Introduction
Diabetic patients have an notably increased
incidence of heart failure according to the Framingham study
(1); however, the molecular
mechanisms underlying the pathogenesis of diabetic cardiomyopathy
are not completely understood. Among the hypothesized mechanisms,
hyperglycemia-induced oxidative stress is recognized as a critical
participant in the pathogenesis and progression of diabetes
(2). Increased levels of oxidative
stress lead to harmful modifications of macromolecules, including
DNA, proteins and lipids (3),
which can result in cardiomyocyte apoptosis, hypertrophy and
fibrosis, ultimately leading to cardiac remodeling and dysfunction
(3). Therefore, developing an
effective treatment to suppress increased oxidative stress levels
and subsequent cardiomyocyte injury in patients with diabetes is
important.
Metformin is widely used as a first-line
hypoglycemic drug (4). Previous
studies identified cardioprotective effects of metformin beyond its
antihyperglycemic effects (5–7). The
United Kingdom Prospective Diabetes Study demonstrated that early
and intensive metformin intervention could reduce the incidence of
myocardial infarction and increase the survival rate in patients
with type 2 diabetes (5).
Metformin exerts cardioprotective effects partly by suppressing
high glucose (HG)-induced excessive reactive oxygen species
production and inflammatory responses (8); however, the exact underlying
mechanisms are not completely understood.
AMP-activated protein kinase (AMPK), a key mediator
of the downstream effects of metformin, can be activated by
cytokines, hormones and oral hypoglycemic agents that are commonly
used to treat type 2 diabetes (9).
Activated AMPK promotes the production of ATP by regulating glucose
and fatty acid metabolism (9),
indicating that AMPK may serve as an innate survival mechanism for
the heart. For example, AMPK activation during myocardial ischemia
reduces the infarct size in the hearts of diabetic model rats
(10). In addition, upregulation
of AMPK signaling in the vasculature improves microvascular
function in the hearts of diabetic model mice (11).
Based on its role in regulating cellular energy
status, AMPK is a major regulator of mitochondrial function.
Hydroxytyrosol, a natural antioxidant, reduces oxidative stress and
improves mitochondrial function, presumably by activating AMPK
signaling in the brain of db/db mice (12). Therefore, it was hypothesized that
AMPK may serve as a critical modulator of mitochondrial biogenesis
and function, and enhance resistance to oxidative stress. However,
the exact role and mechanisms underlying AMPK in the
cardioprotective effects of metformin in response to oxidative
stress are not completely understood.
Mitochondria are the primary energy generating
organelles, but are also the main source of ROS (13). NADH: Ubiquinone oxidoreductase
subunit A13 (NDUFA)13 is an indispensable assembly factor of
complex I (14). The primary role
of NDUFA13 in the antioxidant effect of resveratrol has been
previously reported (15).
Downregulated NDUFA13 expression increases mitochondrial ROS
generation in H9C2 cardiomyocytes (16), and sustained high glucose decreases
NDUFA13 expression levels (15).
Based on the aforementioned studies, it was
hypothesized that NDUFA13 may be associated with alleviating
oxidative stress in diabetic cardiomyopathy, and metformin may
prevent diabetic myocardial injury by suppressing
hyperglycemia-induced ROS overproduction. The present study aimed
to investigate whether the protective effects of metformin were
mediated by promoting mitochondrial biogenesis and NDUFA13
expression. Additionally, the exact role of the AMPK signaling
pathway in metformin-induced effects was investigated.
Materials and methods
Cell culture and treatments
H9C2 cells (The Cell Bank of Type Culture Collection
of the Chinese Academy of Sciences) were cultured in DMEM (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and 100 mg/ml
streptomycin at 37°C in a humidified incubator with 5%
CO2 and 95% air.
H9C2 cells cultured to approximately 80% confluence
were firstly incubated with various concentrations of glucose (5.5,
15, 25, 33.3 40 and 50 mM) for 36 h at 37°C. Cells treated with
various concentrations of glucose and mannitol (Abcam) were then
incubated for 12, 24, 36 and 48 h at 37°C. These included 5.5 and
33.3 mM glucose and 25 mM glucose plus 8.3 mM mannitol. According
to changes in cell viability, 5.5 mM glucose was selected to mimic
normal conditions and 33.3 mM glucose was selected to mimic the
diabetic condition. Subsequently, cells were treated with low
concentrations of metformin (0.5 or 1 mM; Sigma-Aldrich; Merck
KGaA) for 36 h at 37°C. For subsequent experiments, 1 mM metformin
was used, which was proven to be an effective in previous studies
(17,18). The cardiomyocytes were divided into
the following experimental groups: i) Control, medium containing
5.5 mM glucose; ii) HG, medium containing 33.3 mM glucose; iii)
mannitol, medium containing 25 mM glucose + 8.3 mM mannitol; iv)
metformin 0.5 mM, medium containing 33.3 mM glucose + 0.5 mM
metformin; v) Met 1, medium containing 33.3 mM glucose + 1 mM
metformin; and vi) Compound C, medium containing 33.3 mM glucose +
1 mM metformin + 10 µM Compound C (Sigma-Aldrich; Merck KGaA).
Cell Counting Kit-8 (CCK-8) assay for
cell viability
Cell viability was assessed using a CCK-8 assay kit
(Shanghai Yeasen Biotechnology Co., Ltd.) according to the
manufacturer's protocol. H9C2 cells were seeded (5×104
cells/well) into 96-well plates. Subsequently, CCK-8 reagent was
added to each well and incubated for 1 h at 37°C. The absorbance of
each well was measured at a wavelength of 450 nm using a microplate
reader (BioTek Instruments, Inc.).
Lactate dehydrogenase (LDH)
release
Cell death was assessed using an LDH cytotoxicity
assay kit (Shanghai Yeasen Biotechnology Co., Ltd.) according to
the manufacturer's instructions. Following treatment for 36 h, the
cell medium was collected. The absorbance of each sample was
measured at a wavelength of 450 nm using a microplate reader.
Measurement of ROS, malondialdehyde
(MDA) and superoxide dismutase (SOD)
Intracellular ROS levels were determined using a ROS
assay kit (Shanghai Yeasen Biotechnology Co., Ltd.) according to
the manufacturer's protocol. Dichlorofluorescein fluorescence was
detected using an Olympus IX71 fluorescence microscope (Olympus
Corporation; magnification, ×200). MDA levels were assessed using
an MDA assay kit (Beyotime Institute of Biotechnology) according to
the manufacturer's instructions. The absorbance of each sample was
measured at a wavelength of 535 nm using a microplate reader. SOD
activity was measured using a total SOD assay kit (Shanghai Yeasen
Biotechnology Co., Ltd.) according to the manufacturer's protocol.
The absorbance of each sample was measured at a wavelength of 450
nm using a microplate reader.
Western blotting
The H9c2 cells were lysed with RIPA buffer (Beyotime
Institute of Biotechnology) for 45 min at 4°C. Total protein was
quantified using a bicinchoninic acid assay kit (Beyotime Institute
of Biotechnology). Equal amounts of protein were separated via
10–12% SDS-PAGE and transferred electrophoretically to PVDF
membranes (Invitrogen; Thermo Fisher Scientific, Inc.). The
membranes were blocked with Tris buffer (Beyotime Institute of
Biotechnology) containing 0.1% Tween-20 (TBST) in 5% milk for 1 h
at room temperature and then incubated overnight with primary
antibodies targeted against: NDUFA13 (17 kD;1:1,000; cat. no.
ab110240; Abcam), AMPK (62 KD;1:1,000; cat. no. 5831; Cell
Signaling Technology, Inc.), phosphorylated (p)-AMPK (62
KD;1:1,000; cat. no. 50081; Cell Signaling Technology, Inc.) and
GAPDH (37 KD;1:10,000; cat. no. 5174; Cell Signaling Technology,
Inc.). Following primary incubation, the membranes were incubated
with a horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody (1:10,000; cat. no. HA1001; HuaBio Inc.) for 2 h at room
temperature. Protein bands were visualized using an enhanced
chemiluminescence system (EMD Millipore). ImageJ software (version
1.41; National Institutes of Health) was used to quantify the bands
of each protein and GAPDH was used as the loading control.
Immunofluorescence assay
Cells were fixed with 4% paraformaldehyde for 30 min
at room temperature and permeabilized with 0.1% Triton X-100 for 10
min. Subsequently, cells were blocked with 3% bovine serum albumin
(Beyotime Institute of Biotechnology) for 2 h at room temperature
and incubated with an anti-NDUFA13 primary antibody (1:50; cat. no.
ab110240; Abcam) overnight at 4°C. The slides were then washed with
phosphate-buffered saline (PBS, Beyotime Institute of
Biotechnology) and 0.1% Tween-20 (PBST) and incubated with a Texas
Red/Alexa fluor-conjugated secondary antibody for 1 h at room
temperature. The slides were mounted using mounting medium,
counterstained with 6-diamidino-2-phenylindole (Invitrogen; Thermo
Fisher Scientific, Inc.) for 10 min at room temperature and
observed using an IX71 microscope (Olympus Corporation;
magnification, ×200) and Image Pro Plus 3.0 software (Media
Cybernetics, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted using TRIzol®
reagent (Thermo Fisher Scientific, Inc.). Total RNA was reverse
transcribed into cDNA at 42°C for 1 h then 90°C for 5 min using a
cDNA synthesis kit (Takara Biotechnology Co., Ltd.). Subsequently,
qPCR was performed using SYBR green PCR Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The following
thermocycling conditions were used for the qPCR: Initial
denaturation at 95°C for 2 min; and 40 cycles of 95°C for 15 sec
and 60°C for 1 min. The sequences of the primers used for qPCR are
presented in Table I. mRNA
expression levels were quantified using the 2−∆∆Cq
method (19) and normalized to the
internal reference gene β-actin.
 | Table I.Sequences of primers used for reverse
transcription-quantitative PCR. |
Table I.
Sequences of primers used for reverse
transcription-quantitative PCR.
| Gene | Sequence (5′→3′) | Product size
(bp) |
|---|
| β-actin | F:
GCGTCCACCCGCGAGTACAA | 118 |
|
| R:
ACATGCCGGAGCCGTTGTCG |
|
| NDUFA1 | F:
TGCTGCCGGAAGAGCGGTGA | 189 |
|
| R:
TCCTTGCCCCCGTTGGTGAACT |
|
| NDUFA2 | F:
ACTGAGGACTGAACAAGCCCACCA | 223 |
|
| R:
GCGACATCCCAGCGGGTAGC |
|
| NDUFA13 | F:
CTACTGGAGAATAATGAGGTGGAAC | 175 |
|
| R:
CCAGTTGGGCACATCTTTCA |
|
| Mn-SOD | F:
GTGTCTGTGGGAGTCCAAGG | 149 |
|
| R:
TGCTCCCACACATCAATCCC |
|
| PGC-1α | F:
GGGGCACATCTGTTCTTCCA | 156 |
|
| R:
GCTTGACTGGGATGACCGAA |
|
| NRF1 | F:
ACACAGCATAGCCCATCTCG | 226 |
|
| R:
GGTCATTTCACCGCCCTGTA |
|
| NRF2 | F:
AGCAAGACTTGGGCCACTTA | 112 |
|
| R:
TCTGGCTTCTTGCTCTTGGG |
|
Statistical analysis
Data are expressed as the mean ± standard deviation
of at least three independent experiments. Comparisons among
multiple groups were analyzed using one-way ANOVA followed by
Tukey's post hoc test. Statistical analyses were performed using
GraphPad Prism software (version 7; GraphPad Software, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
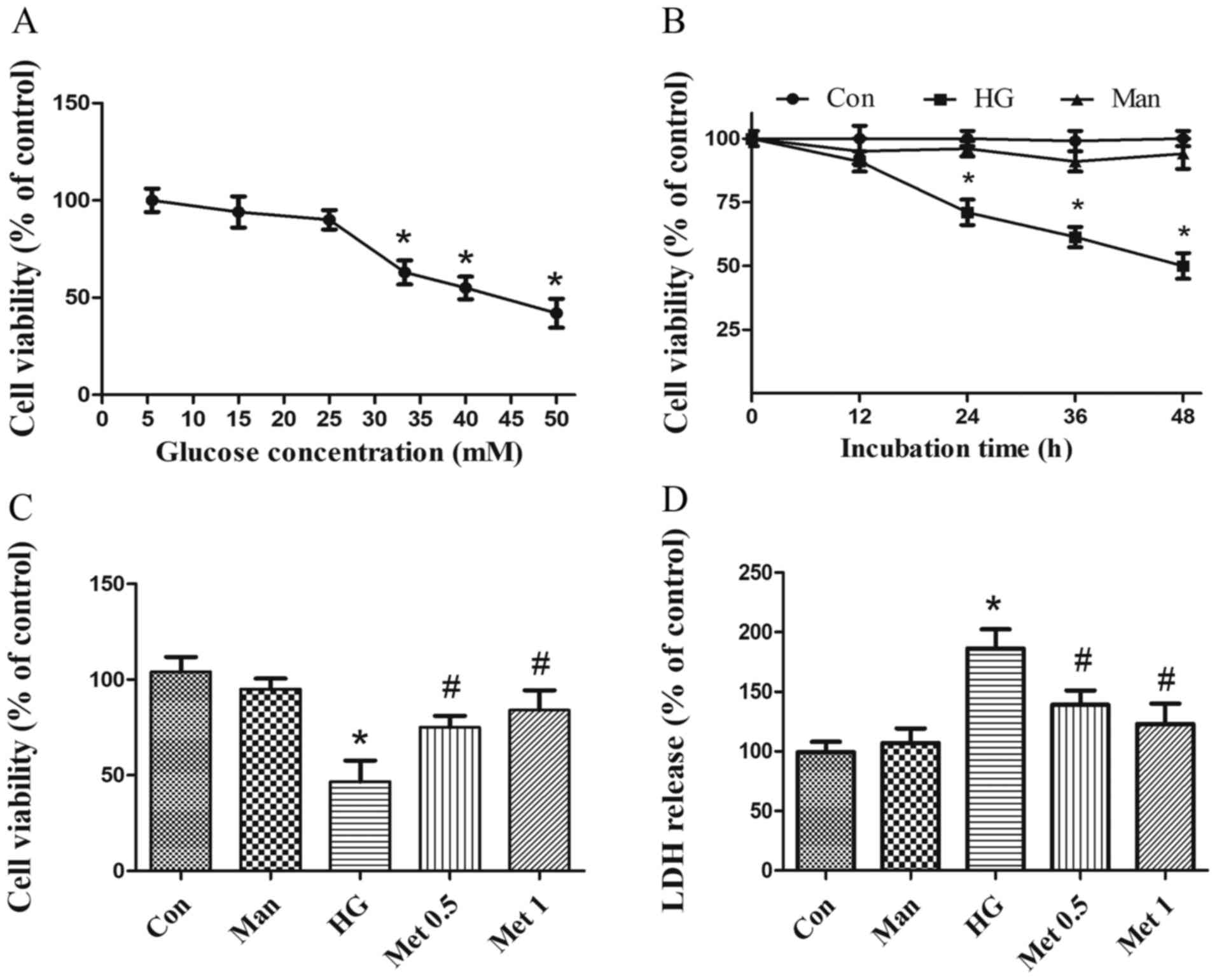
Metformin protects H9C2 cells from
HG-induced cytotoxicity
To investigate the effect of metformin on
cardiomyocyte survival in various concentrations of glucose (5.5–50
mM), H9C2 cell viability was assessed. Cell viability was
significantly decreased following incubation with 33.3 mM glucose
(Fig. 1A) for at least 24 h
(Fig. 1B) compared with the
control group. Subsequently, cardiomyocytes were pretreated with
metformin and then incubated with 33.3 mM glucose for 36 h.
Metformin (0.5 and 1 mM) significantly increased cell viability
(Fig. 1C) and decreased LDH
release (Fig. 1D) under HG
conditions compared with the HG group. The results indicated that
metformin rescued cardiomyocytes from HG-induced injury. Mannitol
was used as an osmotic control and did not mimic the effects of
33.3 mM glucose (Fig. 1). For
subsequent experiments, 33.3 mM glucose was used to induce a HG
situation and 1 mM metformin was applied, unless stated
otherwise.
Clinically, the median plasma concentration of
metformin is 330 µM (20);
however, the concentration of metformin is several times higher in
tissues compared with in the blood (20). Therefore, intracellular metformin
concentrations are 10–15% of the drug present in the medium
(21), meaning the dose used in
the present study was higher than the therapeutic dose.
Metformin suppresses HG-induced
oxidative stress in H9C2 cells
Markers of oxidative stress, including ROS, MDA and
SOD, were detected. Compared with the control and mannitol groups,
ROS and MDA levels were significantly increased in the high glucose
group. However, metformin pretreatment significantly decreased ROS
(Fig. 2A and B) and MDA (Fig. 2C) levels compared with the HG
group. By contrast, HG significantly inhibited SOD activity
compared with the control and mannitol groups, whereas metformin
reversed HG-mediated inhibition of SOD activity in H9C2 cells
(Fig. 2D).
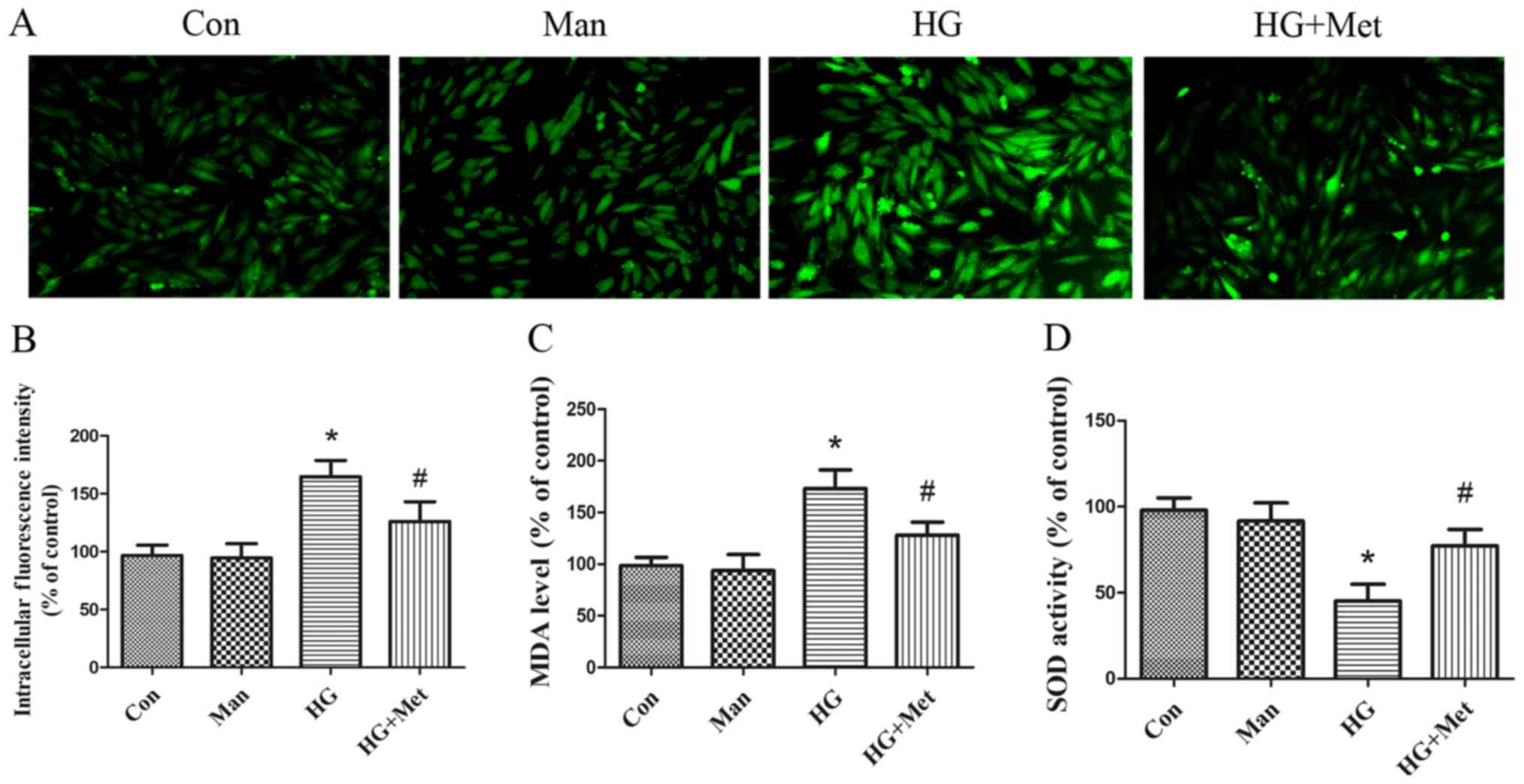 | Figure 2.Effects of metformin (1 mM) on high
glucose-induced oxidative stress in H9C2 cells. Cells were divided
into four groups: i) Con, normal medium; ii) Man, 33.3 mM Man; iii)
HG, 33.3 mM glucose; and iv) HG + Met, 33.3 mM glucose pretreated
with 1 mM Met. ROS levels were detected by (A) dichlorofluorescein
fluorescence intensity and (B) quantified. Magnification, ×200,
Scale bars=50 µm. (C) MDA and (D) SOD levels. *P<0.05 vs. Con;
#P<0.05 vs. HG. Man, mannitol; HG, high glucose; Met,
metformin; ROS, reactive oxygen species; MDA, malondialdehyde; SOD,
superoxide dismutase; Con, control. |
Metformin prevents HG-mediated
downregulation of NDUFA13
To examine the effects of metformin on NDUFA13
expression, immunofluorescence and western blotting assays were
performed. The immunofluorescence results suggested that the
expression of NDUFA13 was significantly decreased in the HG group
compared with the control and mannitol groups, but this effect was
partly reversed by pretreatment with metformin (Fig. 3A and B). The western blotting
results were consistent with the immunofluorescence results
(Fig. 3C and D). Mannitol was used
as the osmotic control and did not mimic the effects of 33.3 mM
glucose (Fig. 3).
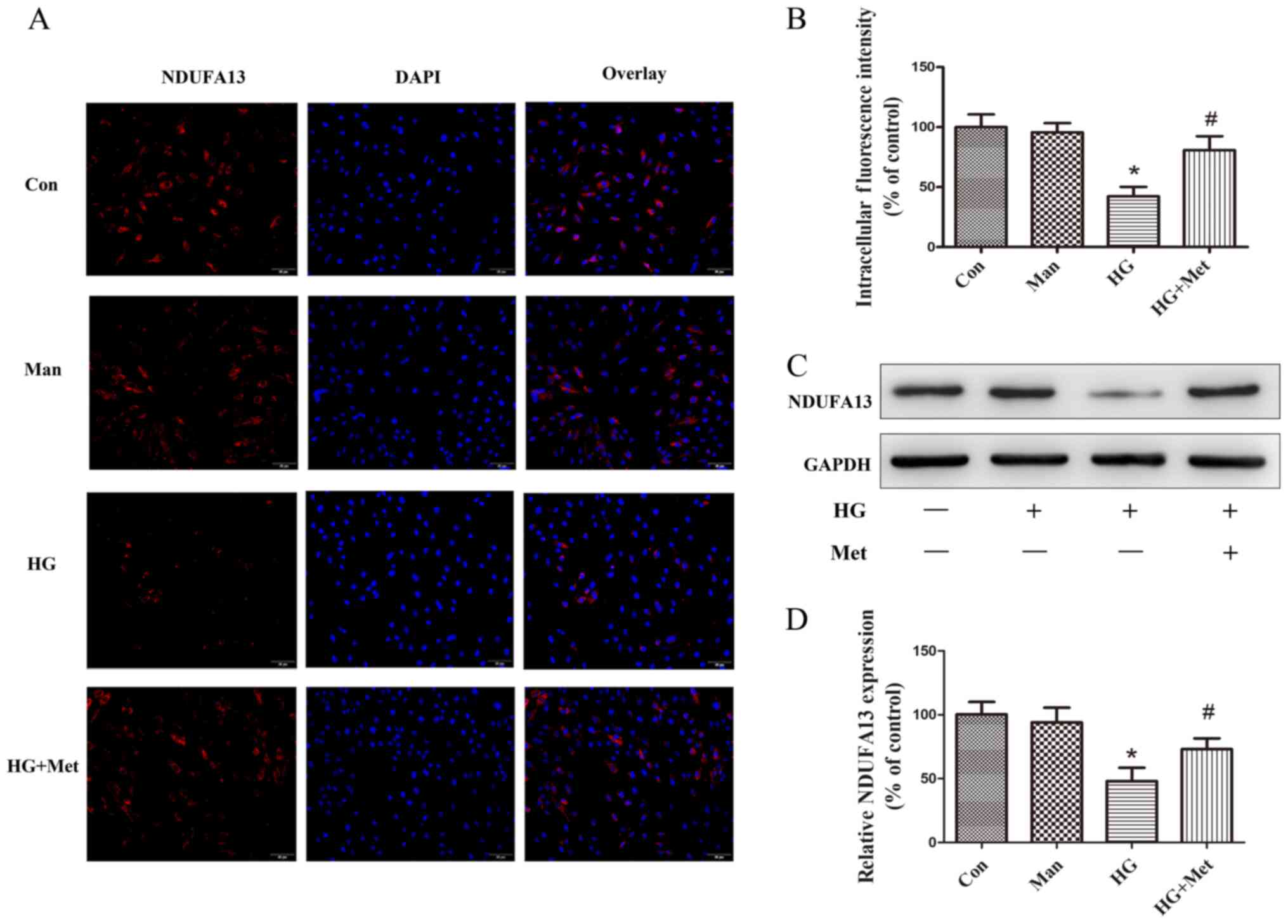 | Figure 3.Effects of metformin (1 mM) on NDUFA13
expression in H9C2 cells under high glucose conditions. Cells were
divided into four groups: i) Con, normal medium; ii) Man, 33.3 mM
Man; iii) HG, 33.3 mM glucose; and iv) HG + Met, 33.3 mM glucose
pretreated with 1 mM Met. NDUFA13 protein expression was (A)
evaluated by immunofluorescence and (B) quantified. Magnification,
×200, Scale bars=50 µm. (C) NDUFA13 protein expression levels were
(C) determined by western blotting and (D) semi-quantified.
*P<0.05 vs. Con; #P<0.05 vs. HG. NDUFA13, NADH:
Ubiquinone oxidoreductase subunit A13; Man, mannitol; HG, high
glucose; Met, metformin; Con, control. |
Metformin activates AMPK and induces
mitochondrial biogenesis
The activity of AMPK was evaluated by detecting the
phosphorylation of Thr-172 (p-AMPK) via western blotting. p-AMPK
expression levels were significantly decreased in the HG group
compared with the control group, but metformin reversed HG-mediated
effects (Fig. 4A and B).
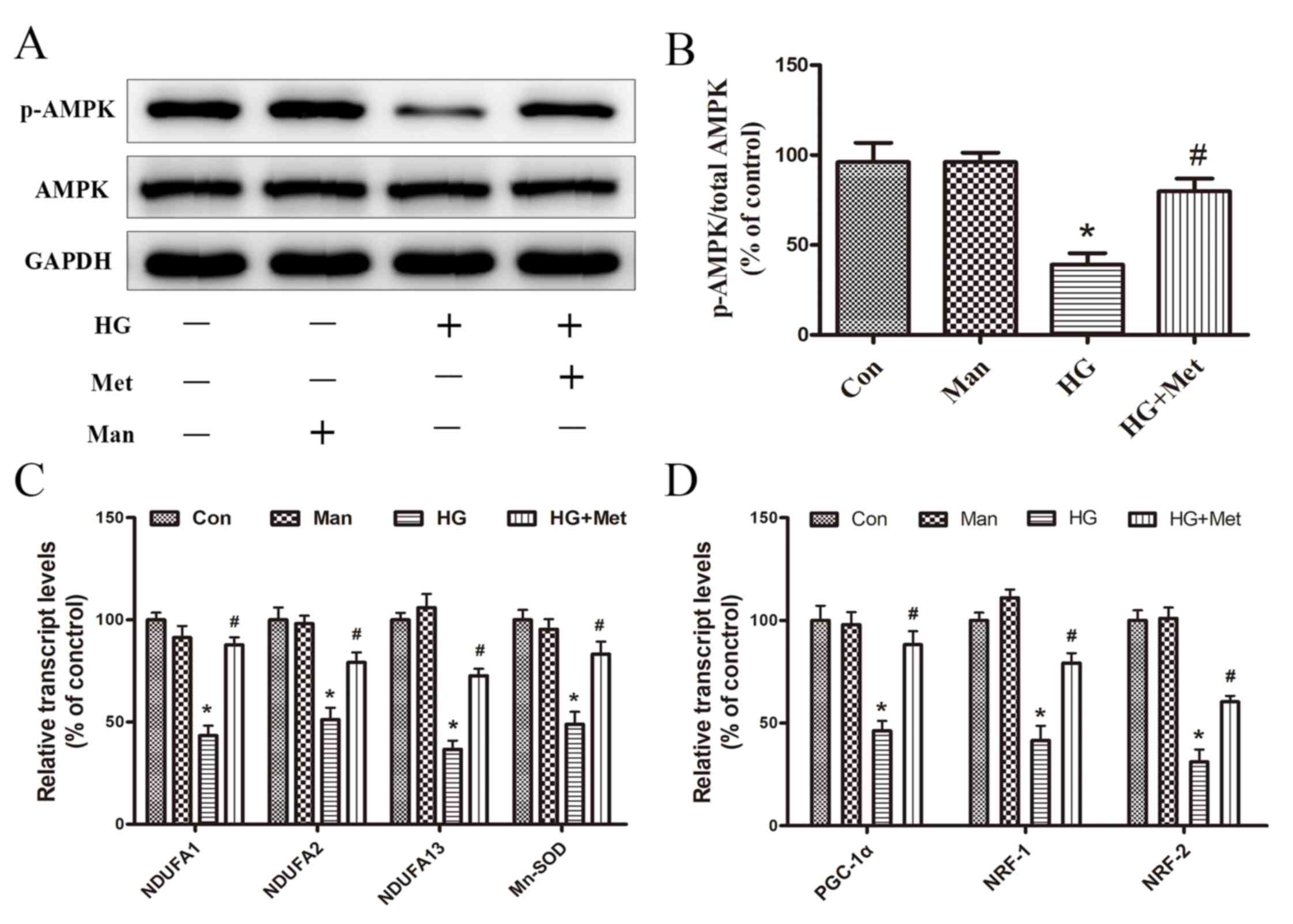 | Figure 4.Effects of metformin (1 mM) on the
expression of AMPK, p-AMPK, and mitochondrial biogenesis genes
under high glucose conditions. Cells were divided into four groups:
i) Con, normal medium; ii) Man, 33.3 mM Man; iii) HG, 33.3 mM
glucose; and iv) HG + Met, 33.3 mM glucose pretreated with 1 mM
Met. (A) Protein expression levels of p-AMPK and AMPK were (A)
determined by western blotting and (B) the ratio of p-AMPK/AMPK was
semi-quantified. mRNA expression levels of (C) NDUFA1, NDUFA2,
NDUFA13, Mn-SOD, (D) PGC-1α, NRF1 and NRF2 were detected via
reverse transcription-quantitative PCR. *P<0.05 vs. Con;
#P<0.05 vs. HG. AMPK, AMP-activated protein kinase;
p, phosphorylated; Man, mannitol; HG, high glucose; Met, metformin;
NDUF, NADH: Ubiquinone oxidoreductase subunit; Mn-SOD, manganese
superoxide dismutase; PGC-1α, peroxisome proliferator-activated
receptor-γ coactivator-1α; NRF, nuclear respiratory factor; Con,
control. |
RT-qPCR was performed to evaluate the effects of
metformin on the mRNA expression levels of mitochondrial genes
(NDUFA1, NDUFA2, NDUFA13 and Mn-SOD) and transcription factors
(PGC-1α, NRF-1 and NRF-2). The results suggested that, compared
with the control group, the expression levels of mitochondrial
genes related to intracellular ROS production were significantly
decreased in the HG group, which was reversed by metformin
(Fig. 4C). Therefore, the results
provided a potential explanation for the ability of metformin to
reduce ROS production. Additionally, the inhibitory effect of HG on
mitochondria biogenesis-related transcription factor expression was
reversed by metformin pretreatment (Fig. 4D), which may be associated with
metformin-mediated promotion of cardiomyocyte survival under high
glucose conditions. Mannitol was used as an osmotic control and did
not mimic the effects of 33.3 mM glucose (Fig. 4).
Compound C reduced the expression of
NDUFA13 and mitochondrial biogenesis via suppressing AMPK
pathway
To examine the dependency of AMPK in
metformin-induced mitochondrial biogenesis and NDUFA13 expression,
a selective AMPK chemical inhibitor (Compound C) was used. The
results indicated that metformin-induced AMPK activation, as
evidenced by an increase in p-AMPK expression levels, was blocked
by pretreatment with Compound C (Fig.
5A and B). Compared with the HG + Met group, mitochondrial gene
and mitochondrial biogenesis-related transcription factor
expression levels were also significantly decreased in the HG + Met
+ Comp C group (Fig. 5D and E). In
addition, metformin-induced NDUFA13 upregulation reversed by
Compound C under HG conditions (Fig.
5A and C). The results suggested that the roles of metformin in
NDUFA13 expression and mitochondrial biogenesis were AMPK signaling
pathway-dependent.
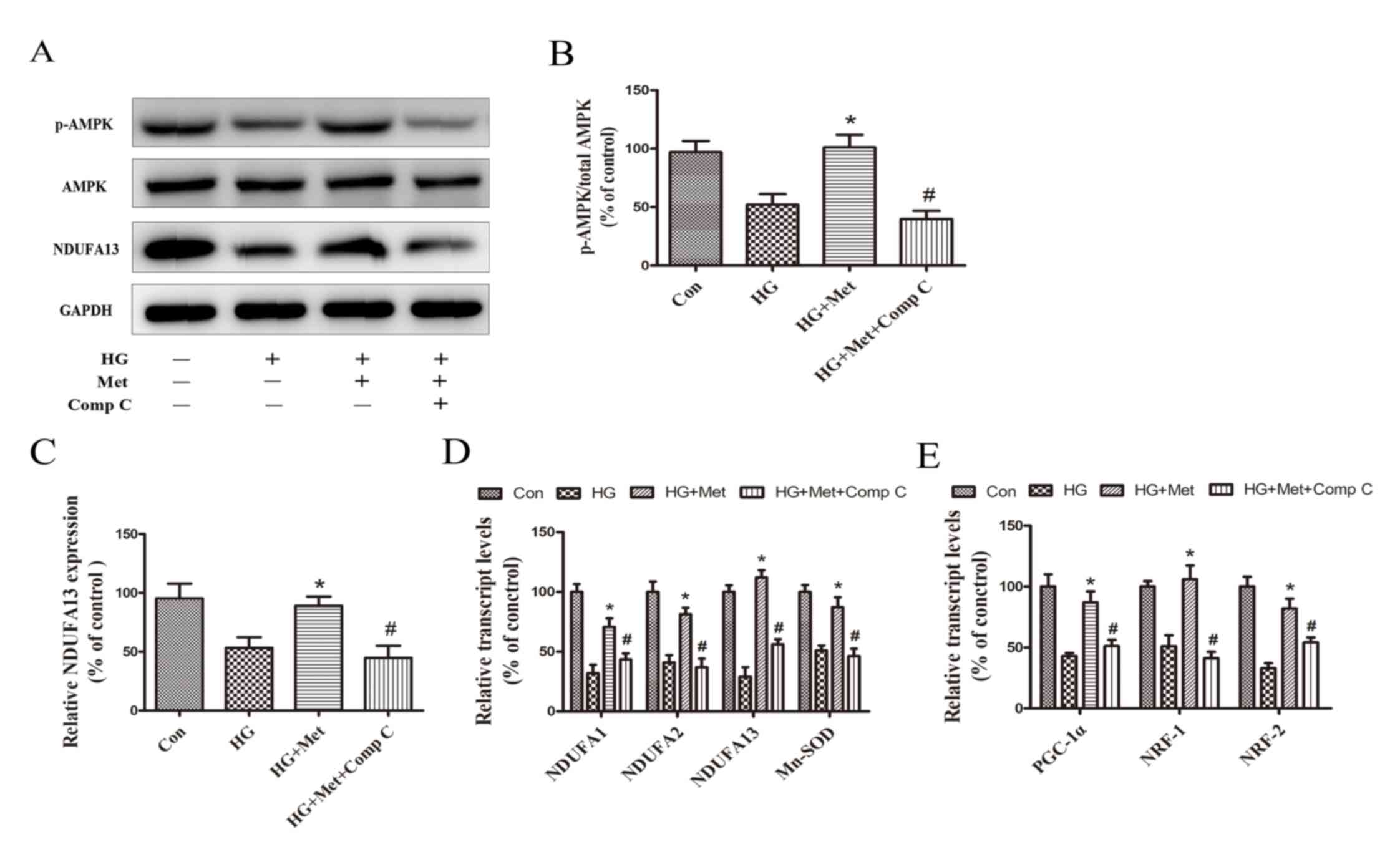 | Figure 5.Role of the AMPK signaling pathway in
the expression NDUFA13 and mitochondrial biogenesis. Cells were
divided into four groups: i) Con, normal medium; ii) HG, 33.3 mM
glucose; iii) HG + Met, 33.3 mM glucose pretreated with 1 mM Met;
and iv) HG + Met + Comp C, 33.3 mM glucose pretreated with 1 mM Met
and Comp C. Protein expression levels were (A) determined by
western blotting and (B) semi-quantified for the ratio of
p-AMPK/AMPK and (C) NDUFA13. mRNA expression levels of (D) NDUFA1,
NDUFA2, NDUFA13, Mn-SOD, (E) PGC-1α, NRF1 and NRF2 were measured
via reverse transcription-quantitative PCR. *P<0.05 vs. HG;
#P<0.05 vs. HG + Met. AMPK, AMP-activated protein
kinase; NDUF, NADH: Ubiquinone oxidoreductase subunit; HG, high
glucose; Met, metformin; Comp C, Compound C; p, phosphorylated;
Mn-SOD, manganese superoxide dismutase; PGC-1α, peroxisome
proliferator-activated receptor-γ coactivator-1α; NRF, nuclear
respiratory factor; Con, control. |
Discussion
In the present study, metformin exerted protective
effects on H9C2 cardiomyocytes by suppressing HG-induced oxidative
stress, as evidenced by ameliorating HG-induced decreases in cell
viability and SOD levels, and increases in ROS and MDA levels. mRNA
expression levels of the mitochondrial genes (NDUFA1, NDUFA2,
NDUFA13 and Mn-SOD) were upregulated by metformin under HG
conditions compared with the HG group. Furthermore, under HG
conditions, the expression levels of the mitochondrial protein
NDUFA13 were increased by metformin compared with the HG group. The
results also indicated that mitochondrial biogenesis-related
transcription factors (PGC-1α, NRF1 and NRF2) were targets of
metformin. The present study identified the role of the AMPK
signaling pathway in the mechanism underlying metformin-mediated
regulation of NDUFA13 and mitochondrial biogenesis based on the
effects of the AMPK inhibitor, Compound C. The results indicated
that metformin protected cardiac cells against HG-induced oxidative
stress via a mechanism involving p-AMPK, NDUFA13 and mitochondrial
biogenesis.
Although the association between mitochondrial
biogenesis and ROS production is not completely understood, it has
been reported that hyperglycemia-induced ROS impairs mitochondrial
biogenesis and mitochondrial function (12,22).
Metformin is an orally administered biguanide that is widely used
to treat type 2 diabetes (4). The
cardioprotective effects of metformin have received increasing
attention and may be related to the ability of metformin to
regulate oxidative stress; however, the exact mechanism is not
completely understood (23).
A previous study identified the role of metformin in
mitochondrial biogenesis, which is an endogenous protective
response to stress (8).
Mitochondrial biogenesis is significantly impaired in
cardiomyocytes from db/db mice, as indicated by reduced
mitochondrial DNA content and mitochondrial respiratory chain
activity (24). Metformin
administration promotes mitochondrial biogenesis and improves
mitochondrial function in the skeletal muscle and human umbilical
vein endothelial cells by activating PGC-1α (25). Activated PGC-1α is a powerful
inducer of NRF-1, NRF-2 and mitochondrial transcription factor A,
which initiates the expression of nuclear and mitochondrial genes
encoding mitochondrial respiratory chain proteins (26). Although the role of metformin in
mitochondrial biogenesis has been investigated, the effects of
metformin in cardiomyocytes have not been fully elucidated.
The results of the current study suggested that
pretreatment with metformin increased the expression of
mitochondrial complex I subcomplexes, including NDUFA1, NDUFA2 and
NDUFA13, under HG conditions. Additionally, the expression levels
of mitochondrial biogenesis-related transcription factors, such as
PGC-1α, NRF-1 and NRF-2, and the antioxidant enzyme, Mn-SOD, were
regulated by metformin under HG conditions. The results suggested
that metformin stimulated cardiomyocyte mitochondrial biogenesis,
and may serve a critical role in cellular defense and cell survival
responses to HG-induced oxidative stress.
The present study investigated the role of AMPK
activation in metformin-mediated effects in cardiomyocytes. AMPK is
a highly conserved sensor of cellular energy homeostasis and is the
most recognized factor that mediates multiple effects of metformin
(27). AMPK activation causes
mitochondrial biogenesis (28).
Mice expressing a dominant-negative form of AMPK in skeletal muscle
are unable to increase mitochondrial biogenesis in response to
energy deprivation (29).
Activated AMPK stimulates PGC-1α, NRF1 and NRF-2, which in turn
promote mitochondrial biogenesis (30). In the present study, metformin
activated AMPK and enhanced the expression of PGC-1α, NRF1 and
NRF-2 under HG conditions compared with the HG group. The
expression of mitochondrial genes NDUFA1, NDUFA2, NDUFA13 and
Mn-SOD was also regulated by metformin. The effects of metformin on
expression were reversed by Compound C; therefore, the results
suggested that AMPK served an important role in promoting
mitochondrial biogenesis and the expression of mitochondrial genes
in response to metformin. Additionally, alternative signaling
pathways involving AMPK, Mn-SOD and PGC-1α, and those independent
of mitochondrial genes should be investigated. Tumor suppressor
p53-mediated PGC-1α upregulation suppresses increased levels of
oxidative stress via nuclear factor (erythroid-derived
2)-like2-mediated expression of Mn-SOD and γ-glutamyl cysteine
ligase without modulating mitochondrial biogenesis (31).
The present study suggested that the expression of
the mitochondrial protein NDUFA13, a newly identified accessory
subunit of mitochondrial complex I, was increased by metformin
under HG conditions. Furthermore, the results indicated that
NDUFA13 was regulated by metformin via an AMPK signal-dependent
pathway, as evidenced by the inhibition of NDUFA13 induction in HG
conditions by Compound C.
NDUFA13 was originally identified as a death
activator in tumor cells and was recognized as an indispensable
subunit of mitochondrial complex I (32). Elimination of NDUFA13 prevents the
assembly and electron transfer activity of complex I, and also
influences other complexes in the mitochondrial respiratory chain
(14). The role of NDUFA13 in H9C2
cardiomyocytes and the relationship among NDUFA13, metformin and
AMPK are not completely understood. However, mutations in the genes
encoding subunits of complex I and complex I increase ROS
generation (33). NDUFA13
downregulation increases basal ROS generation, which may serve as a
survival signal by activating the STAT3/Bcl-2 signaling pathway
(34). In the present study,
NDUFA13 expression levels were decreased in HG-induced H9C2
cardiomyocytes compared with control H9C2 cardiomyocytes.
Therefore, it was hypothesized that reduced expression levels of
NDUFA13 in H9C2 cardiomyocytes were responsible for elevated ROS
production under HG conditions, which may explain the antioxidant
effects of metformin.
Moreover, it was observed that mitochondrial Mn-SOD
was upregulated by metformin under HG conditions. Mn-SOD is a major
antioxidant mitochondrial enzyme that serves as a primary ROS
regulator. Mn-SOD-deficient mitochondria are more susceptible to
oxidative stress and exhibit ultrastructural damage (35). The present study demonstrated that
increased Mn-SOD expression might be involved in the mechanism
underlying metformin-mediated amelioration of oxidative stress.
The present study had several limitations. AMPK
kinase inhibitor Compound C can also inhibit vascular endothelial
growth factor and bone morphogenetic proteins receptors (36,37).
Therefore, further investigations into the effects of AMPK
deficiency, for example, are required to verify the results of the
present study. In addition, only the H9c2 cell line was used in the
present study; therefore, in vivo studies or in vitro
studies involving additional cell lines are required to verify the
conclusions of the present study.
In conclusion, the present study suggested that
metformin protected cardiomyocytes against HG-induced oxidative
stress. The results indicated that metformin-mediated protection
occurred at least in part via promoting mitochondrial biogenesis
and NDUFA13 expression. In addition, the present study indicated
that the AMPK signaling pathway was associated with the mechanisms
underlying metformin-mediated regulation of NDUFA13 and
mitochondrial biogenesis. The results suggested that AMPK may serve
as a potential therapeutic target for normalizing mitochondrial
function in diabetes, and NDUFA13 may serve as a useful target for
designing novel pharmacological approaches to prevent diabetic
complications.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of China (grant no. 81771496), Natural Science
Foundation of China (grant no. 881800331), School of Medicine,
Shanghai Jiao Tong University (grant no. 16XJ21006), Shanghai
Municipal Health Commission (grant no. 201940079) and
Interdisciplinary Program of Shanghai Jiao Tong (grant no.
YG2017QN23).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QYZ, LLW and XDL designed the study. YGL, GYW, YZ,
MLY and YGB performed the experiments. XDL and YGL drafted the
manuscript. MW and XBL helped with the statistical analysis. QYZ,
MW and XBL revised the paper. All authors reviewed the manuscript
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hayat SA, Patel B, Khattar RS and Malik
RA: Diabetic cardiomyopathy: Mechanisms, diagnosis and treatment.
Clin Sci (Lond). 107:539–557. 2004.PubMed/NCBI
|
|
2
|
Cai L and Kang YJ: Oxidative stress and
diabetic cardiomyopathy: A brief review. Cardiovasc Toxicol.
1:181–193. 2001.PubMed/NCBI
|
|
3
|
Khullar M, Al-Shudiefat AA, Ludke A,
Binepal G and Singal PK: Oxidative stress: A key contributor to
diabetic cardiomyopathy. Can J Physiol Pharmacol. 88:233–240.
2010.PubMed/NCBI
|
|
4
|
Sanchez-rangel E and Inzucchi SE:
Metformin: Clinical use in type 2 diabetes. Diabetologia.
60:1586–1593. 2017.PubMed/NCBI
|
|
5
|
Intensive blood-glucose control with
sulphonylureas or insulin compared with conventional treatment and
risk of complications in patients with type 2 diabetes (UKPDS 33).
UK prospective diabetes study (UKPDS) group. Lancet. 352:837–853.
1998.PubMed/NCBI
|
|
6
|
Varjabedian L, Bourji M, Pourafkari L and
Nader ND: Cardioprotection by metformin: Beneficial effects beyond
glucose reduction. Am J Cardiovasc Drugs. 18:181–193.
2018.PubMed/NCBI
|
|
7
|
Calvert JW, Gundewar S, Jha S, Greer JJM,
Bestermann WH, Tian R and Lefer DJ: Acute metformin therapy confers
cardioprotection against myocardial infarction via
AMPK-eNOS-mediated signaling. Diabetes. 57:696–705. 2008.PubMed/NCBI
|
|
8
|
Hu M, Ye P, Liao H, Chen M and Yang F:
Metformin protects H9C2 cardiomyocytes from high-glucose and
hypoxia/reoxygenation injury via inhibition of reactive oxygen
species generation and inflammatory responses: Role of AMPK and
JNK. J Diabetes Res. 2016:29619542016.PubMed/NCBI
|
|
9
|
Kim AS, Miller EJ and Young LH:
AMP-activated protein kinase: A core signalling pathway in the
heart. Acta Physiol (Oxf). 196:37–53. 2009.PubMed/NCBI
|
|
10
|
Paiva MA, Rutter-Locher Z, Gonçalves LM,
Providência LA, Davidson SM, Yellon DM and Mocanu MM: Enhancing
AMPK activation during ischemia protects the diabetic heart against
reperfusion injury. Am J Physiol Heart Circ Physiol.
300:H2123–H2134. 2011.PubMed/NCBI
|
|
11
|
Kusmic C, L'abbate A, Sambuceti G,
Drummond G, Barsanti C, Matteucci M, Cao J, Piccolomini F, Cheng J
and Abraham NG: Improved myocardial perfusion in chronic diabetic
mice by the up-regulation of pLKB1 and AMPK signaling. J Cell
Biochem. 109:1033–1044. 2010.PubMed/NCBI
|
|
12
|
Zheng A, Li H, Xu J, Cao K, Li H, Pu W,
Yang Z, Peng Y, Long J, Liu J and Feng Z: Hydroxytyrosol improves
mitochondrial function and reduces oxidative stress in the brain of
db/db mice: Role of AMP-activated protein kinase activation. Br J
Nutr. 113:1667–1676. 2015.PubMed/NCBI
|
|
13
|
Vakifahmetoglu-norberg H, Ouchida AT and
Norberg E: The role of mitochondria in metabolism and cell death.
Biochem Biophys Res Commun. 482:426–431. 2017.PubMed/NCBI
|
|
14
|
Huang G, Lu H, Hao A, Ng DC, Ponniah S,
Guo K, Lufei C, Zeng Q and Cao X: GRIM-19, a cell death regulatory
protein, is essential for assembly and function of mitochondrial
complex I. Mol Cell Biol. 24:8447–8456. 2004.PubMed/NCBI
|
|
15
|
Li YG, Han BB, Li F, Yu JW, Dong ZF, Niu
GM, Qing YW, Li JB, Wei M and Zhu W: High glucose induces
down-regulated GRIM-19 expression to activate STAT3 signaling and
promote cell proliferation in cell culture. PLoS One.
11:e01536592016.PubMed/NCBI
|
|
16
|
Sazanov LA: Respiratory complex I:
Mechanistic and structural insights provided by the crystal
structure of the hydrophilic domain. Biochemistry. 46:2275–2288.
2007.PubMed/NCBI
|
|
17
|
Zhang Y, Liu X, Zhang L, Li X, Zhou Z,
Jiao L, Shao Y, Li M, Leng B, Zhou Y, et al: Metformin protects
against H2O2-induced cardiomyocyte injury by inhibiting the
miR-1a-3p/GRP94 pathway. Mol Ther Nucleic Acids. 13:189–197.
2018.PubMed/NCBI
|
|
18
|
Chang J, Jung HH, Yang JY, Lee S, Choi J,
Im GJ and Chae SW: Protective effect of metformin against
cisplatin-induced ototoxicity in an auditory cell line. J Assoc Res
Otolaryngol. 15:149–158. 2014.PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Method. 408:402–408. 2001.
|
|
20
|
Foretz M, Guigas B, Bertrand L, Pollak M
and Viollet B: Metformin: From mechanisms of action to therapies.
Cell Metab. 20:953–966. 2014.PubMed/NCBI
|
|
21
|
Dowling RJO, Lam S, Bassi C, Mouaaz S,
Aman A, Kiyota T, Al-Awar R, Goodwin PJ and Stambolic V: Metformin
pharmacokinetics in mouse tumors: Implications for human therapy.
Cell Metab. 23:567–568. 2016.PubMed/NCBI
|
|
22
|
Xie L, Zhu X, Hu Y, Li T, Gao Y, Shi Y and
Tang S: Mitochondrial DNA oxidative damage triggering mitochondrial
dysfunction and apoptosis in high glucose-induced HRECs. Invest
Ophthalmol Vis Sci. 49:4203–4209. 2008.PubMed/NCBI
|
|
23
|
Kukidome D, Nishikawa T, Sonoda K, Imoto
K, Fujisawa K, Yano M, Motoshima H, Taguchi T, Matsumura T and
Araki E: Activation of AMP-activated protein kinase reduces
hyperglycemia-induced mitochondrial reactive oxygen species
production and promotes mitochondrial biogenesis in human umbilical
vein endothelial cells. Diabetes. 55:120–127. 2006.PubMed/NCBI
|
|
24
|
Yan W, Zhang H, Liu P, Wang H, Liu J, Gao
C, Liu Y, Lian K, Yang L, Sun L, et al: Impaired mitochondrial
biogenesis due to dysfunctional adiponectin-AMPK-PGC-1α signaling
contributing to increased vulnerability in diabetic heart. Basic
Res Cardiol. 108:3292013.PubMed/NCBI
|
|
25
|
Karnewar S, Neeli PK, Panuganti D,
Kotagiri S, Mallappa S, Jain N, Jerald MK and Kotamraju S:
Metformin regulates mitochondrial biogenesis and senescence through
AMPK mediated H3K79 methylation: Relevance in age-associated
vascular dysfunction. Biochim Biophys Acta Mol Basis Dis.
1864:1115–1128. 2018.PubMed/NCBI
|
|
26
|
Chen SD, Yang DI, Lin TK, Shaw FZ, Liou CW
and Chuang YC: Roles of oxidative stress, apoptosis, PGC-1α and
mitochondrial biogenesis in cerebral ischemia. Int J Mol Sci.
12:7199–7215. 2011.PubMed/NCBI
|
|
27
|
Adeghate E and Singh J: Structural changes
in the myocardium during diabetes-induced cardiomyopathy. Heart
Fail Rev. 19:15–23. 2014.PubMed/NCBI
|
|
28
|
Nanjaiah H and Vallikannan B: Enhanced
phosphorylation of AMPK by lutein and oxidised lutein that lead to
mitochondrial biogenesis in hyperglycemic HepG2 cells. J Cell
Biochem. 120:15255–15267. 2019.PubMed/NCBI
|
|
29
|
Zong H, Ren JM, Young LH, Pypaert M, Mu J,
Birnbaum MJ and Shulman GI: AMP kinase is required for
mitochondrial biogenesis in skeletal muscle in response to chronic
energy deprivation. Proc Natl Acad Sci USA. 99:15983–15987.
2002.PubMed/NCBI
|
|
30
|
Song P, Kwon Y, Yea K, Moon HY, Yoon JH,
Ghim J, Hyun H, Kim D, Koh A, Berggren PO, et al: Apolipoprotein a1
increases mitochondrial biogenesis through AMP-activated protein
kinase. Cell Signal. 27:1873–1881. 2015.PubMed/NCBI
|
|
31
|
Aquilano K, Baldelli S, Pagliei B, Cannata
SM, Rotilio G and Ciriolo MR: p53 orchestrates the PGC-1α-mediated
antioxidant response upon mild redox and metabolic imbalance.
Antioxid Redox Signal. 18:386–399. 2013.PubMed/NCBI
|
|
32
|
Moreira S, Correia M, Soares P and Máximo
V: GRIM-19 function in cancer development. Mitochondrion.
11:693–699. 2011.PubMed/NCBI
|
|
33
|
Fato R, Bergamini C, Leoni S, Strocchi P
and Lenaz G: Generation of reactive oxygen species by mitochondrial
complex I: Implications in neurodegeneration. Neurochem Res.
33:2487–2501. 2008.PubMed/NCBI
|
|
34
|
Hu H, Nan J, Sun Y, Zhu D, Xiao C, Wang Y,
Zhu L, Wu Y, Zhao J, Wu R, et al: Electron leak from NDUFA13 within
mitochondrial complex I attenuates ischemia-reperfusion injury via
dimerized STAT3. Proc Natl Acad Sci USA. 114:11908–11913.
2017.PubMed/NCBI
|
|
35
|
Williams MD, Van Remmen H, Conrad CC,
Huang TT, Epstein CJ and Richardson A: Increased oxidative damage
is correlated to altered mitochondrial function in heterozygous
manganese superoxide dismutase knockout mice. J Biol Chem.
273:28510–28515. 1998.PubMed/NCBI
|
|
36
|
Yu PB, Hong CC, Sachidanandan C, Babitt
JL, Deng DY, Hoyng SA, Lin HY, Bloch KD and Peterson RT:
Dorsomorphin inhibits BMP signals required for embryogenesis and
iron metabolism. Nat Chem Biol. 4:33–41. 2008.PubMed/NCBI
|
|
37
|
Hao J, Ho JN, Lewis JA, Karim KA, Daniels
RN, Gentry PR, Hopkins CR, Lindsley CW and Hong CC: In vivo
structure-activity relationship study of dorsomorphin analogues
identifies selective VEGF and BMP inhibitors. ACS Chem Biol.
5:245–253. 2010.PubMed/NCBI
|