Introduction
In previous years, the market demand for traditional
Chinese medicine is increasing due to its relatively low economic
cost and fewer adverse effects (1,2).
Paecilomyces tenuipes (P. tenuipes) is one of the
most widely used medicines in Asian countries. Due to its various
biological and pharmacological activities, P. tenuipes can
be used as a substitute for Cordyceps sinensis in health
supplements. This fungus contains numerous biologically active
constituents, in which adenosine is of importance (3–6).
Adenosine displays various pharmacological activities, including
cardioprotective, anti-inflammatory and anticancer properties
(7–10). However, the low adenosine content
in P. tenuipes limits further development and application in
research. Therefore, the adenosine yield of P. tenuipes
needs to be improved using modern breeding technology.
Genetic engineering breeding is an advanced
microbial breeding technology that can effectively improve the
yield of active compounds or eliminate undesirable products
(11). To date, the genomic
resources of P. tenuipes are still limited and only few
nucleotide sequences have been deposited into the National Center
for Biotechnology Information database. However, most metabolic
regulatory genes of adenosine biosynthesis remain unidentified.
Therefore, considering the need to enhance adenosine content of
this fungus, it is particularly important to use transcriptome
sequencing technology to identify the key genes in adenosine
biosynthesis of P. tenuipes.
RNA sequencing (RNA-Seq) technology is a technology
used for mining important functional gene groups (12–14).
Previously, with the rapid development of science and technology,
high-throughput sequencing technology has been widely used in
transcriptome analysis of a variety of organisms, which can provide
accurate information about gene expression and regulation (15–20).
According to the different principles of sequencing, there are
several high-throughput sequencing methods, of which Illumina
technology has received extensive attention due to its large
reading length and novel computational tools (21).
The present study used Illumina sequencing
technology to analyze the transcriptome of P. tenuipes at
different fermentation stages. Following clustering and polishing,
functional annotation was performed using several public databases.
Subsequently, the differentially expressed genes (DEGs) putatively
associated with adenosine biosynthesis were screened. The results
of the present study could be useful in strain breeding, and
provide information on the molecular mechanism of biosynthesis of
active substances in P. tenuipes.
Materials and methods
Determination of the adenosine
accumulation curve of P. tenuipes
P. tenuipes RCEF 4339 was purchased from
Anhui Agricultural University (Anhui, China). This fungus was
originally kept in potato dextrose agar slants and then subcultured
in basal liquid medium (40 g/l glucose, 10 g/l yeast extract powder
and 10 g/l peptone). To determine the change in adenosine content
with fermentation time, laboratory-scale fermentation of P.
tenuipes was performed in a 10-liters fermenter (Biostat,
Sartorius Stedim Biotech; Sartorius AG) at 26°C with a 6 liters
working volume of basal medium (pH 6.8). The other fermentation
conditions were as follows: Rotating speed 100 r/min, inoculation
amount 5% (v/v), seed cultivation time of 4 days, pH 6.0, tank
pressure 0.3 MPa, ventilation volume 0.9 m3/h, culture
time 7 days. Following submerged culture of the fungus at various
time points, the fermentation broth (50 ml) was harvested and
centrifuged (3,000 × g, 4°C, 10 min) to obtain the mycelium. The
dry weight of mycelium and adenosine content were accurately
measured after repeated washing of the mycelium with distilled
water and lyophilization.
Extraction and analytical methods of
intracellular adenosine
Adenosine was extracted from the mycelium of P.
tenuipes by hot water extraction and measured using
high-performance liquid chromatography (HPLC) (22). The detection process was conducted
using a Shimadzu high performance liquid chromatography system,
which was equipped with LC-6AD pump and SPD-A UV-vis detector
(Shimadzu Corporation). C18 column (150×4.6 mm, 5 µm) was used for
separation. The mobile phase was prepared by adding methanol (150
ml) to phosphate buffer solution with pH 6.5 (850 ml). The details
of conditions used for the system were as follows: Column
temperature 35°C, detection wavelength 260 nm, sample quantity 20
µl, flow rate 1 ml/min. Adenosine purchased from Sigma-Aldrich
(Merck KGaA) was used as a standard.
Sample preparation
P. tenuipes was cultured in basal medium. The
fresh mycelium of P. tenuipes cultured for 24 h (PT24), 102
h (PT4102) and 192 h (PT192) were collected. Following freezing in
liquid nitrogen, the mycelia were kept in a refrigerator at −80°C
for later use.
RNA isolation and cDNA synthesis
TRIzol® reagent (Takara Biotechnology
Co., Ltd.) was used to extract total RNA from the three samples.
The integrity, quantity and quality of extracted RNA were detected
to ensure RNA availability via agarose gel electrophoresis,
NanoDrop ND-1000 Spectrometer (Thermo Fisher Scientific, Inc.) and
Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.),
respectively.
Poly (A)+ mRNA was isolated from total
RNA using oligo(dT) magnetic beads and then fragmented into short
fragments using Fragmentation Buffer (Illumina, Inc.).
First-stranded cDNA was synthesized using N6 random primers and
Reverse Transcriptase SuperScript II (Takara Biotechnology Co.,
Ltd.) using short fragments of RNA as the template. Subsequently,
second chain cDNA was synthesized using DNA polymerase I and RNase
H (Takara Biotechnology Co., Ltd.). cDNAs were purified using the
RNeasy RNA Cleanup kit (Qiagen GmbH).
Transcriptome sequencing, functional
annotation and expression analysis
Three constructed cDNA libraries were sequenced
using an 100 base pair (bp) double-ended sequencing on an Illumina
HiSeq™ 2500 platform at Shanghai Biotechnology Corporation. Raw
RNA-Seq data were screened. Low quality sequences, adaptor
sequences and reads <20 bp were separated from clean reads
(23). Unigenes were generated
from the clean reads using CLC Genomics Workbench (version 6.0.4;
Qiagen Sciences, Inc.).
The assembled unigenes were functionally annotated
against the UniProt database using BLASTx
(E-value≤10−5). For Gene Ontology (GO) annotations
(24), all best hit alignments
were imported into Blast2GO software (v.2.8.0) (25). Subsequently, GO functional
classifications were determined using WEGO software (v.2.0)
(26). Additionally, Eukaryotic
Orthologous Groups (KOG) (27) and
Kyoto Encyclopedia of Genes and Genomes (KEGG) (28) databases were used to classify
function and analyze metabolic pathways.
Once gene expression levels of transcripts were
normalized using the reads per kilobase transcriptome per million
mapped reads (RPKM) method, the DEGs were identified using the
DESeq package (29). The threshold
of DEG screening was set as follows: Fold-change ≥2 and false
discovery rate ≤0.05. To illustrate differences between the groups,
Volcano plots were employed using Origin v.8.5 (30). All DEGs between compared sample
were subjected to GO and KEGG enrichment analysis as
aforementioned.
Changes in putative gene expression in
different samples
To analyze the association between the expression of
the putative genes and adenosine content, the mycelium of P.
tenuipes was harvested at different growth stages (24, 54, 102,
120 and 192 h). Reverse transcription-quantitative PCR (RT-qPCR)
was employed to detect the relative expression of target genes in
mycelium. These genes were likely associated with adenosine
production according to previous studies (31–38)
and the present experimental results. Following extraction of total
RNA from the mycelium of the five samples as aforementioned, 1,000
ng RNA was reverse transcribed using the PrimeScript RT reagent kit
(Takara Biotechnology Co., Ltd.). According to the manufacturer's
protocols, the genomic DNA removal reaction system was incubated at
42°C for 2 min and cDNA was obtained at 37°C for 15 min followed by
85°C for 15 sec. Primers of putative genes used in the present
study are presented in Table SI.
To normalize expression levels, the translation elongation factor
gene 1 alpha (tef-1α) of P. tenuipes was selected as
the internal standard. Meanwhile, adenosine content in the five
samples was determined by HPLC as aforementioned.
RT-qPCR was performed on a Mx3000P thermocycler
(Agilent Technologies, Inc.) using the SYBR Premix Ex Taq II kit
(Takara Biotechnology, Inc.). The following thermocycling
conditions were used for qPCR: 94°C for 2 min, followed by 40
cycles of 94°C for 20 sec and 60°C for 34 sec. Relative expression
levels of unigenes in three samples were analyzed using the
comparative threshold method (2−ΔΔCt) (39).
Statistical analysis
Data are presented as the mean ± SD. Statistical
analysis was performed using one-way ANOVA followed by Tukey's
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Samples of transcriptome sequencing
were determined according to the intracellular adenosine
accumulation curve
To select transcriptome sequencing samples, P.
tenuipes was cultured in a 10 liter fermenter to evaluate the
curve of intracellular adenosine accumulation with culture time. As
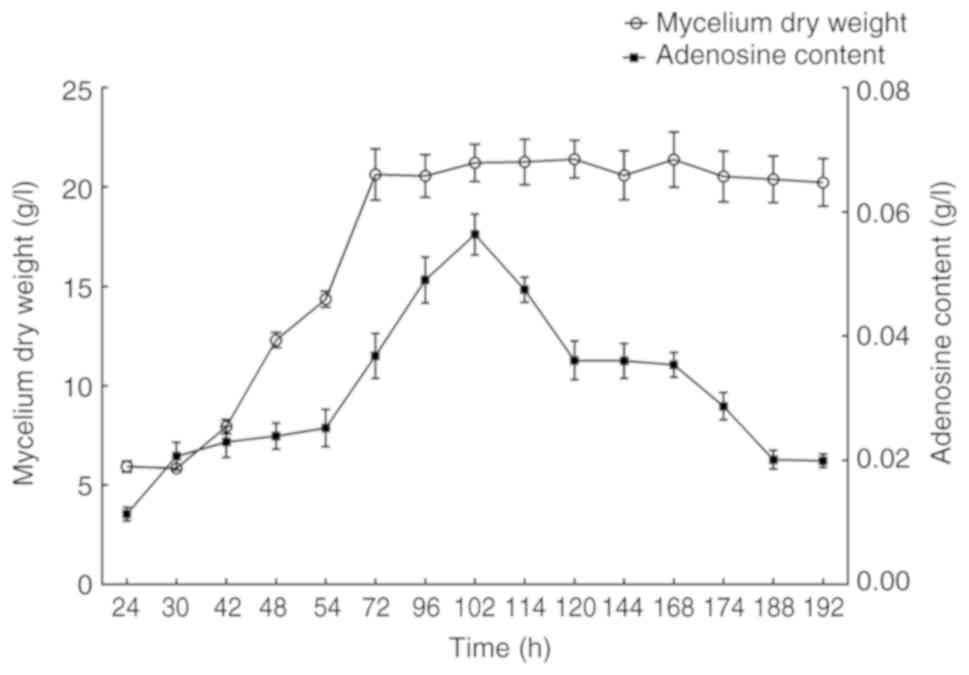
shown in Fig. 1, mycelium dry
weight and adenosine production increased gradually at the
beginning until a maximum value was reached at 102 h, following
which, the mycelium dry weight remained nearly constant. However,
there was a decrease in adenosine production after 102 h. To
analyze the key genes related to adenosine biosynthesis by
transcriptome sequencing, the mycelium of P. tenuipes in the
early stage (24 h), the highest adenosine production stage (102 h)
and the late stage of fermentation (192 h) were chosen according to
the adenosine accumulation curve. The adenosine production of P.
tenuipes cultured for 24, 102 and 192 h were 0.011, 0.056 and
0.020 g/l, respectively.
Transcriptome sequencing and
assembly
To study the transcriptional profile of P.
tenuipes at different growth stages, the Illumina HiSeq
platform was used to sequence the three constructed cDNA libraries.
A total of 32,061,948, 37,531,698 and 34,766,538 raw reads were
obtained from PT24, PT102 and PT192, respectively. The raw
sequences were filtered and 27,207,076 (84.86% of the raw reads),
31,708,556 (84.48% of the raw reads) and 29,265,588 (84.18% of the
raw reads) clean reads were generated for PT24, PT102 and PT192,
respectively (Table SII). The
sequence data sets are available in the Sequence Read Archive
database (accession no. PRJNA640721). After assembling the clean
reads, 13,353 unigenes with an average length of 1,804 bp were
obtained (Table I). The results of
unigene distribution is shown in Fig.
2. Most unigenes (34.52%) were >2,000 bp long, followed
closely by unigenes (23.10%) distributed within the range of
200–400 bp. The longest unigene contained 18,413 bp.
 | Table I.Results of the Paecilomyces
tenuipes transcriptome sequencing and assembly. |
Table I.
Results of the Paecilomyces
tenuipes transcriptome sequencing and assembly.
| Parameters | Number |
|---|
| Total reads | 104,360,184 |
| Contigs | 20,484 |
| Average length of
contigs, bp | 1,140 |
| N50 of contig set,
bp | 2,197 |
| Unigenes | 13,353 |
| Average length of
unigenes, bp | 1,804 |
| N50 of unigene set,
bp | 3,171 |
Functional annotation and
classification of unigenes
Sequence similarity analyses were performed in
various databases, such as Uniprot, KOG, GO and KEGG. Out of 13,353
unigenes, 8,099 were annotated in the Uniprot database. The
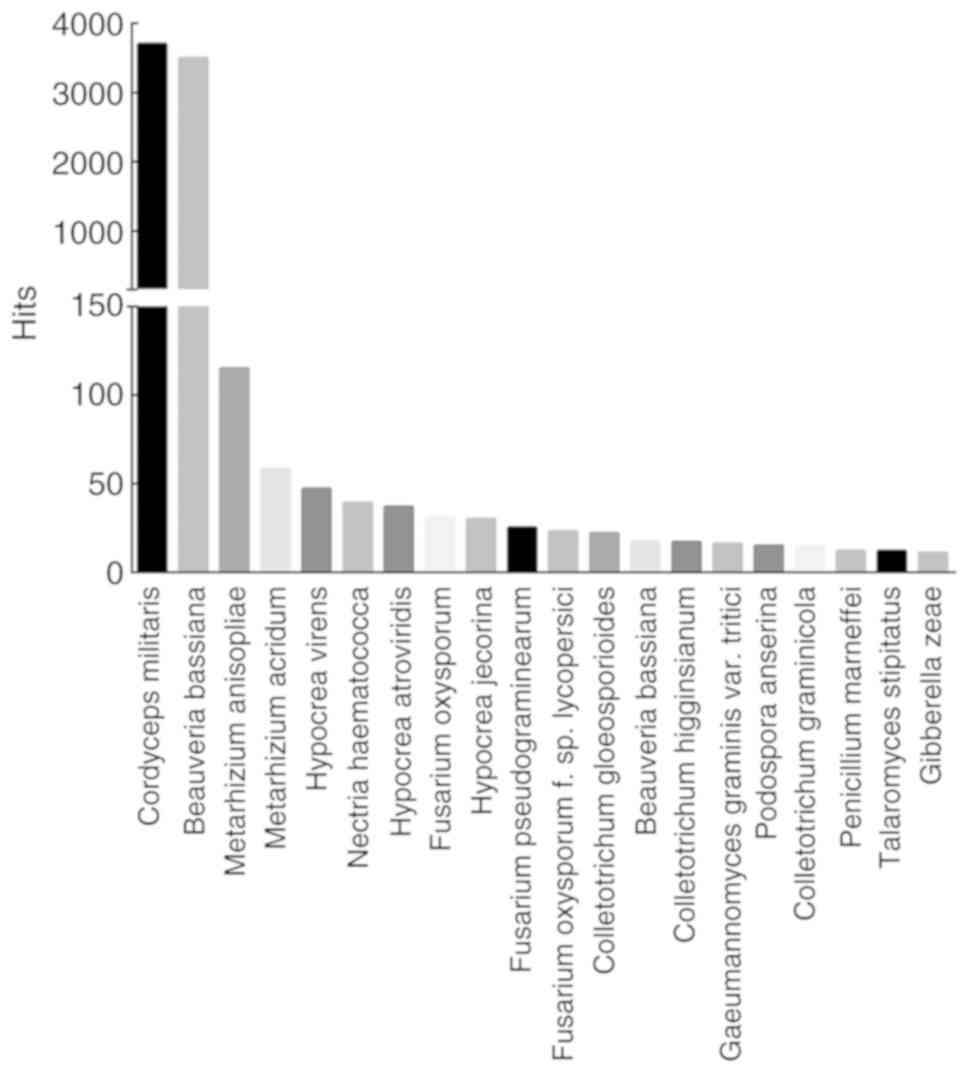
similarity of P. tenuipes genes was compared with other
species whose complete or draft genomes are available, and the top
20 species are shown in Fig. 3. A
total of 3,703 unigenes (45.72%) showed similarity to Cordyceps
militaris (strain CM01) genes, 3,496 (43.17%) were similar to
the genes of Beauveria bassiana (strain ARSEF2860), whereas
only 115 (1.42%) unigenes corresponded to Metarhizium
anisopliae.
The GO database is widely used for the analysis of
transcriptome data, which includes three main functional categories
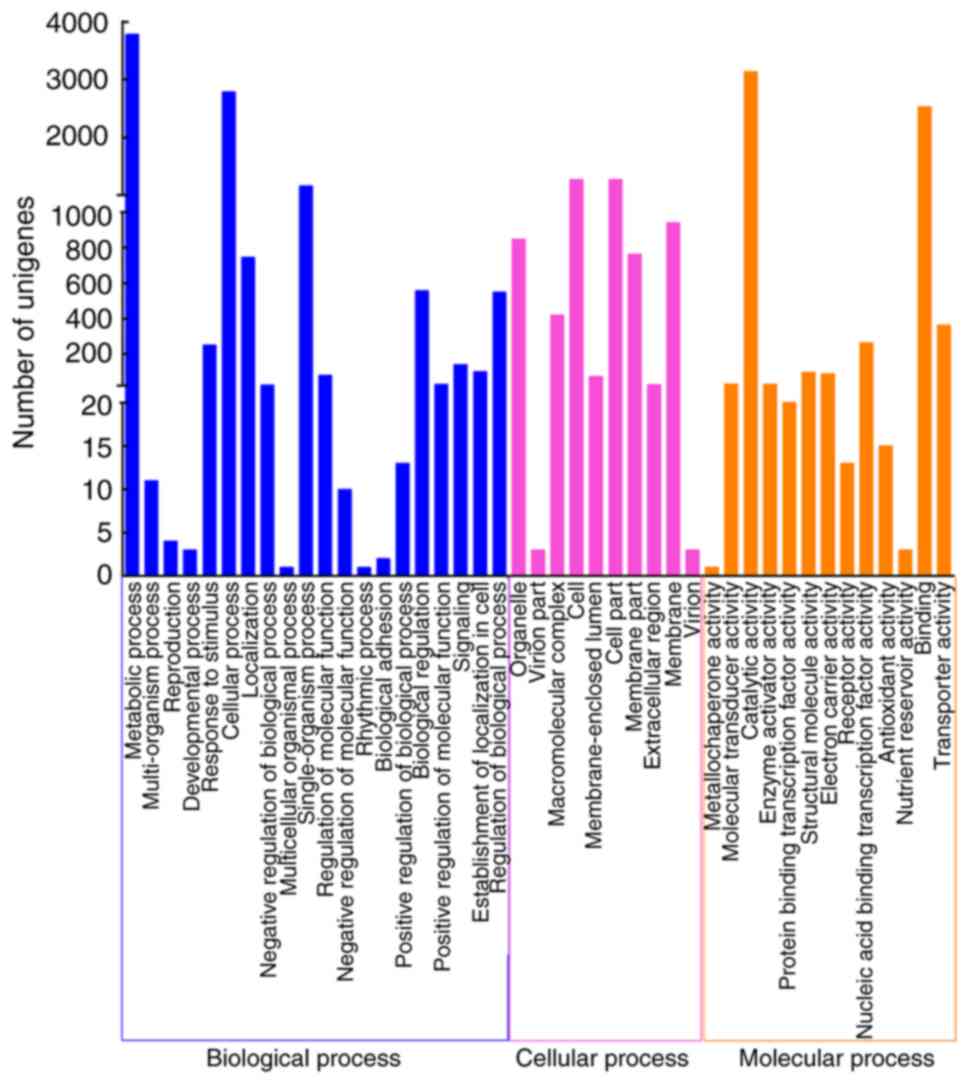
(40,41). With regard to GO analysis, 5,123
out of 13,353 unigenes were mapped to 43 functional groups
(Fig. 4). In the ‘cellular
process’ group, ‘cell part’ (1,278; 21.56%) and ‘cell’ (1,278;
21.56%) were represented the most, followed by ‘membrane’ (940;
15.85%). Under ‘molecular process’, genes involved in ‘catalytic
activity’ (3,139; 47.37%) and ‘binding’ (2,534; 38.24%) were highly
represented. In addition, ‘metabolic process’ (3,781; 34.68%) and
‘cellular process’ (2,790; 25.59%) were the majority of the
assignments in the ‘biological process’ ontology.
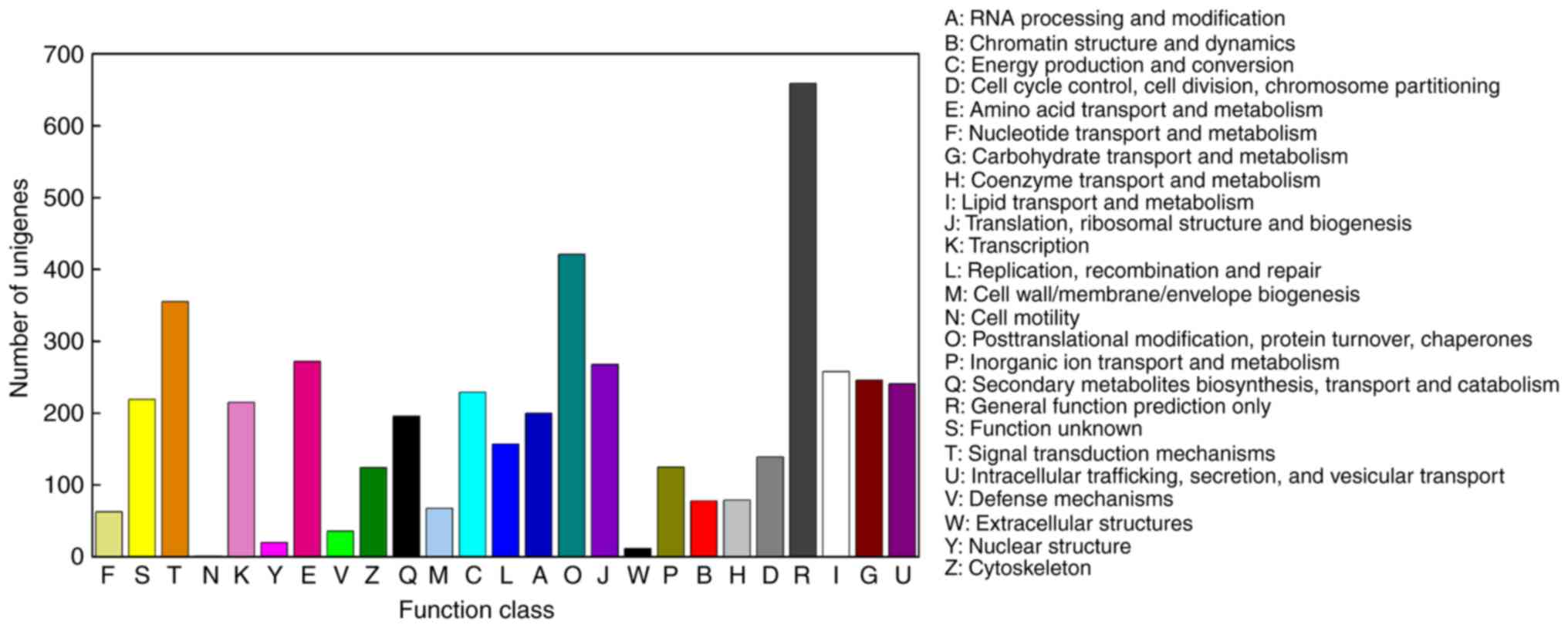
To improve our understanding of the unigenes, KOG
analysis was performed to classify orthologous gene products. In
total, 4,158 unigenes were functionally categorized into 25 KOG
categories (Fig. 5). A total of
659 unigenes and 421 unigenes were classified into the categories
of ‘general function prediction only’ (15.85%) and
‘posttranslational modification, protein turnover, chaperones’
(10.13%), respectively, which represented the two largest groups.
The categories ‘extracellular structures’ (12 members; 0.29%) and
‘cell motility’ (1 member; 0.02%) represented the smallest
groups.
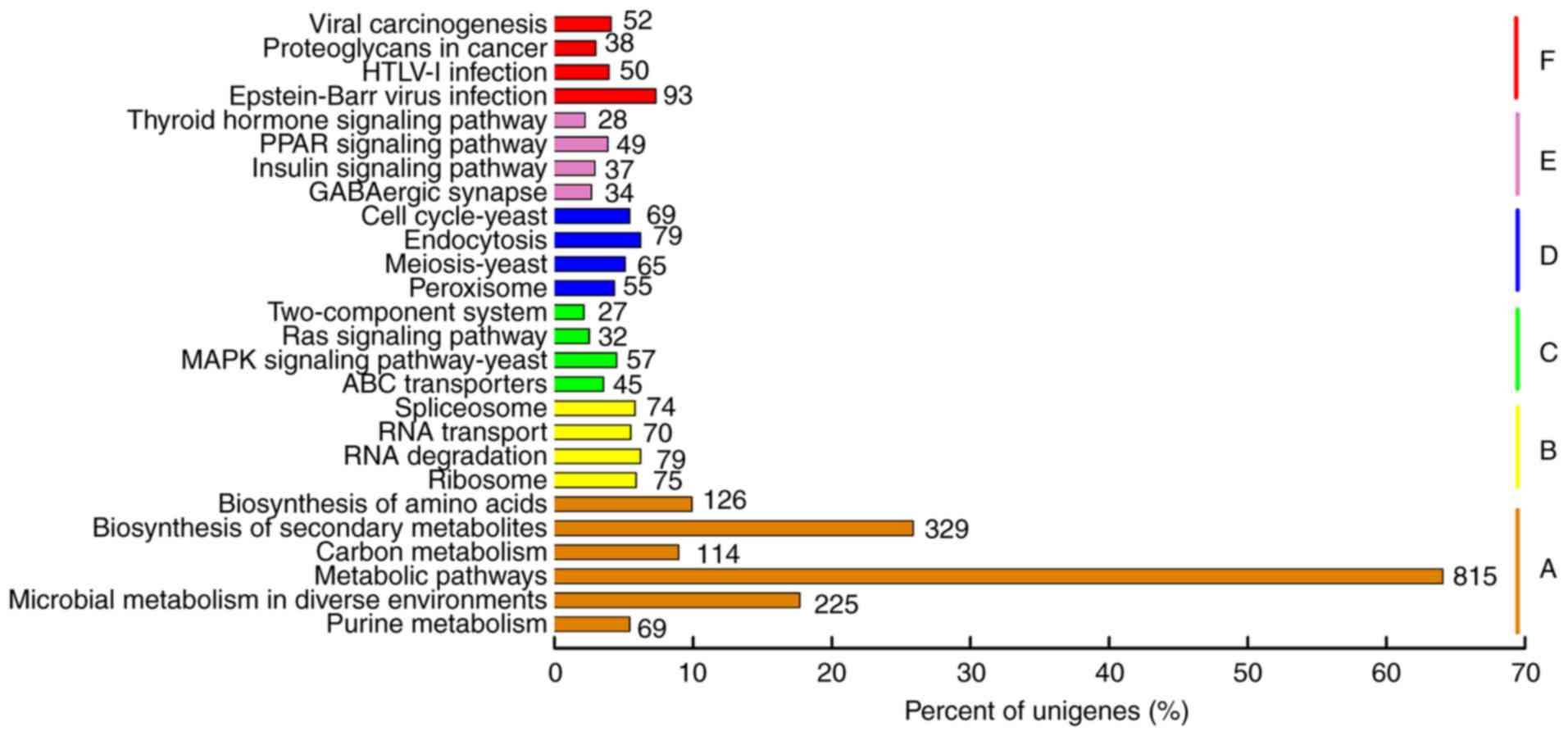
To find genes related to adenosine biosynthesis in
P. tenuipes, the sequences were searched against the KEGG
database. Consequently, 1,272 sequences were enriched in 281 KEGG
pathways (Fig. 6). The most
representative pathways were ‘metabolic pathways’ (815; 39.49%),
followed by ‘biosynthesis of secondary metabolites’ (329; 15.94%)
and ‘microbial metabolism in diverse environments’ (225; 10.90%).
Using KEGG, information relating to genes of the purine metabolism
pathway (Ko00230) was obtained, which is the primary pathway of
adenosine biosynthesis (Table
SIII). These results showed that most of the enzymes involved
in this pathway were expressed in P. tenuipes. This
information may provide valuable resources for further research of
this fungus.
Identification of DEGs
DEGs in the P. tenuipes transcriptome were
analyzed and identified based on RNA-Seq data. The gene expression
abundance was calculated using RPKM, which was used to estimate the
significance of the DEGs between the three samples. The expression
levels of most unigenes ranged between 0–100 RPKM (Table II). Thousands of DEGs were
identified, demonstrating the substantial changes at the three
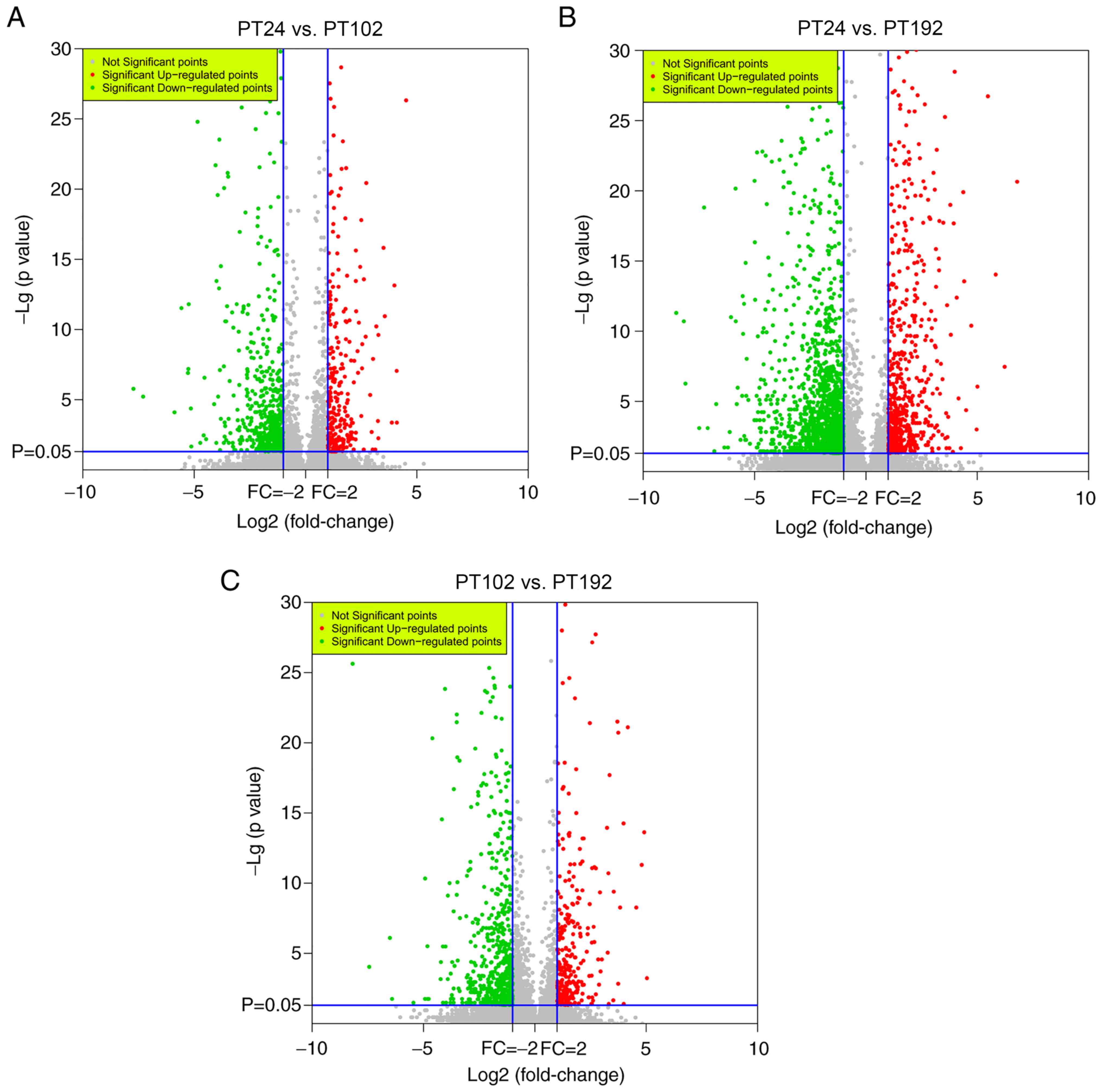
different developmental stages. According to the volcano plots
(Fig. 7), between PT24 and PT102
libraries, 601 DEGs were found, of which 230 genes were upregulated
and 371 were downregulated. PT24 with PT192 were compared and it
was found that a total of 1,658 DEGs were identified, of which 557
genes were upregulated and 1,101 genes were downregulated.
Similarly, 217 upregulated genes and 411 downregulated genes (a
total of 628 DEGs) were found in PT102 corresponding to PT192.
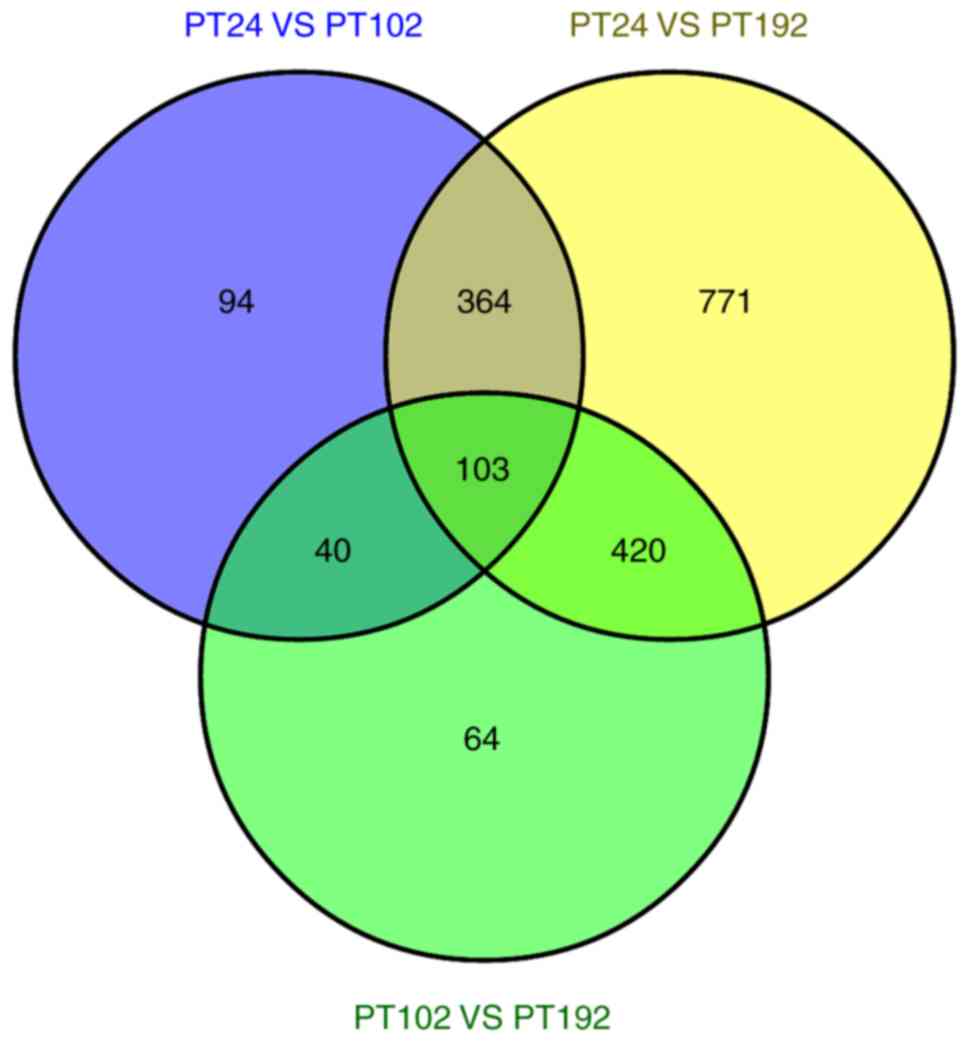
Based on our datasets, a total of 103 DEGs were commonly expressed
in the three libraries. It was identified that 94, 64 and 771 genes
were expressed only in PT24, PT102 and PT192, respectively
(Fig. 8). In addition, all the
DEGs were searched against the GO and KEGG database, and the
results were included in Figs. S1
and S2. In the GO biological
process category and cellular process category,
‘oxidation-reduction process’ and ‘integral to membrane’ were the
most highly enriched terms respectively in PT24 vs. PT102, PT24 vs.
PT192 and PT102 vs. PT192. In the GO molecular process category,
‘nucleotide binding’ was significantly enriched in PT24 vs. PT192
and PT102 vs. PT192, while ‘oxidoreductase activity’ was enriched
in PT24 vs. PT102. KEGG enrichment analysis of DEGs between the
compared samples (PT24 vs. PT102, PT24 vs. PT192, PT102 vs. PT192)
showed that the most representative pathways were ‘metabolic
pathways’.
| Table II.Expression levels of unigenes. |
Table II.
Expression levels of unigenes.
|
| Number of
unigenes |
|---|
|
|
|
|---|
| RPMK value | PT24 | PT102 | PT192 |
|---|
| >1,000 | 47 | 42 | 31 |
|
>100<999.9 | 704 | 724 | 755 |
| >10<99.9 | 4,572 | 5,141 | 5,221 |
| >1<9.9 | 4,023 | 4,131 | 4,905 |
| <0.99 | 4,007 | 3,315 | 2,441 |
Determination of adenosine
biosynthesis-related genes via in silico analyses
Several genes putatively involved in the
biosynthesis of adenosine were identified from the DEG analyses of
the transcriptome sequence data. Among them, various DEGs (such as
nucleoside-diphosphate kinase, ADP-ribose pyrophosphatase,
adenylate kinase, pyruvate kinase, 3′,5′-cyclic-nucleotide
phosphodiesterase and IMP dehydrogenase) were differentially
expressed in PT24 vs. PT192. Ten DEGs were found in PT102 vs.
PT192. According to the KEGG analysis, these DEGs coded enzymes
that play a role in purine metabolism. The results also showed that
there were 4 upregulated (Unigene 1296, Unigene 1606, Unigene 5708,
Unigene 571) and 6 downregulated unigenes (Unigene 1591, Unigene
1707, Unigene 2933, Unigene 3749, Unigene 3962, Unigene 9544) in
the ‘purine metabolism’ pathway in PT102 (PT102 vs. PT192) samples.
In addition, 1 upregulated (Unigene 814) and 5 downregulated
(Unigene 2010, Unigene 1606, Unigene 571, Unigene 5708, Unigene
5138) adenosine biosynthesis genes were found in PT24 (PT24 vs.
PT102). Overall, based on the transcriptome sequencing results and
reports, 14 DEGs that were selected to verify the accuracy of
transcriptome sequencing (Table
III).
| Table III.Differentially expressed genes
putatively related to adenosine metabolism in Paecilomyces
tenuipes. |
Table III.
Differentially expressed genes
putatively related to adenosine metabolism in Paecilomyces
tenuipes.
|
|
| Relative mRNA level
(RPKM) |
|
|---|
|
|
|
|
|
|---|
| Gene ID | Length | PT24 | PT102 | PT192 | Annotation |
|---|
| contig_114 | 2,785 | 46.2494 | 80.1168 | 127.9236 | 5-hydroxyisourate
hydrolase |
| contig_1296 | 2,172 | 352.3985 | 271.8885 | 125.6277 |
Nucleoside-diphosphate kinase |
| contig_1591 | 5,255 | 29.6818 | 27.9372 | 61.6834 | DNA-directed RNA
polymerase I subunit A1 |
| contig_1606 | 3,383 | 70.4259 | 143.3399 | 69.0219 |
Ribonucleoside-diphosphate reductase
subunit M1 |
| contig_1707 | 2,638 | 16.8871 | 24.9292 | 101.7325 |
3′,5′-cyclic-nucleotide
phosphodiesterase |
| contig_2933 | 4,675 | 12.6516 | 9.2906 | 35.7920 |
3′,5′-cyclic-nucleotide
phosphodiesterase |
| contig_3519 | 2,127 | 38.0276 | 25.6053 | 15.0668 | ADP-ribose
pyrophosphatase |
| contig_4264 | 2,833 | 18.3151 | 4.9743 | 3.6178 | Urease subunit
α |
| contig_5708 | 2,014 | 25.6055 | 215.9969 | 101.0778 | Sulfate
adenylyltransferase |
| contig_571 | 1,643 | 227.0272 | 527.1583 | 221.3372 |
Ribonucleoside-diphosphate reductase
subunit M2 |
| contig_753 | 2,297 | 152.4581 | 163.3336 | 315.4079 | IMP
dehydrogenase |
| contig_814 | 2,500 | 220.1618 | 101.5178 | 85.6467 | Pyruvate
kinase |
| contig_901 | 1,352 | 339.3854 | 185.8043 | 103.8499 | Adenylate
kinase |
| contig_9544 | 2,117 | 1.8926 | 3.7142 | 23.4699 |
5′-nucleotidase |
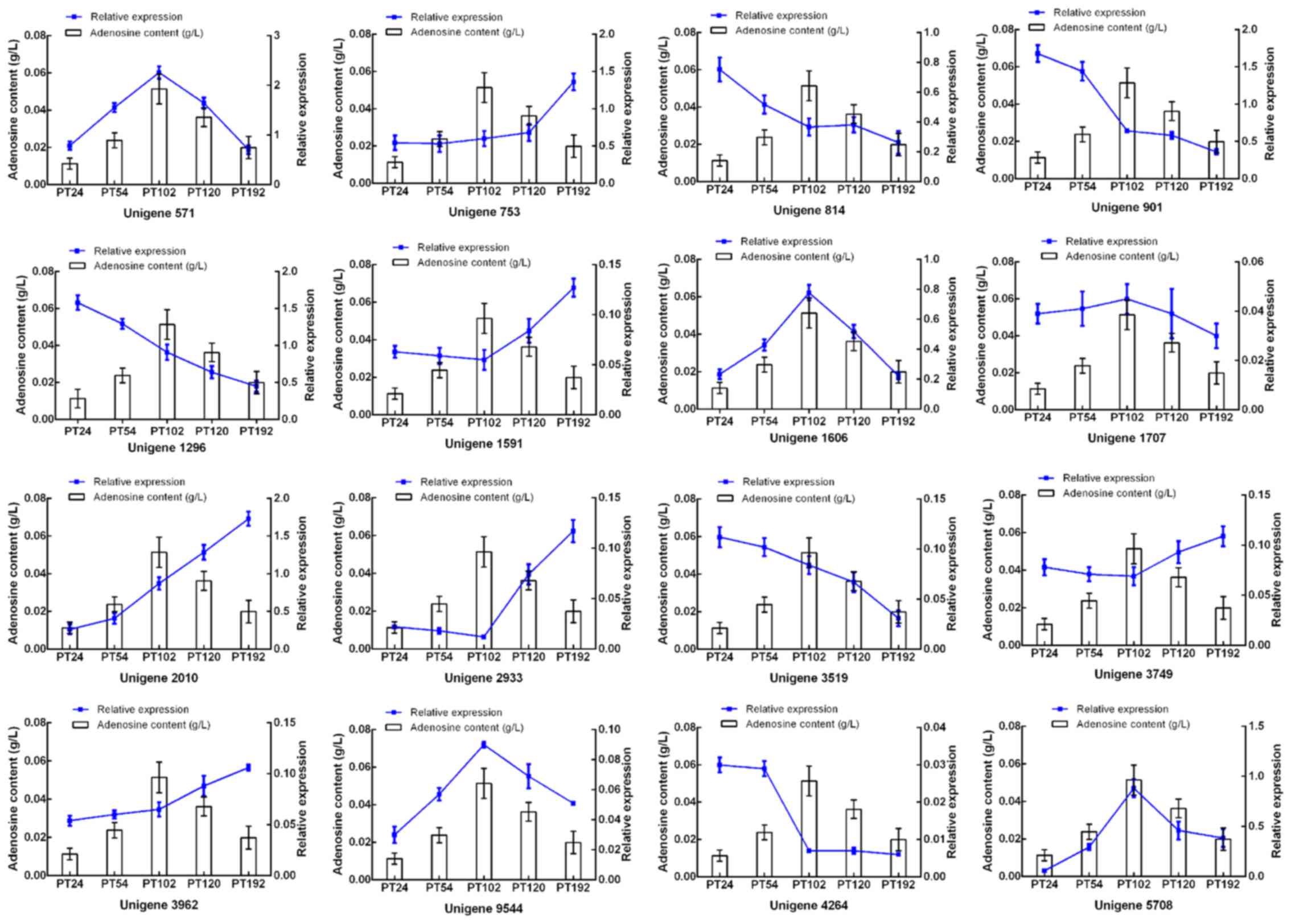
RT-qPCR analysis of adenosine-related
gene expression
To determine whether the 14 candidate DEGs were
closely related to adenosine biosynthesis of P. tenuipes,
RT-qPCR was performed to analyze the expression of these genes in
the mycelium at different fermentation times. Concurrently,
adenosine production in the mycelium was also analyzed. The
expression of 2 DEGs (Unigene 9544 and Unigene 1606) were
positively associated with adenosine accumulation (Fig. 9).
Discussion
P. tenuipes, a Chinese medicinal fungus, is a
source of pharmaceutically active compounds, but research into its
genetic information is lacking. Transcriptome sequencing technology
is widely used for mining DEGs in different samples (42,43).
In the present study, Illumina sequencing was employed to analyze
the transcriptome of P. tenuipes and discover key genes in
adenosine biosynthesis.
In the present study, 88,181,220 clean reads
(27,207,076, 31,708,556 and 29,265,588 clean reads in PT24, PT102
and PT192, respectively) were obtained and assembled into 13,353
unigenes using de novo assembly. All unigenes matched the
major pubic databases, including UniProt, KOG, GO and KEGG. A total
of 8,099 unigenes were consistent with known gene sequences in the
Uniprot databases. In the GO classification, the largest groups in
the three different categories were ‘cell’ (the number of genes
enriched in ‘cell part’ was the same as that in ‘cell’), ‘catalytic
activity’ and ‘metabolic process’. In KEGG pathway analysis, 1,272
unigenes were categorized into 272 KEGG pathways. These results
provide data for further studies investigating the biosynthesis of
active substances in this fungus.
Differential expression analysis was conducted by
comparing between three samples. The present study screened out
601, 1,658 and 628 unigenes that were differentially expressed in
PT24 vs. PT102, PT24 vs. PT192 and PT102 vs. PT192, respectively.
Most of the DEGs were involved in oxidation-reduction process and
integral to membrane according to GO enrichment. Meanwhile,
according to KEGG enrichment analysis, ‘metabolic pathways’ was the
most enriched term. These findings indicated that the metabolic
activity of P. tenuipes was altered by the culture time.
Based on KEGG enrichment analysis of DEGs, multiple
DEGs were enriched in the purine metabolism pathway, of which the
5′-nucleotidase gene was of interest. Previous studies have
reported that 5′-nucleotidase is a key enzyme in the
nucleoside/nucleotide metabolic pathway, which is widely
distributed in fungi, bacteria and numerous other cell types. This
enzyme may catalyze the formation of nucleoside or deoxynucleoside
from various nucleosides-5′-phosphates and
deoxynucleoside-5′-phosphates (44–47).
Covarrubias et al (48)
found that 5′-nucleotidase was a key molecule in the regulation of
adenosine monophosphate dephosphorylation and adenosine synthesis
in mice. Meanwhile, previous studies on Ophiocordycepssinensis
and Cordyceps militaris found that 5′-nucleotidase played an
important role in the phosphorylation and dephosphorylation of
adenosine (49,50). According to the results of a study
that investigated its expression pattern, upregulating the
transcription levels of 5′-nucleotidase can significantly affect
the accumulation of nucleoside compounds, including adenosine
(51). Therefore, in view of its
important role in the biosynthesis of nucleoside compounds, more
attention has been paid to 5′-nucleotidase. The 5′-nucleotidase
gene of numerous other species has been cloned and its expression
pattern has been investigated (52,53).
However, to the best of our knowledge, there are no reports
regarding the 5′-nucleotidase of P. tenuipes. The present
study only analyzed the expression levels of these genes so further
research is required to establish their functions.
To the best of our knowledge, this is the first
transcriptomic analysis of the P. tenuipes. The present
findings provided novel insight into the molecular mechanisms of
adenosine biosynthesis in order to aid further studies into this
topic. The present data is also beneficial in the investigation of
other medicinal fungal species.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This research was supported by the Scientific
Research Foundation of Jilin Agricultural University of China
(grant no. 2015014).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the NCBI SRA repository (https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA640721)
with the accession number PRJNA640721 (release date
15/06/2024).
Authors' contributions
HL, JY and LD guided the design of the whole
experiment and manuscript revision. LH and YL wrote the manuscript
and analyzed the experimental results. MN was mainly responsible
for language editing and for the fermentation of P.
tenuipes; YG, XM and GC completed the sample preparation,
adenosine content determination, gene expression analysis and
manuscript revision. YD helped in the process of sequencing data
analysis. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang M, Guan X, Chi Y, Robinson N and Liu
JP: Chinese herbal medicine as adjuvant treatment to chemotherapy
for multidrug-resistant tuberculosis (MDR-TB): A systematic review
of randomized clinical trials. Tuberculosis (Edinb). 95:364–372.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen SY, Lin YH, Huang JW and Chen YC:
Chinese herbal medicine network and core treatments for allergic
skin diseases: Implications from a nationwide database. J
Ethnopharmacol. 168:260–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu CP, Kim SW, Hwang HJ and Yun JW:
Production of exopolysaccharides by submerged culture of an
enthomopathogenic fungus, Paecilomyces tenuipes C240 in
stirred-tank and airlift reactors. Bioresource Technol. 97:770–777.
2006. View Article : Google Scholar
|
|
4
|
Du LN, Liu CG, Teng MY, Meng QF, Lu JH,
Zhou YL, Liu Y, Cheng YK, Wang D and Teng LS: Anti-diabetic
activities of Paecilomyces tenuipes N45 extract in
alloxan-induced diabetic mice. Mol Med Rep. 13:1701–1708. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sapkota K, Moon SM, Choi BS, Kim S, Kim YS
and Kim SJ: Enhancement of IL-18 expression by Paecilomyces
tenuipes. Mycoscience. 52:260–267. 2011. View Article : Google Scholar
|
|
6
|
Kim HC, Choi BS, Sapkota K, Kim S and Lee
HJ: Purification and characterization of a novel, highly potent
fibrinolytic enzyme from Paecilomyces tenuipes. Process
Biochem. 46:1545–1553. 2011. View Article : Google Scholar
|
|
7
|
Moezi L, Akbarian R, Niknahad H and
Shafaroodi H: The interaction of adenosine and morphine on
pentylenetetrazole-induced seizure threshold in mice.
Neuropharmacology. 72:1–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang CH, Tsai SK, Chiang SC, Lai CC and
Weng ZC: The role of adenosine in preconditioning by brief pressure
overload in rats. J Formos Med Assoc. 114:756–763. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ondrackova P, Kovaru H, Kovaru F, Leva L
and Faldyna M: Adenosine modulates LPS-induced cytokine production
in porcine monocytes. Cytokine. 61:953–961. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang DQ, Song JY, Wu LJ, Ma YF, Song CH,
Dovat S, Nishizaki T and Liu J: Induction of senescence by
adenosine suppressing the growth of lung cancer cells. Biochem
Biophys Res Commun. 440:62–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Magocha TA, Zabed H, Yang MM, Yun JH,
Zhang HH and Qi XH: Improvement of industrially important microbial
strains by genome shuffling: Current status and future prospects.
Bioresource Technol. 257:281–289. 2018. View Article : Google Scholar
|
|
12
|
Li DM, Wu W, Zhang D, Liu XR, Liu XF and
Lin YJ: Floral transcriptome analyses of four Paphiopedilum
Orchids with distinct flowering behaviors and development of
simple sequence repeat markers. Plant Mol Biol Rep. 33:1928–1952.
2015. View Article : Google Scholar
|
|
13
|
Cândido Ede S, Fernandes Gda R, de Alencar
SA, Cardoso M, Lima SM, Miranda Vde J, Porto WF, Nolasco DO, de
Oliveira-Júnior NG, Barbosa AE, et al: Shedding some light over the
floral metabolism by arum lily (Zantedeschia aethiopica)
spathe de novo transcriptome assembly. PLoS One. 9:e904872014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shakeel A, Zhan CS, Yang YY, Wang XK, Yang
TW, Zhao ZY, Zhang QY, Li XH and Hu XB: The transcript profile of a
traditional Chinese medicine, Atractylodes lancea, revealing
its sesquiterpenoid biosynthesis of the major active components.
PLoS One. 11:e01519752016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sangwan RS, Tripathi S, Singh J, Narnoliya
LK and Sangwan NS: de novo sequencing and assembly of Centella
asiatica leaf transcriptome for mapping of structural,
functional and regulatory genes with special reference to secondary
metabolism. Gene. 525:58–76. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao X, Han J, Lu Z, Li Y and He C: De novo
assembly and characterization of spotted seal Phoca largha
transcriptome using Illumina paired-end sequencing. Comp Comp
Biochem Physiol Part D Genomics Proteomics. 8:103–110. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bräutigam A, Mullick T, Schliesky S and
Weber APM: Critical assessment of assembly strategies for non-model
species mRNA-Seq data and application of next-generation sequencing
to the comparison of C3 and C4 species. J Exp
Bot. 62:3093–3102. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y, Jiang R, Wu H, Liu P, Xie J, He Y
and Pang H: Next-generation sequencing-based transcriptome analysis
of Cryptolaemus montrouzieri under insecticide stress
reveals resistance-relevant genes in laybirds. Genomics. 100:35–41.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Farlora R, Araya-Garay J and
Gallardo-Escárate C: Discovery of sex-related genes through
high-throughput transcriptome sequencing from the salmon louse
Caligus rogercresseyi. Mar Genom. 15:85–93. 2014. View Article : Google Scholar
|
|
20
|
Liu G, Wei X, Chen R, Zhou H, Li X, Sun Y,
Xie S, Zhu Q, Qu N, Yang G, et al: A novel mutation of the SLC25A13
gene in a Chinese patient with citrin deficiency detected by target
next-generation sequencing. Gene. 533:547–553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang S, Sui Z, Chang L, Kang K, Ma J,
Kong F, Zhou W, Wang JG, Guo L, Geng H, et al: Transcriptome de
novo assembly sequencing and analysis of the toxic dinoflagellate
Alexandrium catenella using the Illumina platform. Gene.
537:285–293. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Du L, Liu Y, Liu C, Meng Q, Song J, Wang
D, Lu J and Teng L, Zhou Y and Teng L: Acute and subchronic
toxicity studies on safety assessment of Paecilomyces
tenuipes N45 extracts. Comb Chem High Throughput Screen.
18:809–818. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li G, Zhao Y, Liu Z, Gao C, Yan F, Liu B
and Feng J: De novo assembly and characterization of the spleen
transcriptome of common carp (Cyrinus carpio) using Illumina
paired-end sequencing. Fish Shellfish Immunol. 44:420–129. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
The Gene Ontology Consortium, . The gene
ontology resource: 20 years and still going strong. Nucleic Acids
Res. 47:D330–D338. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zheng Y, Xu QF, Chen HY, Chen QP, Gong ZJ
and Lai W: Transcriptome analysis of ultraviolet A-induced photo
aging cells with deep sequencing. J Dermatol. 45:175–181. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ye J, Fang L, Zheng HK, Zhang Y, Chen J,
Zhang ZJ and Wang J, Li ST, Li RQ, Bolund L and Wang J: WEGO: A web
tool for plotting GO annotations. Nucleic Acids Res. 34:W293–W297.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sello C, Liu C, Sun Y, Msuthwana P, Hu J,
Sui Y, Chen S, Zhou Y, Lu H, Xu C, et al: De novo assembly and
comparative transcriptome profiling of Anser and Anser cygnodies
geese species' embryonic skin feather follicles. Genes (Basel).
10:3512019. View Article : Google Scholar
|
|
28
|
Kanehisa M, Sato Y, Furumichi M, Morishima
K and Tanabe M: New approach for understanding genome variations in
KEGG. Nucleic Acids Res. 47:D590–D595. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Anders S and Huber W: Differential
expression analysis for sequence count data. Genome Biol.
11:R1062010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li WT: Volcano plots in analyzing
differential expressions with mRNA microarrays. J Bioinf Comput
Biol. 10:12310032012. View Article : Google Scholar
|
|
31
|
Forconi M, Biscotti MA, Barucca M,
Buonocore F, Moro GD, Fausto AM, Fausto AM, Gerdol M, Pallavicini
G, Schartl M, et al: Characterization of purine catabolic pathway
genes in coelacanths. J Exp Zool B Mol Dev Evol. 322:334–341. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Losenkova K, Zuccarini M, Karikoski M,
Laurila J, Boison D, Jalkanen S and Yegutkin GG:
Compartmentalization of adenosine metabolism in cancer cells and
its modulation during acute hypoxia. J Cell Sci. 133:jcs2414632020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Striepen B, Pruijssers AJ, Huang JL, Li C,
Gubbels MJ, Umejiego NN, Hedstrom L and Kissinger JC: Gene transfer
in the evolution of parasite nucleotide biosynthesis. Proc Natl
Acad Sci USA. 101:3154–3159. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stasolla C, Katahira R, Thorpe TA and
Ashihara H: Purine and pyrimidine nucleotide metabolism in higher
plants. J Plant Physiol. 160:1271–95. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Layland J, Carrick D, Lee M, Oldroyd K and
Berry C: Adenosine: Physiology, pharmacology, and clinical
applications. JACC Cardiovasc Interv. 7:581–591. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pettersson J, Schrumpf ME, Raffel SJ,
Porcella SF, Guyard C, Lawrence K, Gherardini FC and Schwan TG:
Purine salvage pathways among Borrelia species. Infect Immun.
75:3877–3884. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sasaki Y, Goto H, Wake T and Sasaki R:
Purine ribonucleotide homopolymer formation activity of RNA
polymerase from cauliflower. Biochim Biophys Acta. 366:443–453.
1974. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lunt SY, Muralidhar V, Hosios AM,
Israelsen WJ, Gui DY, Newhouse L, Ogrodzinski M, Hecht V, Xu K,
Acevedo PM, et al: Pyruvate kinase isoform expression alters
nucleotide synthesis to impact cell proliferation. Mol Cell.
57:95–107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang JJ, Bai WW, Zhou W, Liu J, Chen J,
Liu XY, Xiang TT, Liu RH, Wang WH, Zhang BL and Wan YJ:
Transcriptomic analysis of two Beauveria bassiana strains
grown on cuticle extracts of the silkworm uncovers their different
metabolic response at early infection stage. J Invertebr Pathol.
145:45–54. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang B, Zhou J, Liu HZ and Zheng FR:
Analysis of transcriptome profiling from the brain at maturation
and regression phases in starry flounder (Platichthys
stellatus). Gene Rep. 4:45–52. 2016. View Article : Google Scholar
|
|
41
|
Sun L, Wang Q, Wang Q, Dong K, Xiao Y and
Zhang YJ: Identification and characterization of odorant binding
proteins in the forelegs of Adelphocoris lineolatus (Goeze).
Front Physiol. 8:7352017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li JY, Pan LQ, Miao JJ, Xu RY and Xu WJ:
De novo assembly and characterization of the ovarian transcriptome
reveal mechanisms of the final maturation stage in Chinese scallop
Chlamys farreri. Comp Biochem Physiol Part D Genomics
Proteomics. 20:118–124. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fan XD, Wang JQ, Yang N, Dong YY, Liu L,
Wang FW, Wang N, Chen H, Liu WC, Sun YP, et al: Gene expression
profiling of soybean leaves and roots under salt, saline-alkali and
drought stress by high-throughput Illumina sequencing. Gene.
512:392–402. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Stutzer C, Mans BJ, Gaspar ARM, Neitz AWH
and Maritz-Olivier C: Ornithodoros savignyi: Soft tick
apyrase belongs to the 5′-nucleotidase family. Exp Parasitol.
122:318–327. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Santos CA, Saraiva AM, Toledo MAS, Beloti
LL, Crucello A, Favaro MTP, Horta MAC, Santiago AS, Mendes JS,
Souza AA and Souza AP: Initial biochemical and functional
characterization of a 5′-nucleotidase from Xylella
fastidiosa related to the human cytosolic 5′-nucleotidase I.
Microb Pathog. 59-60:1–6. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fenckova M, Hobizalova R, Fric ZF and
Dolezal T: Functional characterization of ecto-5′-nucleotidases and
apyrases in Drosophila melanogaster. Insect Biochem Mol
Biol. 41:956–967. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hunsucker SA, Mitchell BS and Spychala J:
The 5′-nucleotidases as regulators of nucleotide and drug
metabolism. Pharmacol Ther. 107:1–30. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Covarrubias R, Chepurko E, Reynolds A,
Huttinger ZM, Huttinger R, Stanfill K, Wheeler DG, Novitskaya T,
Robson SC, Dwyer KM, et al: Role of the CD39/CD37 purinergic
pathway in modulating arterial thrombosis in mice. Arterioscler
Thromb Vasc Biol. 36:1809–1820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xiang L, Li Y, Zhu Y, Luo H, Li C, Xu X,
Sun C, Song J, Shi L, He L, et al: Transcriptome analysis of the
Ophiocordyceps sinensis fruiting body reveals putative genes
involved in fruiting body development and cordycepin biosynthesis.
Genomics. 103:154–159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zheng P, Xia Y, Xiao G, Xiong C, Hu X,
Zhang S, Zheng H, Huang Y, Zhou Y, Wang S, et al: Genome sequence
of the insect pathogenic fungus Cordyceps militaris, a
valued traditional Chinese medicine. Genome Biol. 12:R1162011.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sakai Y, Tamao Y, Shimamoto T, Hama H,
Tsuda M and Tsuchiya T: Cloning and expression of the
5′-nucleotidase gene of Vibrio parahaemolyticus in Escherichia
coli and overproduction of the enzyme. J Biochem. 105:841–846.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Rampazzo C, Mazzon C, Reichard P and
Bianchi V: 5′-Nucleotidases: Specific assays for five different
enzymes in cell extracts. Biochem Biophys Res Commun. 293:258–263.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ubeidat M, Eristi CM and Rutherford CL:
Expression pattern of 5′-nucleotidase in Dictyostelium. Mech
Dev. 110:237–239. 2002. View Article : Google Scholar : PubMed/NCBI
|