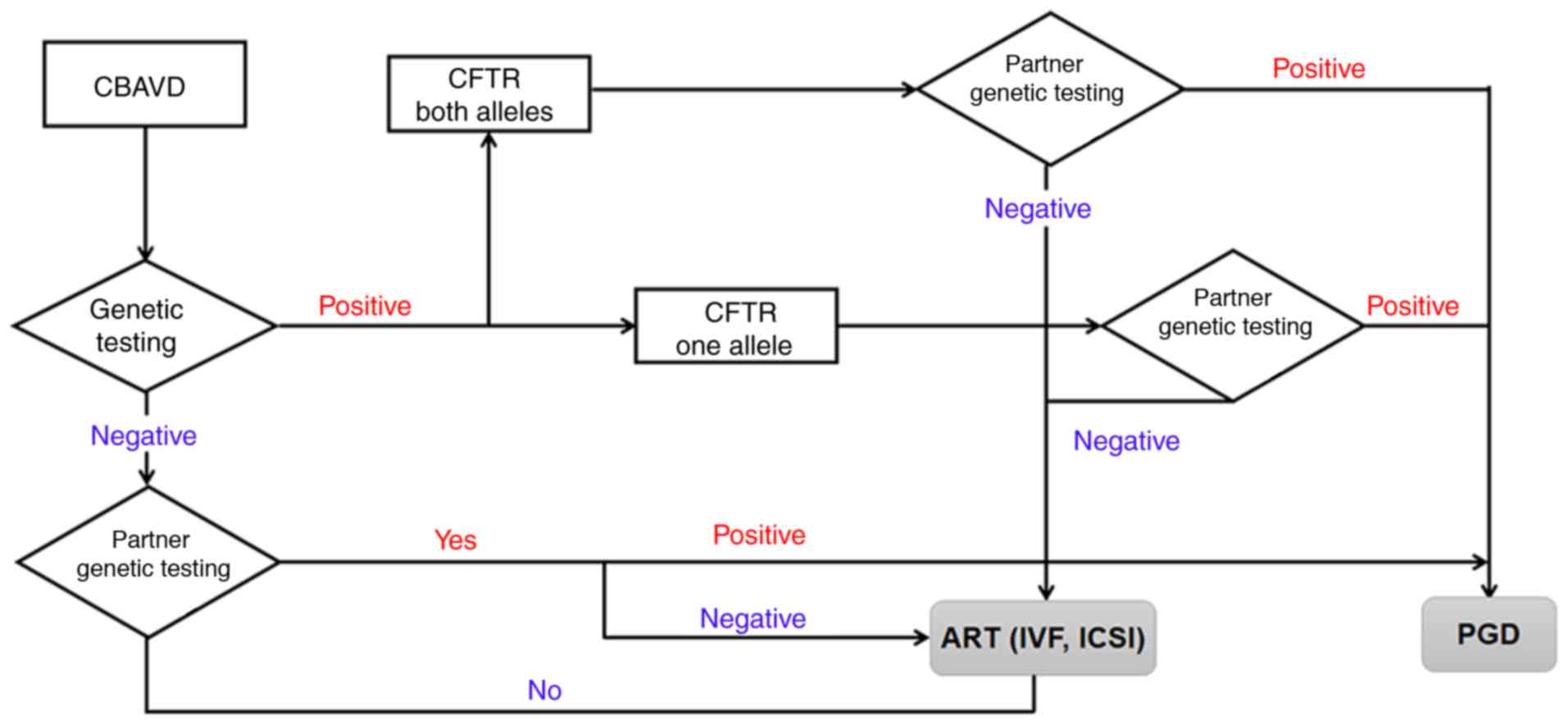 | Figure 4.Systematic genotyping strategy for
couples with CBAVD. Genetic counseling should therefore be provided
to CBAVD couples, and the male partner should be tested for CFTR
mutations in order to determine the risk of having a CBAVD child.
In addition, for all couples with positive results, PGD and PND are
recommended. CBAVD, congenital bilateral absence of the vas
deferens; CTFR, cystic fibrosis transmembrane conductance
regulator; ART, assisted reproductive techniques; IVF, in
vitro fertilization; ICSI, intracytoplasmic sperm injection;
PGD, pre-implantation genetic diagnosis; PND, pre-natal
diagnosis. |
Conclusions
CBAVD is an atypical manifestation of CF caused by
mutations in the CFTR gene, which frequently leads to male
infertility. The present review summarized recent findings linking
the CFTR gene to male fertility and highlighted the progress made
in understanding the role of CFTR in reproductive events pertinent
to male fertility. Genetic counseling for couples affected by CBAVD
remains challenging. Characterizing the mutation spectrum of the
CFTR gene and its association with the pathogenesis of CBAVD may
prove conducive to disease diagnosis and genetic risk assessment
prior to IVF, thereby reducing the incidence of this disease or the
rate of CFTR mutations in infants resulting from ART. Consistent
guidelines are also required concerning the extent of mutational
analysis for the CFTR gene during the diagnosis of CBAVD, as well
as options available for couples affected by CBAVD in association
with PND and PGD.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Scientific
Research Project of Shanxi Provincial Department of Health (grant
no. 201601070), the Initial Scientific Research Fund of PhD at
Shanxi Provincial People's Hospital (grant no. b201635), the
Natural Science Foundation of Shanxi (grant nos. 201901D211519 and
201901D211546),the Research Project Supported by Shanxi Scholarship
Council of China (grant no. HGKY2019092) and China Postdoctoral
Science Foundation (grant no. 2020M670703).
Availability of data and materials
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the present
study.
Authors' contributions
XC and XJ designed the study. XW and QL interpreted
the data. All authors wrote, read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Radpour R, Gourabi H, Dizaj AV, Holzgreve
W and Zhong XY: Genetic investigations of CFTR mutations in
congenital absence of vas deferens, uterus, and vagina as a cause
of infertility. J Androl. 29:506–513. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gajbhiye R, Kadam K, Khole A, Gaikwad A,
Kadam S, Shah R, Kumaraswamy R and Khole V: Cystic fibrosis
transmembrane conductance regulator (CFTR) gene abnormalities in
Indian males with congenital bilateral absence of vas deferens
& renal anomalies. Indian J Med Res. 143:616–623. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li CY, Jiang LY, Chen WY, Li K, Sheng HQ,
Ni Y, Lu JX, Xu WX, Zhang SY and Shi QX: CFTR is essential for
sperm fertilizing capacity and is correlated with sperm quality in
humans. Hum Reprod. 25:317–327. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ferlin A and Stuppia L: Diagnostics of
CFTR-negative patients with congenital bilateral absence of vas
deferens: Which mutations are of most interest? Expert Rev Mol
Diagn. 20:265–267. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gallego A, Rogel R, Perez-Ardavin J,
Lorenzo L, Lujan S, Oltra S, Molina I and Broseta E: Congenital
bilateral absence of the vas deferens (CBAVD): Do genetic disorders
modify assisted reproductive technologies outcomes? Arch Esp Urol.
72:1038–1042. 2019.(In Spanish). PubMed/NCBI
|
|
6
|
Diao R, Fok KL, Zhao L, Chen H, Tang H,
Chen J, Zheng A, Zhang X, Gui Y, Chan HC and Cai Z: Decreased
expression of cystic fibrosis transmembrane conductance regulator
impairs sperm quality in aged men. Reproduction. 146:637–645. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mak V, Zielenski J, Tsui LC, Durie P, Zini
A, Martin S, Longley TB and Jarvi KA: Proportion of cystic fibrosis
gene mutations not detected by routine testing in men with
obstructive azoospermia. JAMA. 281:2217–2224. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu J, Chen Z, Ni Y and Li Z: CFTR
mutations in men with congenital bilateral absence of the vas
deferens (CBAVD): A systemic review and meta-analysis. Hum Reprod.
27:25–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Riordan JR, Rommens JM, Kerem B, Alon N,
Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et
al: Identification of the cystic fibrosis gene: Cloning and
characterization of complementary DNA. Science. 245:1066–1073.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsui LC and Dorfman R: The cystic fibrosis
gene: A molecular genetic perspective. Cold Spring Harb Perspect
Med. 3:a0094722013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guillot L, Beucher J, Tabary O, Le Rouzic
P, Clement A and Corvol H: Lung disease modifier genes in cystic
fibrosis. Int J Biochem Cell Biol. 52:83–93. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Linsdell P: Cystic fibrosis transmembrane
conductance regulator chloride channel blockers: Pharmacological,
biophysical and physiological relevance. World J Biol Chem.
5:26–39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Poroca DR, Amer N, Li A, Hanrahan JW and
Chappe VM: Changes in the R-region interactions depend on
phosphorylation and contribute to PKA and PKC regulation of the
cystic fibrosis transmembrane conductance regulator chloride
channel. FASEB Bioadv. 2:33–48. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jarosz-Griffiths HH, Scambler T, Wong CH,
Lara-Reyna S, Holbrook J, Martinon F, Savic S, Whitaker P,
Etherington C, Spoletini G, et al: Different CFTR modulator
combinations downregulate inflammation differently in cystic
fibrosis. Elife. 9:e545562020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jaworska J, Marach-Mocarska A and Sands D:
Uncommon clinical presentation of cystic fibrosis in a patient
homozygous for a rare CFTR mutation: A case report. BMC Pediatr.
20:902020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen JH: Protein kinase A phosphorylation
potentiates cystic fibrosis transmembrane conductance regulator
gating by relieving autoinhibition on the stimulatory C terminus of
the regulatory domain. J Biol Chem. 295:4577–4590. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
NandyMazumdar M, Yin S, Paranjapye A,
Kerschner JL, Swahn H, Ge A, Leir SH and Harris A: Looping of
upstream cis-regulatory elements is required for CFTR expression in
human airway epithelial cells. Nucleic Acids Res. 48:3513–3524.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Laselva O, Stone TA, Bear CE and Deber CM:
Anti-infectives restore ORKAMBI((R)) rescue of
F508del-CFTR function in human bronchial epithelial cells infected
with clinical strains of P. aeruginosa. Biomolecules. 10:3342020.
View Article : Google Scholar
|
|
19
|
McCarron A, Cmielewski P, Reyne N,
McIntyre C, Finnie J, Craig F, Rout-Pitt N, Delhove J, Schjenken
JE, Chan HY, et al: Phenotypic characterization and comparison of
cystic fibrosis Rat models generated using CRISPR/Cas9 gene
editing. Am J Pathol. 190:977–993. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Froux L, Elbahnsi A, Boucherle B, Billet
A, Baatallah N, Hoffmann B, Alliot J, Zelli R, Zeinyeh W,
Haudecoeur R, et al: Targeting different binding sites in the CFTR
structures allows to synergistically potentiate channel activity.
Eur J Med Chem. 190:1121162020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Choi JY, Muallem D, Kiselyov K, Lee MG,
Thomas PJ and Muallem S: Aberrant CFTR-dependent HCO3- transport in
mutations associated with cystic fibrosis. Nature. 410:94–97. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Van Mourik P, van Haaren P, Kruisselbrink
E, Korkmaz C, Janssens HM, de Winter-de Groot KM, van der Ent CK,
Hagemeijer MC and Beekman JM: R117H-CFTR function and response to
VX-770 correlate with mRNA and protein expression in intestinal
organoids. J Cyst Fibros. 2020.(Epub ahead of print). View Article : Google Scholar
|
|
23
|
De Santi C, Fernandez Fernandez E, Gaul R,
Vencken S, Glasgow A, Oglesby IK, Hurley K, Hawkins F, Mitash N, Mu
F, et al: Precise targeting of miRNA sites restores CFTR activity
in CF bronchial epithelial cells. Mol Ther. 28:1190–1199. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jain R, Middleton PG and Rowe SM: Triple
therapy for cystic fibrosis with a phe508del CFTR mutation. Reply.
N Engl J Med. 382:6842020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Morris-Rosendahl DJ, Edwards M, McDonnell
MJ, John S, Alton EWFW, Davies JC and Simmonds NJ: Whole gene
sequencing of CFTR reveals a high prevalence of the intronic
variant c.3874-4522A>G in cystic fibrosis. Am J Respir Crit Care
Med. 201:1438–1441. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gaillard DA, Carre-Pigeon F and Lallemand
A: Normal vas deferens in fetuses with cystic fibrosis. J Urol.
158:1549–1552. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ferec C and Cutting GR: Assessing the
disease-liability of mutations in CFTR. Cold Spring Harb Perspect
Med. 2:a0094802012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tamburino L, Guglielmino A, Venti E and
Chamayou S: Molecular analysis of mutations and polymorphisms in
the CFTR gene in male infertility. Reprod Biomed Online. 17:27–35.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Patel B, Parets S, Akana M, Kellogg G,
Jansen M, Chang C, Cai Y, Fox R, Niknazar M, Shraga R, et al:
Comprehensive genetic testing for female and male infertility using
next-generation sequencing. J Assist Reprod Genet. 35:1489–1496.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cuppens H and Cassiman JJ: CFTR mutations
and polymorphisms in male infertility. Int J Androl. 27:251–256.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hinzpeter A, Aissat A, Sondo E, Costa C,
Arous N, Gameiro C, Martin N, Tarze A, Weiss L, de Becdelièvre A,
et al: Alternative splicing at a NAGNAG acceptor site as a novel
phenotype modifier. PLoS Genet. 6:e10011532010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kopito RR: Biosynthesis and degradation of
CFTR. Physiol Rev. 79 (1 Suppl):S167–S173. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schwiebert EM, Egan ME, Hwang TH, Fulmer
SB, Allen SS, Cutting GR and Guggino WB: CFTR regulates outwardly
rectifying chloride channels through an autocrine mechanism
involving ATP. Cell. 81:1063–1073. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Seibert FS, Linsdell P, Loo TW, Hanrahan
JW, Riordan JR and Clarke DM: Cytoplasmic loop three of cystic
fibrosis transmembrane conductance regulator contributes to
regulation of chloride channel activity. J Biol Chem.
271:27493–27499. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Highsmith WE, Burch LH, Zhou Z, Olsen JC,
Boat TE, Spock A, Gorvoy JD, Quittel L, Friedman KJ, Silverman LM,
et al: A novel mutation in the cystic fibrosis gene in patients
with pulmonary disease but normal sweat chloride concentrations. N
Engl J Med. 331:974–980. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Green DM, McDougal KE, Blackman SM, Sosnay
PR, Henderson LB, Naughton KM, Collaco JM and Cutting GR: Mutations
that permit residual CFTR function delay acquisition of multiple
respiratory pathogens in CF patients. Respir Res. 11:1402010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Haardt M, Benharouga M, Lechardeur D,
Kartner N and Lukacs GL: C-terminal truncations destabilize the
cystic fibrosis transmembrane conductance regulator without
impairing its biogenesis. A novel class of mutation. J Biol Chem.
274:21873–21877. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fulmer SB, Schwiebert EM, Morales MM,
Guggino WB and Cutting GR: Two cystic fibrosis transmembrane
conductance regulator mutations have different effects on both
pulmonary phenotype and regulation of outwardly rectified chloride
currents. Proc Natl Acad Sci USA. 92:6832–6836. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bombieri C, Claustres M, De Boeck K,
Derichs N, Dodge J, Girodon E, Sermet I, Schwarz M, Tzetis M,
Wilschanski M, et al: Recommendations for the classification of
diseases as CFTR-related disorders. J Cyst Fibros. 10 (Suppl
2):S86–S102. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Claustres M, Guittard C, Bozon D,
Chevalier F, Verlingue C, Ferec C, Girodon E, Cazeneuve C, Bienvenu
T, Lalau G, et al: Spectrum of CFTR mutations in cystic fibrosis
and in congenital absence of the vas deferens in France. Hum Mutat.
16:143–156. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cuppens H, Lin W, Jaspers M, Costes B,
Teng H, Vankeerberghen A, Jorissen M, Droogmans G, Reynaert I,
Goossens M, et al: Polyvariant mutant cystic fibrosis transmembrane
conductance regulator genes. The polymorphic (Tg)m locus explains
the partial penetrance of the T5 polymorphism as a disease
mutation. J Clin Invest. 101:487–496. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kosova G, Pickrell JK, Kelley JL, McArdle
PF, Shuldiner AR, Abney M and Ober C: The CFTR Met 470 allele is
associated with lower birth rates in fertile men from a population
isolate. PLoS Genet. 6:e10009742010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pompei F, Ciminelli BM, Bombieri C,
Ciccacci C, Koudova M, Giorgi S, Belpinati F, Begnini A, Cerny M,
Des Georges M, et al: Haplotype block structure study of the CFTR
gene. Most variants are associated with the M470 allele in several
European populations. Eur J Hum Genet. 14:85–93. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Du Q, Li Z, Pan Y, Liu X, Pan B and Wu B:
The CFTR M470V, intron 8 poly-T, and 8 TG-repeats detection in
Chinese males with congenital bilateral absence of the vas
deferens. Biomed Res Int. 2014:6891852014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kumar S, Tana A and Shankar A: Cystic
fibrosis--what are the prospects for a cure? Eur J Intern Med.
25:803–807. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Leonardi S, Pratico AD, Rotolo N, Di Dio
G, Lionetti E and La Rosa M: Early acute pancreatitis in a child
with compound heterozygosis F508/R1438W/Y1032C cystic fibrosis: A
case report. J Med Case Rep. 7:1882013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu CC, Hsieh-Li HM, Lin YM and Chiang HS:
Cystic fibrosis transmembrane conductance regulator gene screening
and clinical correlation in Taiwanese males with congenital
bilateral absence of the vas deferens. Hum Reprod. 19:250–253.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zvereff VV, Faruki H, Edwards M and
Friedman KJ: Cystic fibrosis carrier screening in a North American
population. Genet Med. 16:539–546. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kornreich R, Ekstein J, Edelmann L and
Desnick RJ: Premarital and prenatal screening for cystic fibrosis:
Experience in the Ashkenazi Jewish population. Genet Med.
6:415–420. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sugarman EA, Rohlfs EM, Silverman LM and
Allitto BA: CFTR mutation distribution among U.S. Hispanic and
African American individuals: Evaluation in cystic fibrosis patient
and carrier screening populations. Genet Med. 6:392–399. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Massie RJ, Poplawski N, Wilcken B,
Goldblatt J, Byrnes C and Robertson C: Intron-8 polythymidine
sequence in Australasian individuals with CF mutations R117H and
R117C. Eur Respir J. 17:1195–1200. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kiesewetter S, Macek M Jr, Davis C,
Curristin SM, Chu CS, Graham C, Shrimpton AE, Cashman SM, Tsui LC,
Mickle J, et al: A mutation in CFTR produces different phenotypes
depending on chromosomal background. Nat Genet. 5:274–278. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Feng J, Wu X and Zhang Y, Yang X, Ma G,
Chen S, Luo S and Zhang Y: A novel mutation (−195C>A) in the
promoter region of CFTR gene is associated with Chinese congenital
bilateral absence of vas deferens (CBAVD). Gene. 719:1440072019.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yoshimura K, Nakamura H, Trapnell BC,
Dalemans W, Pavirani A, Lecocq JP and Crystal RG: The cystic
fibrosis gene has a ‘housekeeping’-type promoter and is expressed
at low levels in cells of epithelial origin. J Biol Chem.
266:9140–9144. 1991.PubMed/NCBI
|
|
55
|
McCarthy VA and Harris A: The CFTR gene
and regulation of its expression. Pediatr Pulmonol. 40:1–8. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Giordano S, Amato F, Elce A, Monti M,
Iannone C, Pucci P, Seia M, Angioni A, Zarrilli F, Castaldo G and
Tomaiuolo R: Molecular and functional analysis of the large
5′promoter region of CFTR gene revealed pathogenic mutations in CF
and CFTR-related disorders. J Mol Diagn. 15:331–340. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chillon M, Casals T, Mercier B, Bassas L,
Lissens W, Silber S, Romey MC, Ruiz-Romero J, Verlingue C,
Claustres M, et al: Mutations in the cystic fibrosis gene in
patients with congenital absence of the vas deferens. N Engl J Med.
332:1475–1480. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mak V, Jarvi KA, Zielenski J, Durie P and
Tsui LC: Higher proportion of intact exon 9 CFTR mRNA in nasal
epithelium compared with vas deferens. Hum Mol Genet. 6:2099–2107.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hwang TC and Sheppard DN: Gating of the
CFTR Cl- channel by ATP-driven nucleotide-binding domain
dimerisation. J Physiol. 587:2151–2161. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Radpour R, Gilani MA, Gourabi H, Dizaj AV
and Mollamohamadi S: Molecular analysis of the IVS8-T splice
variant 5T and M470V exon 10 missense polymorphism in Iranian males
with congenital bilateral absence of the vas deferens. Mol Hum
Reprod. 12:469–473. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Anzai C, Morokawa N, Okada H, Kamidono S,
Eto Y and Yoshimura K: CFTR gene mutations in Japanese individuals
with congenital bilateral absence of the vas deferens. J Cyst
Fibros. 2:14–18. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Disset A, Michot C, Harris A, Buratti E,
Claustres M and Tuffery-Giraud S: A T3 allele in the CFTR gene
exacerbates exon 9 skipping in vas deferens and epididymal cell
lines and is associated with congenital bilateral absence of vas
deferens (CBAVD). Hum Mutat. 25:72–81. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nam MH, Hijikata M, Tuan LA, Lien LT,
Shojima J, Horie T, Nakata K, Matsushita I, Ohashi J, Tokunaga K
and Keicho N: Variations of the CFTR gene in the Hanoi-Vietnamese.
Am J Med Genet A. 136:249–253. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ranum LP and Cooper TA: RNA-mediated
neuromuscular disorders. Annu Rev Neurosci. 29:259–277. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Czajka-Oraniec I, Zgliczynski W,
Kurylowicz A, Mikula M and Ostrowski J: Association between
gynecomastia and aromatase (CYP19) polymorphisms. Eur J Endocrinol.
158:721–727. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Martin P, Makepeace K, Hill SA, Hood DW
and Moxon ER: Microsatellite instability regulates transcription
factor binding and gene expression. Proc Natl Acad Sci USA.
102:3800–3804. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lopez E, Viart V, Guittard C, Templin C,
René C, Méchin D, Des Georges M, Claustres M, Romey-Chatelain MC
and Taulan M: Variants in CFTR untranslated regions are associated
with congenital bilateral absence of the vas deferens. J Med Genet.
48:152–159. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Silber SJ, Balmaceda J, Borrero C, Ord T
and Asch R: Pregnancy with sperm aspiration from the proximal head
of the epididymis: A new treatment for congenital absence of the
vas deferens. Fertil Steril. 50:525–528. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Nicopoullos JD, Gilling-Smith C, Almeida
PA and Ramsay JW: The results of 154 ICSI cycles using surgically
retrieved sperm from azoospermic men. Hum Reprod. 19:579–585. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Liu J, Lissens W, Silber SJ, Devroey P,
Liebaers I and Van Steirteghem A: Birth after preimplantation
diagnosis of the cystic fibrosis delta F508 mutation by polymerase
chain reaction in human embryos resulting from intracytoplasmic
sperm injection with epididymal sperm. JAMA. 272:1858–1860. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Rechitsky S, Verlinsky O and Kuliev A: PGD
for cystic fibrosis patients and couples at risk of an additional
genetic disorder combined with 24-chromosome aneuploidy testing.
Reprod Biomed Online. 26:420–430. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Girardet A, Ishmukhametova A, Willems M,
Coubes C, Hamamah S, Anahory T, Des Georges M and Claustres M:
Preimplantation genetic diagnosis for cystic fibrosis: The
montpellier center's 10-year experience. Clin Genet. 87:124–132.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
McCallum TJ, Milunsky JM, Cunningham DL,
Harris DH, Maher TA and Oates RD: Fertility in men with cystic
fibrosis: An update on current surgical practices and outcomes.
Chest. 118:1059–1062. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chiang HS, Wu CC, Wu YN, Lu JF, Lin GH and
Hwang JL: CFTR mutation analysis of a Caucasian father with
congenital bilateral absence of vas deferens, a Taiwanese mother,
and twins resulting from ICSI procedure. J Formos Med Assoc.
107:736–740. 2008. View Article : Google Scholar : PubMed/NCBI
|