Introduction
As a broad-spectrum anthracycline antitumour drug,
doxorubicin (DOX) is widely used in chemotherapy to treat numerous
types of tumour including solid tumours, transplantable leukaemia
and lymphomas (1). However, the
clinical application of DOX is limited by its cardiotoxicity, which
manifests as congestive heart failure (2). Cardiomyocyte apoptosis, necrosis and
other modes of cell death may be primary mechanisms underlying
DOX-induced deterioration of cardiac function (3). In addition, excessive oxidative
stress, lipid peroxidation, DNA damage and autophagy are also
involved in this pathological process (4). Nevertheless, the progression and
mechanisms underlying this process are still unclear. Investigating
the molecular mechanism of DOX may help to identify a suitable
strategy for the prevention and treatment of DOX-induced myocardial
injury.
As a member of the papain family, Cathepsin B (CTSB)
is a widely expressed lysosomal cysteine endopeptidase (5). High levels of CTSB are found in
macrophages, osteoclasts and different types of cancer cells,
including lung, colon, prostate, breast and gastric cancer
(6). Moreover, CTSB is also
expressed in cardiomyocytes, and increased CTSB expression levels
and activity in the myocardium are reported to be induced by DOX
(7,8), angiotensin II (9) and isoproterenol (10) and in patients with dilated
cardiomyopathy (11).
Additionally, CTSB is associated with apoptosis (12,13)
and oxidative stress (14,15), which serve key roles in the process
of DOX-induced myocardial injury. Our previous study demonstrated
that CTSB was upregulated in the heart following pressure overload,
and functions as a modulator of the hypertrophic response via
regulating the TNF-α/apoptosis signal-regulating kinase 1
(ASK1)/JNK pathway (9). However,
the mechanism by which CTSB regulates DOX-induced cardiotoxicity
remains unverified. The present study demonstrated that CTSB
exacerbated cardiomyocyte apoptosis and oxidative stress induced by
DOX, and that the underlying mechanism was due to the activation of
NF-κB signalling.
Materials and methods
Cell culture
H9C2 cells were obtained from the Cell Bank of the
Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) containing 10% FBS at 37°C
in a humidified atmosphere with 5% CO2. After culturing
for 24 h, the cells were treated with 1 µM DOX or PBS. The specific
NF-κB inhibitor JSH-23 (10 µmol/l) (16) was administered to the H9C2 cells to
inhibit NF-κB activation for 24 h at 37°C.
Cell counting kit (CCK)-8 assay
H9C2 cell viability was determined via CCK-8 assay
according to the manufacturer's instructions (Beyotime Institute of
Biotechnology). Briefly, the cells were seeded in 96-well plates.
Different concentrations (0.25, 0.50, 1.00, 2.50 or 5.00 µM) of DOX
were used to treat the H9C2 cells for 24 h at 37°C. Then, 10 µl
CCK-8 was added to each well and incubated at 37°C for 1 h. The
optical density values were obtained at 450 nm.
Cell transfection
In order to overexpress CTSB, H9C2 cells were plated
in 6-well plates and transfected with adenovirus (Ad)-CTSB (MOI,
100) or Ad-negative control (NC) for 6 h and then stimulated with 1
µM DOX or PBS for 24 h at 37°C. In order to knock down CTSB
expression levels, H9C2 cells were transfected with small
interfering (si)RNA targeting CTSB (siCTSB; 50 nM) or scrambled
siRNA (50 nM) for 24 h using 1X riboFECT™ CP Reagent according to
the manufacturer's protocol. and then stimulated with 1 µM DOX or
PBS for 24 h. The siCTSB sequence was 5′-GGACGACATGATTAACTAT-3′.
The siNC sequence was 5′-TTCTCCGAACGTGTCACGTdTdT-3′ (Guangzhou
RiboBio Co., Ltd.).
Reverse transcription-quantitative
(RT-q)PCR
RT-qPCR was performed as previously described
(9). Briefly, TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) was used to
extract total RNA. A Transcriptor First-Strand cDNA Synthesis kit
(Roche Diagnostics) was used to reverse transcribe the total RNA
into cDNA. PCR amplifications were quantified using a LightCycler
480 SYBR-Green 1 Master Mix (Roche Diagnostics). The thermocycling
conditions for qPCR were as follows: 95°C for 30 sec; 40 cycles of
95°C for 10 sec, 60°C for 30 sec and 95°C for 15 sec; 60°C for 1
min; 95°C for 15 sec. The results were normalized to GAPDH gene
expression levels. Relative gene expression levels were calculated
using the 2−ΔΔcq method (17). The following primers were used:
GAPDH: Forward, 5′-GACATGCCGCCTGGAGAAAC-3′ and reverse,
5′-AGCCCAGGATGCCCTTTAGT-3′; GP91: Forward,
5′-GACCATTGCAAGTGAACACCC-3′ and reverse,
5′-AAATGAAGTGGACTCCACGCG-3′; P67: Forward,
5′-CGAGGGAACCAGCTGATAGA-3′ and reverse, 5′-CATAGGCACGCTGAGCTTCA-3′;
glutathione peroxidase (GPx): Forward, 5′-GAGAATGGCAAGAATGAAGAG-3′
and reverse, 5′-GAAGGTAAAGAGCGGGTGA-3′.
Western blot analysis
Proteins were extracted from H9C2 cells, and the
concentration was measured by a BCA protein assay kit (Thermo
Fisher Scientific, Inc.) as previously described (9). Protein samples (50 µg) were separated
by 10% SDS-PAGE (Wuhan Servicebio Technology Co., Ltd.) and then
transferred to PVDF Immobilon-P transfer membrane (EMD Millipore).
The membrane was blocked with 5% skimmed milk in Tris-buffered
saline Tween-20 (Sigma-Aldrich) for 1 h at room temperature and
then incubated overnight at 4°C with the indicated primary
antibodies. Primary antibodies against Bax (cat. no. 2772; Cell
Signaling Technology, Inc.), CTSB (cat. no. 31718; Cell Signaling
Technology, Inc.), GAPDH (cat. no. 2118; Cell Signaling Technology,
Inc.), caspase-3 (cat. no. 9662; Cell Signaling Technology, Inc.),
cleaved (c)-caspase-3 (cat. no. 9661; Cell Signaling Technology,
Inc.), superoxide dismutase (SOD)1 (cat. no. ab16831; Abcam), Bcl-2
(cat. no. ab196495; Abcam), SOD2 (cat. no. ab68155; Abcam), NF-κB
p65 (cat. no. ab16502; Abcam), phosphorylated (p)-NF-κB p65 (cat.
no. ab194726; Abcam), IκBα (cat. no. ab7217; Abcam), and p-IκBα
(cat. no. ab133462; Abcam) were used for western blotting. The
dilution of all primary antibodies was 1:1,000. Then the membrane
was incubated with goat anti-rabbit IgG secondary antibody
(1:10,000; cat. no. A21020; Abbkine Scientific Co., Ltd.) for 1 h
at room temperature. The blots were visualised using ECL Plus
(Wuhan Servicebio Technology Co., Ltd.) reagent and a ChemiDoc™
Imaging System (Bio-Rad Laboratories, Inc.). Image Lab software 3.0
(Bio-Rad Laboratories, Inc.) was used to perform
quantification.
Oxidative stress detection
Detection of reactive oxygen species (ROS) in the
H9C2 cells of each group was performed using a ROS Assay kit
according to the manufacturer's instructions (Beyotime Institute of
Biotechnology). Briefly, after treatment, DCFH-DA (1:1,000) was
added to H9C2 cells cultured in serum-free medium for 30 min in the
dark at 37°C, then cells were washed with PBS three times and
observed under a fluorescence microscope at ×200 magnification.
Dihydroethidium (DHE; Beyotime Institute of Biotechnology) was used
to detect the superoxide anion levels in cells. Briefly, 2 µM DHE
was added to the H9C2 cells after treatment, and the cells were
incubated at 37°C for 30 min, then washed with PBS for three
times.
Immunofluorescence staining
Immunofluorescence staining was performed as
previously described (9). Briefly,
following transfection with Ad-CTSB/Ad-NC or siCTSB/siNC and
stimulation with DOX, the cells were fixed, permeabilized and
blocked. Then, cells were incubated with primary antibodies against
cathepsin B and p-NF-κB p65 at 4°C overnight. The next day, the
cells were washed with PBS three times and incubated with Alexa
Fluor 488-conjugated goat anti-rat IgG (1:200; cat. no. A21090;
Invitrogen; Thermo Fisher Scientific, Inc.) for 1 h at 37°C. Then,
the cells were observed with a fluorescence microscope at ×400
magnification.
Apoptosis assessment
TUNEL staining was performed according to the
manufacturer's instructions with a ApopTag® Fluorescein
Direct In Situ Apoptosis Detection Kit (EMD Millipore),
Briefly, Cells were fixed in 1% paraformaldehyde for 10 min at room
temp and post-fixed with precooled ethanol and Acetic acid (2:1)
for 5 min at −20°C. Next, the equilibration buffer was added for
~10 sec at room temperature, followed by 55 µl/5 cm2 of
working Strength TdT enzyme for 1 h at 37°C. The specimens were
then placed in a coplin jar containing stop/wash buffer, agitated
for 15 sec and incubated for 10 min at room temperature. Then
mounting medium containing 0.5–1.0 µg/ml propidium iodide was added
for 1 min at room temperature and the images were captured (>50
fields per coverslip) via fluorescence microscopy at ×200
magnification.
Statistical analysis
SPSS software (version 23.0; IBM Corp.) was used for
analysis. The results are expressed as the mean ± SD of three
independent repeats. Differences between two groups were analysed
via the Student's t-test. One-way ANOVA was used to evaluate
differences between multiple groups, followed by post hoc Tukey's
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
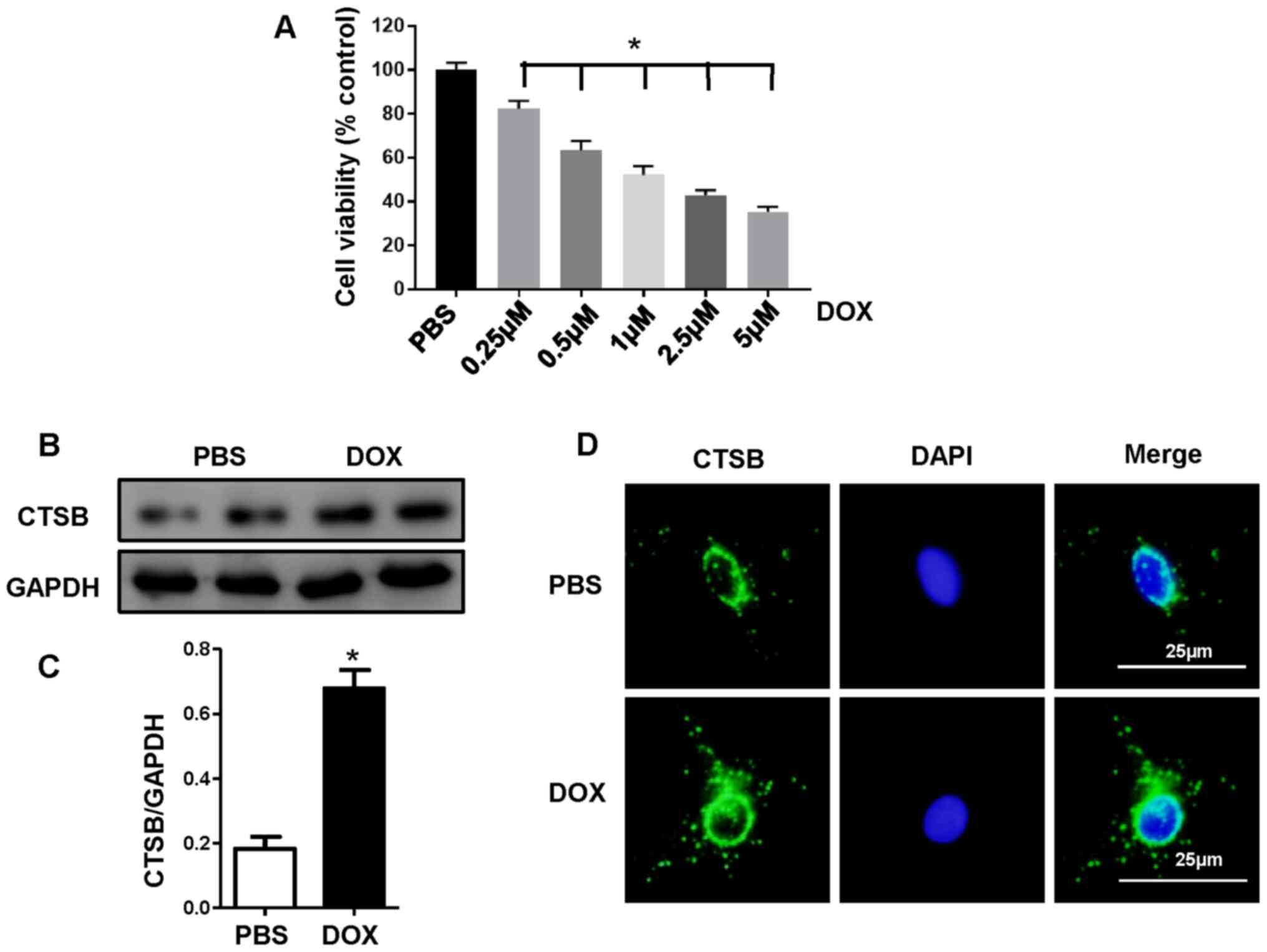
CTSB is upregulated in H9C2 cells
treated with DOX
H9C2 cells were treated with different
concentrations of DOX (0.25, 0.50, 1.00, 2.50 or 5.00 µmol/l), and
cell viability was detected (Fig.
1A). Treatment with DOX at a dose of 1 µmol/l for 24 h
decreased the cell viability to 52.11±4.14%. Thus, further
experiments were performed with 1 µmol/DOX, which was consistent
with the dose used in previous studies (18,19).
In order to investigate whether CTSB is involved in DOX-induced
cardiac injury, CTSB expression levels were assessed in an in
vitro model following treatment with DOX. The western blotting
results showed that CTSB expression levels were increased following
DOX treatment compared with PBS treatment (Fig. 1B and C). Localization of CTSB was
determined via immunofluorescence staining, which demonstrated that
CTSB was distributed in the cytoplasm of cardiomyocytes and
upregulated following DOX treatment (Fig. 1D).
CTSB deficiency attenuates DOX-induced
apoptosis and oxidative stress in H9C2 cells
In order to investigate whether CTSB exerts an
effect on DOX-mediated cardiac injury, CTSB siRNA was used to knock
down CTSB expression levels (Fig. S1A
and B). Cardiomyocyte apoptosis and oxidative stress serve key
roles in DOX-induced cardiotoxicity (18). In the present study, TUNEL staining
(Fig. 2A and B) showed that CTSB
knockdown significantly decreased apoptosis in H9C2 cells treated
with DOX, and the expression levels of the apoptosis-associated
proteins Bax and c-caspase3 also decreased, whereas the expression
levels of Bcl-2 increased after CTSB knockdown treated with DOX
(Fig. 2D-G). In addition, ROS
generation was assessed via DHE, which demonstrated that DOX
treatment resulted in increased oxidative stress in H9C2 cells and
that CTSB knockdown notably inhibited ROS production (Fig. 2B). In addition, western blot
analysis showed that the protein expression levels of SOD1 and SOD2
were also increased in the siCTSB+DOX group compared with the
siNC+DOX group (Fig. 2D, H and I).
CTSB knockdown also decreased the mRNA expression levels of the
NADPH oxidase subunits p67phox and GP91 and increased the
expression levels of GPx in DOX-treated H9C2 cells (Fig. 2J-L).
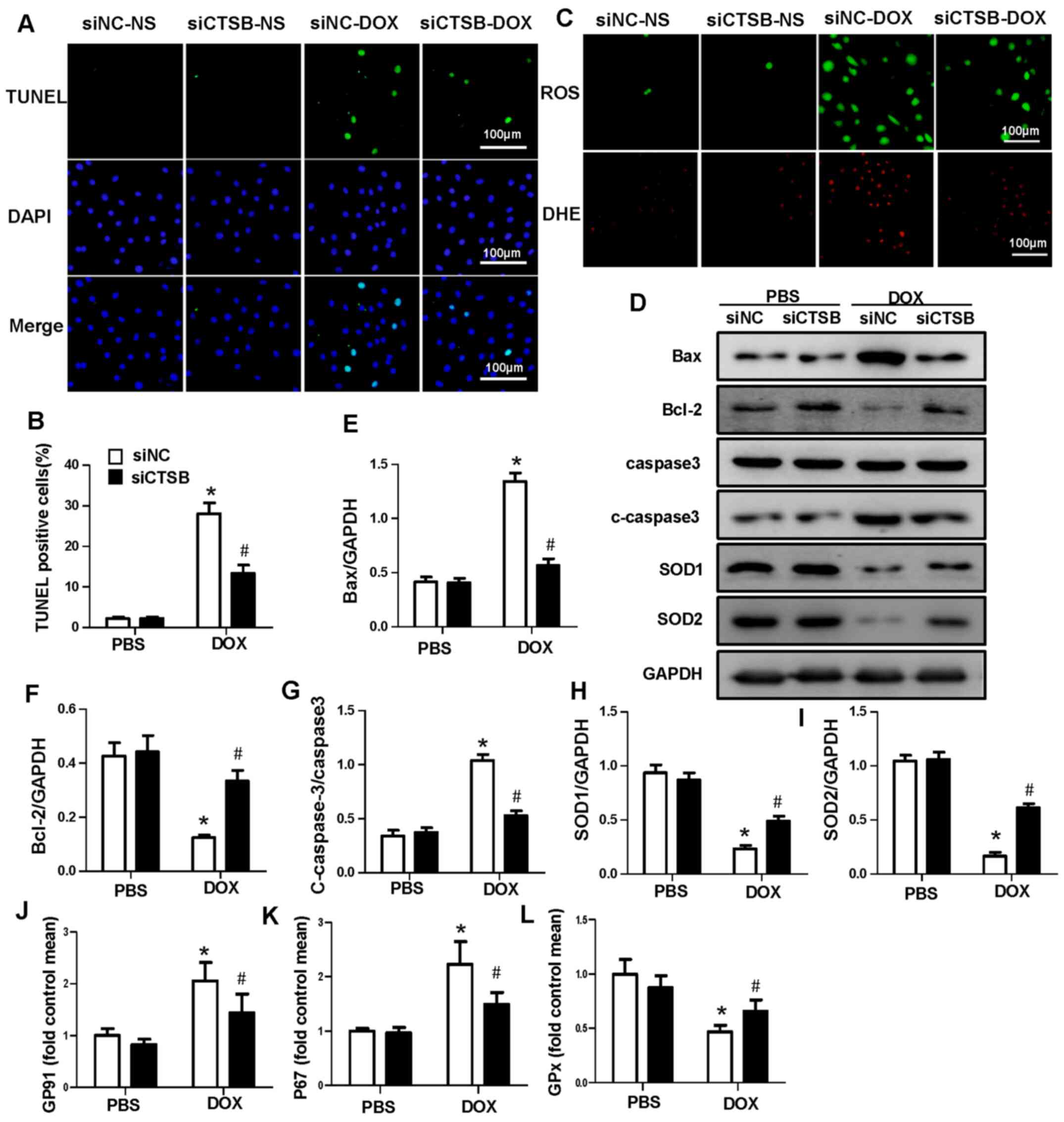 | Figure 2.CTSB deficiency attenuates
DOX-induced apoptosis and oxidative stress in H9C2 cells. (A)
Representative images and (B) quantification of TUNEL staining. (C)
Representative images of ROS and DHE staining. (D) Western blotting
and quantification of expression levels of the apoptosis- and
oxidative stress-associated proteins (E) Bax, (F) Bcl-2, (G)
caspase-3, c-caspase-3, (H) SOD1 and (I) SOD2. mRNA expression
levels of (J) GP91, (K) P67 and (L) GPx. n=6. *P<0.05 vs.
siNC+PBS; #P<0.05 vs. siNC+DOX. CTSB, cathepsin B;
DOX, doxorubicin; ROS, reactive oxygen species; DHE,
dihydroethidium; c-caspase-3, cleaved caspase-3; SOD, superoxide
dismutase; GPx, glutathione peroxidase; siNC, small interfering
negative control. |
CTSB overexpression aggravates
DOX-induced apoptosis and oxidative stress in H9C2 cells
Next, it was investigated whether increased CTSB
levels affected H9C2 apoptosis and oxidative stress in response to
DOX. Ad-CTSB was used to overexpress CTSB in H9C2 cells (Fig. S1C and D). Fluorescence staining
showed that CTSB overexpression exacerbated apoptosis and oxidative
stress in vitro (Fig.
3A-C). The protein expression levels of Bax and c-caspase-3
increased and those of Bcl-2, SOD1 and SOD2 decreased in the
Ad-CTSB+DOX group compared with the Ad-NC+DOX group (Fig. 3D-I). CTSB overexpression also
increased the mRNA expression levels of the NADPH oxidase subunits
p67phox and GP91 and decreased the expression levels of GPx in
DOX-treated H9C2 cells (Fig.
3J-L).
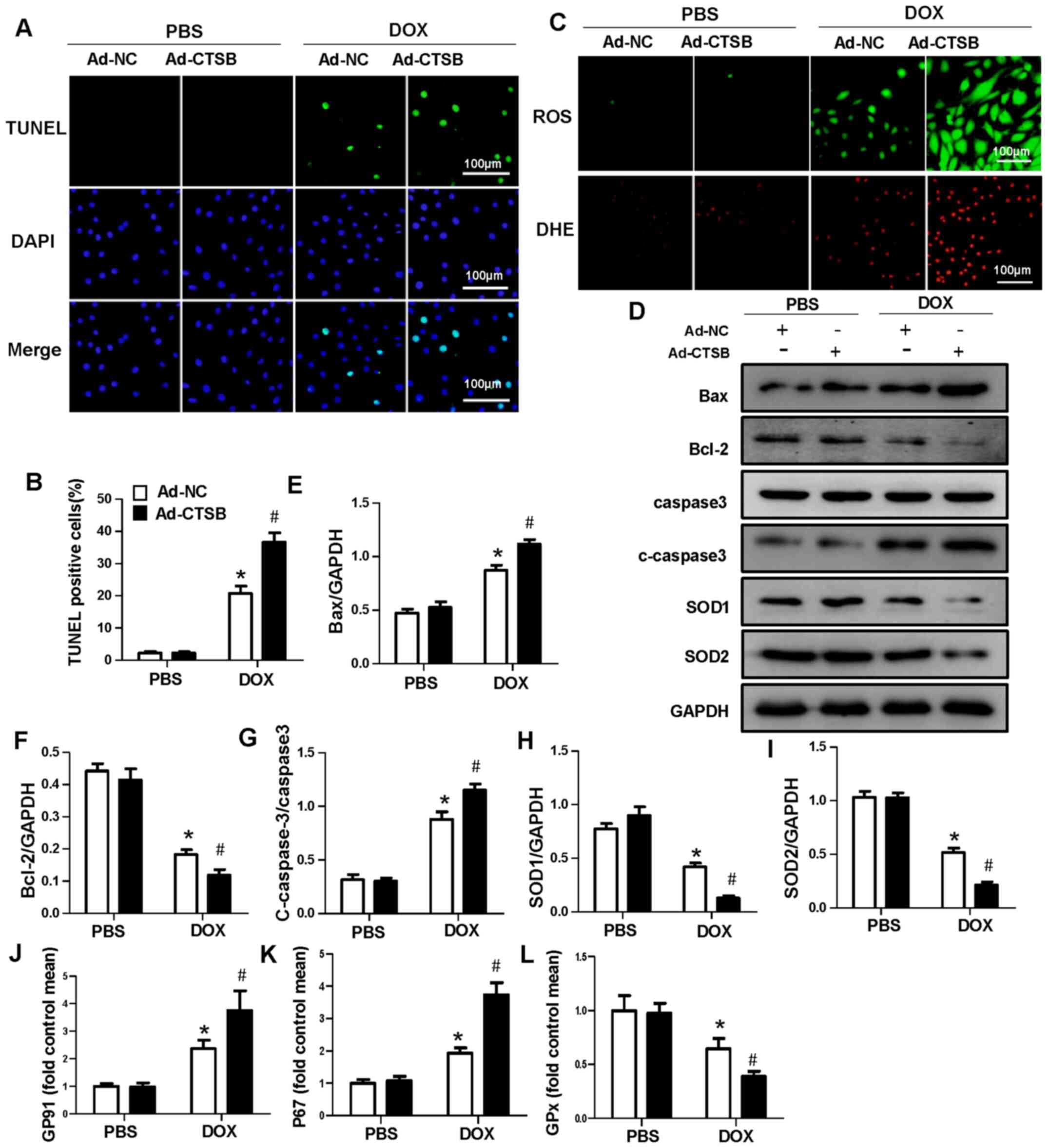 | Figure 3.CTSB overexpression aggravates
DOX-induced apoptosis and oxidative stress in H9C2 cells. (A)
Representative images and (B) quantification of TUNEL staining. (C)
Representative images of ROS and DHE staining. (D) Western blotting
and quantification of expression levels of the apoptosis- and
oxidative stress-associated proteins (E) Bax, (F) Bcl-2, (G)
caspase-3, c-caspase-3, (H) SOD1 and (I) SOD2. mRNA expression
levels of (J) GP91, (K) P67 and (L) GPx. n=6. *P<0.05 vs.
Ad-NC+PBS; #P<0.05 vs. Ad-NC+DOX. CTSB, cathepsin B;
DOX, doxorubicin; ROS, reactive oxygen species; DHE,
dihydroethidium; c-caspase-3, cleaved caspase-3; SOD, superoxide
dismutase; GPx, glutathione peroxidase; Ad-NC, adenovirus-negative
control. |
CTSB mediates activation of the NF-κB
pathway in response to DOX
The NF-κB pathway is associated with the apoptotic
pathway, and it has dual regulatory effects in inhibiting and
promoting apoptosis (20). CTSB
has been found to regulate NF-κB in numerous types of cell
(21,22). Thus, the NF-κB pathway was
investigated. The results showed that DOX treatment notably
enhanced NF-κB activation. CTSB did not affect NF-κB activation at
baseline, but CTSB knockdown decreased NF-κB activation in response
to DOX and decreased the levels of p-NF-κB p65 and p-IκBa.
Moreover, nuclear translocation of p-NF-κB p65 also decreased in
response to DOX following CTSB knockdown (Fig. 4A-C and G). CTSB overexpression
increased NF-κB activation and nuclear translocation following DOX
treatment (Fig. 4D-F and H).
Subsequently, it was determined whether CTSB lost its pro-apoptotic
and pro-oxidative stress effects when NF-κB was inhibited. As
expected, the NF-κB inhibitor JSH-23 mitigated DOX-induced
apoptosis and oxidative stress in CTSB-overexpressing H9C2 cells,
which was reflected by decreased protein expression levels of Bax
and c-caspase-3 and increased protein expression levels of Bcl-2,
SOD1 and SOD2 (Fig. 5A-F).
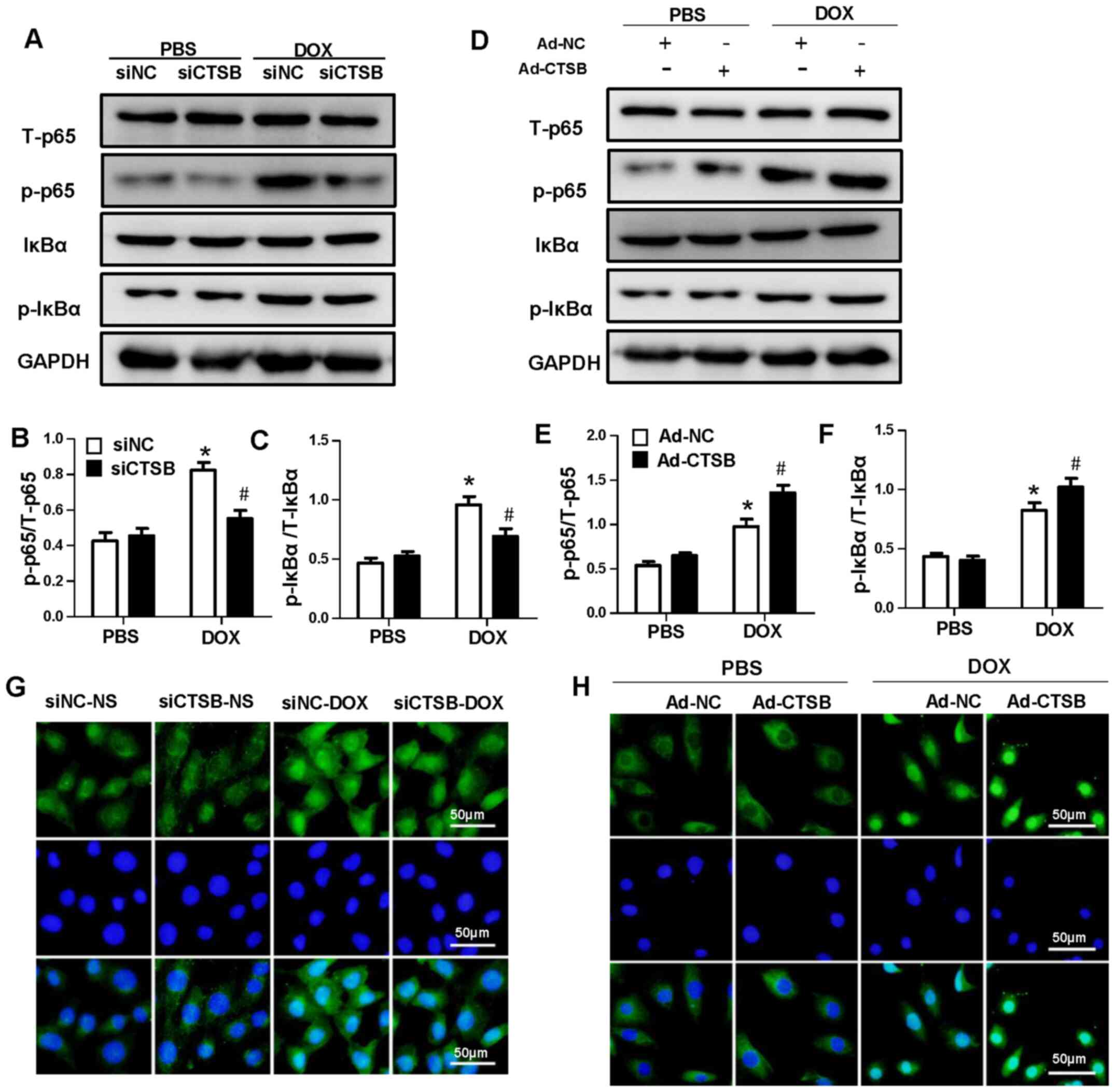 | Figure 4.CTSB mediates the activation of the
NF-κB pathway in response to DOX. (A) Representative western
blotting and quantification analysis of (B) NF-κB p65, p-NF-κB p65,
(C) IκBα and p-IκBα. *P<0.05 vs. siNC+PBS; #P<0.05
vs. siNC+DOX. (D) Representative western blotting and
quantification analysis of (E) NF-κB p65, p-NF-κB p65, (F) IκBα and
p-IκBα. n=6. *P<0.05 vs. Ad-NC+PBS; #P<0.05 vs.
Ad-NC+DOX. Immunofluorescence staining of p-NF-κB p65 nuclear
translocation in cells transfected with (G) siNC, siCTSB, (H) Ad-NC
and Ad-CTSB. CTSB, cathepsin B; DOX, doxorubicin; p-,
phosphorylated; si, small interfering; NC, negative control; Ad,
adenovirus. |
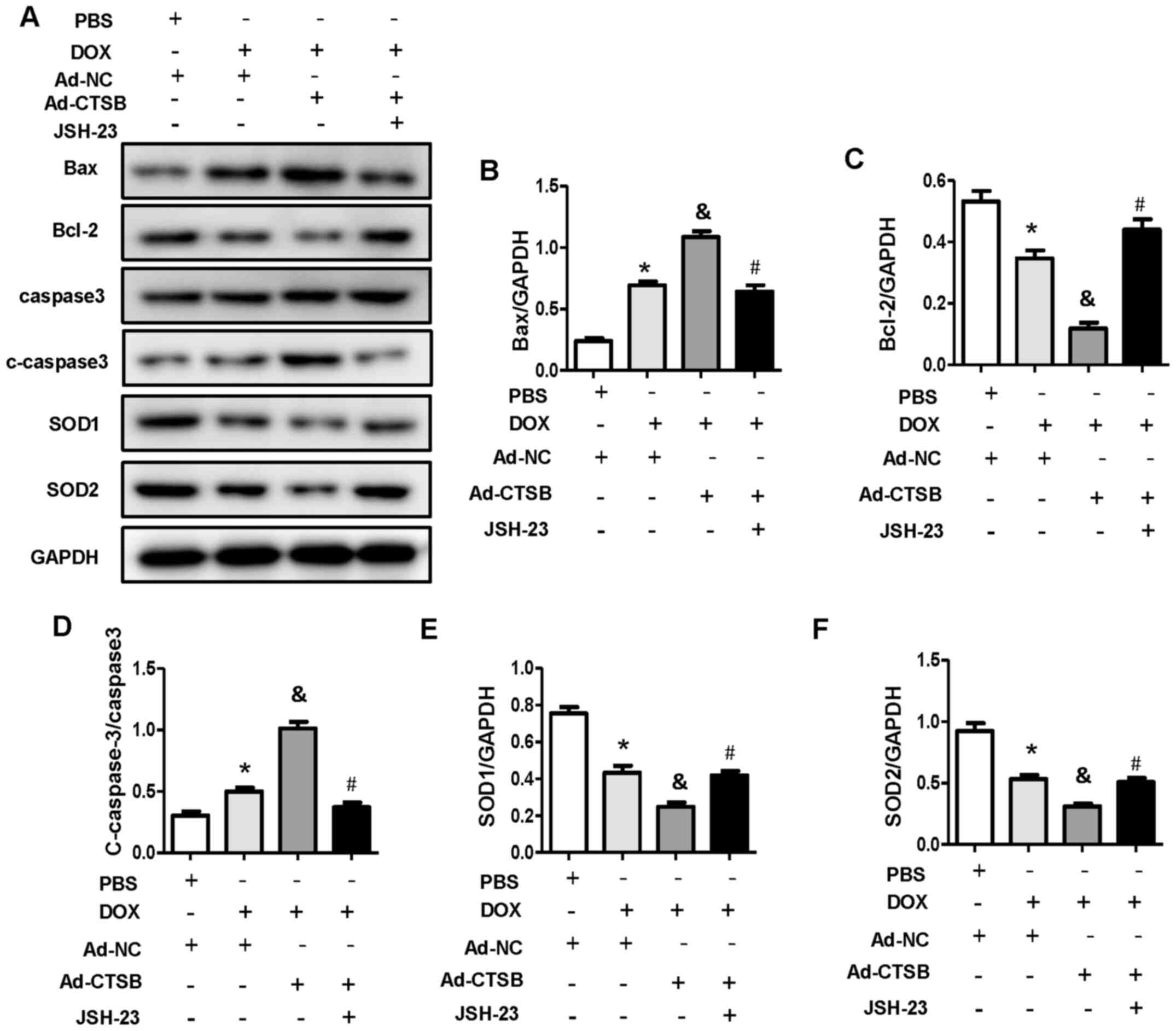 | Figure 5.NF-κB inhibitor blocks the
pro-apoptotic and pro-oxidative stress effects of CTSB
overexpression in response to DOX. (A) Western blotting and
quantification of expression levels of the apoptosis- and oxidative
stress-associated proteins (B) Bax, (C) Bcl-2, (D) caspase-3,
c-caspase-3, (E) SOD1 and (F) SOD2. (n=6). *P<0.05 vs.
Ad-NC+PBS; &P<0.05 vs. Ad-NC+DOX;
#P<0.05 vs. Ad-CTSB+DOX. CTSB, cathepsin B; DOX,
doxorubicin; ROS, reactive oxygen species; DHE, dihydroethidium;
c-caspase-3, cleaved caspase-3; SOD, superoxide dismutase; Ad,
adenovirus; NC, negative control. |
Discussion
In the present study, DOX upregulated the protein
levels of CTSB in H9C2 cells, indicating that CTSB may serve a
certain role in DOX-induced cardiotoxicity. Proteomic profiling of
H9C2 cells in response to DOX treatment showed that CTSB was
upregulated, which may be associated with NF-κB (7), but the exact mechanisms have not
previously been clarified. Thus, Ad-CTSB and siCTSB were used to
transfect H9C2 cells to investigate the specific role of CTSB in
response to DOX, which demonstrated that CTSB overexpression
exacerbated apoptosis and oxidative stress induced by DOX and that
CTSB knockdown reversed the exacerbated phenotype of DOX-induced
H9C2 injury.
Cardiomyocyte apoptosis is a notable contributor to
DOX-induced cell death and can be mediated by different mechanisms,
such as the AMPKα/UCP2 and FDNC5/AKT pathways (18,23,24).
Growing evidence has shown that different proteolytic enzymes are
involved in the regulation of apoptosis (25,26).
CTSB is a protease that is localized in lysosomes under
physiological conditions, and is released from lysosomes into the
cytoplasm and trigger cell apoptosis via different pathways,
including the activation of caspases or the release of
pro-apoptotic factors from the mitochondria in response to certain
stresses (27). The increase in
mitochondrial membrane permeability mediates the release of
cytochrome c, and it has been demonstrated that CTSB induces loss
of mitochondrial membrane potential, triggers the release of
cytochrome c from the mitochondria into the cytosol and activates
caspase-3 in coelomocytes (28).
Additionally, CTSB cleaves the pro-apoptotic Bcl-2 family member
Bid, and truncated-Bid translocates to mitochondria to induce the
release of cytochrome c, which triggers the activation of the
apoptotic cascade (29). CTSB has
been shown to be involved in apoptosis in several systems, such as
hepatocytes, neurons and immune cells, and a
lysosomal-mitochondrial axis theory of cell death has been proposed
to indicate CTSB-regulated apoptosis (30). CTSB is widely expressed in the
myocardium (11). Wu et al
(9) observed that CTSB
participates in the regulation of stress-induced cardiomyocyte
apoptosis, cardiac hypertrophy and remodelling via the
TNF-α/ASK1/JNK pathway. In the present study, CTSB regulated
DOX-induced H9C2 cell apoptosis, which was consistent with previous
studies (7,13).
Due to its high energetic metabolic rate, the heart
has the highest rate of ROS production and is susceptible to
oxidative stress-associated injury. Additionally, the heart has
lower levels of antioxidants and total antioxidant enzyme activity
than other organs (31). Cardiac
oxidative stress is associated with fibrosis, hypertrophy and
decreased cardiac performance and contractility, which leads to
severe cardiac dysfunction and potentially fatal cardiac events
(32). CTSB mediates the
regulation of oxidative stress (14). Inhibition of CTSB activity
maintains the function of mitochondria and decreases the generation
of ROS during in vitro ageing of oocytes (33). Genetic ablation of CTSB in mice
significantly decreases the generation of ROS during
neuroinflammation and improves cognitive impairment during ageing.
In cultured microglia, inhibition of CTSB significantly decreases
mitochondria-derived ROS and proinflammatory mediators induced by
L-leucyl-L-leucine methyl ester (LLOMe), which is a
lysosome-destabilizing agent (34). Overexpression of CTSB in microglia
following treatment with LLOMe increases generation of ROS and
proinflammatory mediators via impairment of mtDNA biosynthesis
(34). CTSB regulates the
expression levels of collagens III and IV via the toll-like
receptor 2/NF-κB pathway and subsequent oxidative damage in
fibroblasts (21). The present
study investigated the association between CTSB and ROS in H9C2
cells and found that CTSB deficiency significantly decreased ROS
generation and that CTSB overexpression increased ROS generation in
DOX-induced H9C2 cytotoxicity.
The present study has demonstrated that CTSB
regulates DOX-induced H9C2 cell apoptosis and oxidative stress. The
underlying mechanism by which CTSB participates in DOX-induced
cardiac injury was further investigated. NF-κB is a pleiotropic
transcription factor that is present in almost all types of cell
and is involved in numerous biological processes, such as
inflammation, immunity, differentiation, cell growth, tumorigenesis
and apoptosis (20). CTSB is
responsible for NF-κB activation (35,36).
CA-074Me, a specific CTSB inhibitor, prevents the activation of
NF-κB via autophagic-dependent pathways in cultured microglia
(35). CTSB inhibition decreases
nuclear p65-NF-κB- and κB-dependent gene expression levels
following lipopolysaccharide or TNF stimulation via enhancing
sirtuin 1 expression levels in primary parenchymal and
non-parenchymal hepatic cell types and cell lines (36). Activation of the NF-κB pathway
serves a key role in the pathophysiology of multiple types of
injury factor-associated cardiac dysfunction and cardiomyopathy
(37,38). In the present study, DOX induced
phosphorylation of IκBα and translocation of p65 NF-κB to the
nucleus in H9C2 cells. Moreover, CTSB overexpression increased the
phosphorylation of IκBα and the nuclear translocation of p65 NF-κB,
and CTSB knockdown decreased the activation of the NF-κB pathway.
The NF-κB inhibitor JSH-23 blocked the pro-apoptotic and
pro-oxidative stress effects of CTSB overexpression in response to
DOX. Thus, the present investigation indicated that CTSB may
mediate NF-κB activation to regulate H9C2 cell apoptosis and
oxidative stress. However, NF-κB is also sensitive to ROS; it has
been proven that ROS activate IKK, thus promoting the activation of
NF-κB (39). ROS and NF-κB
activation may mutually regulate the mechanism of DOX-induced
cardiotoxicity.
In conclusion, CTSB may be a potential therapeutic
agent for the treatment of DOX-induced cardiotoxicity.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National
Natural Science Foundation of China (grant no. 81530012), the
National Key R&D Program of China (grant no. 2018YFC1311300),
the Development Center for Medical Science and Technology National
Health and Family Planning Commission of the People's Republic of
China (the prevention and control project of cardiovascular
disease, grant no. 2016ZX-008-01), the Fundamental Research Funds
for the Central Universities (grant no. 2042018kf1032), and the
National Natural Science Foundation of Hubei Province (grant no.
2017CFB320).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
CL and ZC conceptualised the study design. LZ
designed the experiments. CL analysed the data. CL, ZC, TH and QY
performed the experiments. CL drafted the manuscript. LZ and ZC
reviewed and revised the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rivankar S: An overview of doxorubicin
formulations in cancer therapy. J Cancer Res Ther. 10:853–858.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Swain SM, Whaley FS and Ewer MS:
Congestive heart failure in patients treated with doxorubicin: A
retrospective analysis of three trials. Cancer. 97:2869–2879. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carvalho FS, Burgeiro A, Garcia R, Moreno
AJ, Carvalho RA and Oliveira PJ: Doxorubicin-induced
cardiotoxicity: From bioenergetic failure and cell death to
cardiomyopathy. Med Res Rev. 34:106–135. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shabalala S, Muller CJF, Louw J and
Johnson R: Polyphenols, autophagy and doxorubicin-induced
cardiotoxicity. Life Sci. 180:160–170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mort JS, Buttle DJ and Cathepsin B: Int J
Biochem Cell Biol. 29:715–720. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aggarwal N and Sloane BF: Cathepsin B:
Multiple roles in cancer. Proteomics Clin Appl. 8:427–437. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bao GY, Wang HZ, Shang YJ, Fan HJ, Gu ML,
Xia R, Qin Q and Deng AM: Quantitative proteomic study identified
cathepsin B associated with doxorubicin-induced damage in H9c2
cardiomyocytes. Biosci Trends. 6:283–287. 2012.PubMed/NCBI
|
|
8
|
Moreira AC, Branco AF, Sampaio SF,
Cunha-Oliveira T, Martins TR, Holy J, Oliveira PJ and Sardão VA:
Mitochondrial apoptosis-inducing factor is involved in
doxorubicin-induced toxicity on H9c2 cardiomyoblasts. Biochim
Biophys Acta. 1842:2468–2478. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu QQ, Xu M, Yuan Y, Li FF, Yang Z, Liu Y,
Zhou MQ, Bian ZY, Deng W, Gao L, et al: Cathepsin B deficiency
attenuates cardiac remodeling in response to pressure overload via
TNF-α/ASK1/JNK pathway. Am J Physiol Heart Circ Physiol.
308:H1143–H1154. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brindha E and Rajasekapandiyan M:
Preventive effect of phytic acid on lysosomal hydrolases in normal
and isoproterenol-induced myocardial infarction in Wistar rats.
Toxicol Mech Methods. 25:150–154. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ge J, Zhao G, Chen R, Li S, Wang S, Zhang
X, Zhuang Y, Du J, Yu X, Li G and Yang Y: Enhanced myocardial
cathepsin B expression in patients with dilated cardiomyopathy. Eur
J Heart Fail. 8:284–289. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sendler M, Maertin S, John D, Persike M,
Weiss FU, Krüger B, Wartmann T, Wagh P, Halangk W, Schaschke N, et
al: Cathepsin B activity initiates apoptosis via digestive protease
activation in pancreatic acinar cells and experimental
pancreatitis. J Biol Chem. 291:14717–14731. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hsu SF, Hsu CC, Cheng BC and Lin CH:
Cathepsin B is involved in the heat shock induced cardiomyocytes
apoptosis as well as the anti-apoptosis effect of HSP-70.
Apoptosis. 19:1571–1580. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bai H, Yang B, Yu W, Xiao Y, Yu D and
Zhang Q: Cathepsin B links oxidative stress to the activation of
NLRP3 inflammasome. Exp Cell Res. 362:180–187. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liow KY and Chow SC: The cathepsin B
inhibitor z-FA-CMK induces cell death in leukemic T cells via
oxidative stress. Naunyn Schmiedebergs Arch Pharmacol. 391:71–82.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang XW, Zhang FX, Yang F, Ding ZF,
Agarwal N, Guo ZK and Mehta JL: Effects of linagliptin and
liraglutide on glucose- and angiotensin II-induced collagen
formation and cytoskeleton degradation in cardiac fibroblasts in
vitro. Acta Pharmacol Sin. 37:1349–1358. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang X, Hu C, Kong CY, Song P, Wu HM, Xu
SC, Yuan YP, Deng W, Ma ZG and Tang QZ: FNDC5 alleviates oxidative
stress and cardiomyocyte apoptosis in doxorubicin-induced
cardiotoxicity via activating AKT. Cell Death Differ. 27:540–555.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang X, Zhu JX, Ma ZG, Wu HM, Xu SC, Song
P, Kong CY, Yuan YP, Deng W and Tang QZ: Rosmarinic acid alleviates
cardiomyocyte apoptosis via cardiac fibroblast in
doxorubicin-induced cardiotoxicity. Int J Biol Sci. 15:556–567.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oeckinghaus A, Hayden MS and Ghosh S:
Crosstalk in NF-κB signaling pathways. Nat Immunol. 12:695–708.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li X, Wu Z, Ni J, Liu Y, Meng J, Yu W,
Nakanishi H and Zhou Y: Cathepsin B regulates collagen expression
by fibroblasts via prolonging TLR2/NF-κB activation. Oxid Med Cell
Longev. 2016:78942472016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sendler M, Weiss FU, Golchert J, Homuth G,
van den Brandt C, Mahajan UM, Partecke LI, Döring P, Gukovsky I,
Gukovskaya AS, et al: Cathepsin B-mediated activation of
trypsinogen in endocytosing macrophages increases severity of
pancreatitis in mice. Gastroenterology. 154:704–718.e10. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang H, Tao A, Song J, Liu Q, Wang H and
Rui T: Doxorubicin-induced cardiomyocyte apoptosis: Role of
mitofusin 2. Int J Biochem Cell Biol. 88:55–59. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu C, Zhang X, Wei W, Zhang N, Wu H, Ma Z,
Li L, Deng W and Tang Q: Matrine attenuates oxidative stress and
cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via
maintaining AMPK α/UCP2 pathway. Acta Pharm Sin B. 9:690–701. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Turk B and Stoka V: Protease signalling in
cell death: Caspases versus cysteine cathepsins. FEBS Lett.
581:2761–2767. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cohen GM: Caspases: The executioners of
apoptosis. Biochem J. 326:1–16. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chwieralski CE, Welte T and Bühling F:
Cathepsin-regulated apoptosis. Apoptosis. 11:143–149. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen H, Lv M, Lv Z, Li C, Zhang W, Zhao X,
Duan X, Jin C, Xiong J, Xu F and Li Y: Divergent roles of three
cytochrome c in CTSB-modulating coelomocyte apoptosis in
Apostichopus japonicus. Dev Comp Immunol. 76:65–76. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tardy C, Codogno P, Autefage H, Levade T
and Andrieu-Abadie N: Lysosomes and lysosomal proteins in cancer
cell death (new players of an old struggle). Biochim Biophys Acta.
1765:101–125. 2006.PubMed/NCBI
|
|
30
|
Terman A, Gustafsson B and Brunk UT: The
lysosomal-mitochondrial axis theory of postmitotic aging and cell
death. Chem Biol Interact. 163:29–37. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Münzel T, Camici GG, Maack C, Bonetti NR,
Fuster V and Kovacic JC: Impact of oxidative stress on the heart
and vasculature: Part 2 of a 3-part series. J Am Coll Cardiol.
70:212–229. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Faria A and Persaud SJ: Cardiac oxidative
stress in diabetes: Mechanisms and therapeutic potential. Pharmacol
Ther. 172:50–62. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liang S, Jiang H, Shen XH, Zhang JB and
Kim NH: Inhibition of cathepsin B activity prevents deterioration
in the quality of in vitro aged porcine oocytes. Theriogenology.
116:103–111. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ni J, Wu Z, Stoka V, Meng J, Hayashi Y,
Peters C, Qing H, Turk V and Nakanishi H: Increased expression and
altered subcellular distribution of cathepsin B in microglia induce
cognitive impairment through oxidative stress and inflammatory
response in mice. Aging Cell. 18:e128562019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ni J, Wu Z, Peterts C, Yamamoto K, Qing H
and Nakanishi H: The critical role of proteolytic relay through
cathepsins B and E in the phenotypic change of
microglia/macrophage. J Neurosci. 35:12488–12501. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
de Mingo Á, de Gregorio E, Moles A,
Tarrats N, Tutusaus A, Colell A, Fernandez-Checa JC, Morales A and
Mari M: Cysteine cathepsins control hepatic NF-κB-dependent
inflammation via sirtuin-1 regulation. Cell Death Dis. 7:e24642016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang XQ, Tang R, Li L, Szucsik A, Javan
H, Saegusa N, Spitzer KW and Selzman CH: Cardiomyocyte-specific p65
NF-κB deletion protects the injured heart by preservation of
calcium handling. Am J Physiol Heart Circ Physiol. 305:H1089–H1097.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang Z, Gao L, Xiao L, Kong L, Shi H, Tian
X and Zhao L: Bakuchiol protects against pathological cardiac
hypertrophy by blocking NF-κB signaling pathway. Biosci Rep.
38:BSR201810432018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hashimoto Y, Kakegawa H, Narita Y, Hachiya
Y, Hayakawa T, Kos J, Turk V and Katunuma N: Significance of
cathepsin B accumulation in synovial fluid of rheumatoid arthritis.
Biochem Biophys Res Commun. 283:334–339. 2001. View Article : Google Scholar : PubMed/NCBI
|